





Abstract
The incorporation of the paternal genome into the zygote during fertilization requires chromatin remodeling. The maternal haploid (mh) mutation in Drosophila affects this process and leads to the formation of haploid embryos without the paternal genome. mh encodes the Drosophila homolog of SPRTN, a conserved protease essential for resolving DNA–protein cross-linked products. Here we characterize the role of MH in genome maintenance. It is not understood how MH protects the paternal genome during fertilization, particularly in light of our finding that MH is present in both parental pronuclei during zygote formation. We showed that maternal chromosomes in mh mutant embryos experience instabilities in the absence of the paternal genome, which suggests that MH is generally required for chromosome stability during embryogenesis. This is consistent with our finding that MH is abundantly present on chromatin throughout the cell cycle. Remarkably, MH is prominently enriched at the 359-bp satellite repeats during interphase, which becomes unstable without MH. This dynamic localization and specific enrichment of MH at the 359 repeats resemble that of Topoisomerase 2 (Top2), suggesting that MH regulates Top2, possibly as a protease for the resolution of Top2-DNA intermediates. We propose that maternal MH removes proteins specifically enriched on sperm chromatin. In the absence of that function, paternal chromosomes are precipitously lost. This mode of paternal chromatin remodeling is likely conserved and the unique phenotype of the Drosophila mh mutants represents a rare opportunity to gain insights into the process that has been difficult to study.
CHROMATIN is the building block of eukaryotic genomes. The process of chromatin remodeling is essential for a variety of biological processes such as transcription, DNA repair, and replication. Chromatin remodeling at the largest scale likely occurs during animal germline development which produces gametes, and fertilization which gives rise to the zygotes. In many animal species, including flies and humans, the process of spermatogenesis results in the removal of the bulk of the histone molecules and their replacement with highly charged sperm-specific proteins such as protamines in mammals and the MST-HMG-Box proteins in Drosophila (Rathke et al. 2014; Doyen et al. 2015). This process of whole-genome histone removal reverses during fertilization in which maternal histones and other chromosomal proteins are redeposited onto the paternal genome, resulting in similar chromatin states between the two parental genomes. Neither the process of paternal protein removal nor that of maternal protein redeposition is well understood.
Several maternal-effect mutations have been identified in Drosophila melanogaster that impair zygote formation, leading to lethal gynohaploid embryos that develop with only the maternal chromosomes. Mutations of the Drosophila histone chaperone Hira and the CHD1 chromatin remodeling ATPase prevent deposition of the histone variant H3.3 onto paternal chromatin, which is required for male pronucleus formation (Konev et al. 2007; Loppin et al. 2015).
Another gynohaploid mutation called maternal haploid (mh) manifests a unique phenotype that, in embryos from mh homozygous females, even when fertilized with wild-type males, the paternal genome decondenses initially but fails to condense properly in metaphase of the first zygotic mitosis; leading to the formation of chromatin bridges during division. Consequently, the majority of mh embryos arrest development after a few rounds of aberrant divisions producing aneuploid nuclei, but ∼20% of mh embryos develop as gynohaploids (Loppin et al. 2001). Recently, Delabaere et al. (2014) reported that mh1 is a point mutation in the CG9203 gene, which encodes the ortholog of mammalian SPRTN/DVC1, a metalloprotease whose proteolytic activity has recently been demonstrated (Lopez-Mosqueda et al. 2016; Stingele et al. 2016; Vaz et al. 2016).
SPRTN, conserved from Caenorhabditis elegans to humans (Centore et al. 2012; Davis et al. 2012; Ghosal et al. 2012; Mosbech et al. 2012), is characterized by a conserved SprT-like N-terminal domain, which contains a predicted HEXXH protease active-site motif and several C-terminal protein–protein interaction domains (Davis et al. 2012; Ghosal et al. 2012; Mosbech et al. 2012). Earlier studies in human cell models showed that SPRTN binds ubiquitinated PCNA and recruits VCP/p97 protein segregase to stalled replication forks, and suggested that SPRTN plays a crucial role in translesion synthesis (Centore et al. 2012; Davis et al. 2012; Ghosal et al. 2012; Juhasz et al. 2012; Mosbech et al. 2012; Kim et al. 2013). Knocking out SPRTN causes early embryonic lethality in mice, and conditional knockout in mouse embryonic fibroblasts leads to incomplete DNA replication and chromatin bridges (Maskey et al. 2014). Mice with a hypomorphic SPRTN mutation are viable but show progeroid phenotypes (Maskey et al. 2014). Similarly, patients with the Ruijs–Aalfs syndrome were identified with mutations in the SPRTN gene, which causes genome instability, premature aging, and early-onset hepatocellular carcinoma (Lessel et al. 2014). Phenotypes in flies, mice, and humans suggest that SPRTN plays a more general role in replication regulation, independent of translesion synthesis (Lessel et al. 2014; Maskey et al. 2014; Stingele et al. 2016).
It was discovered that yeast Wss1 displays proteolytic activity required for DNA–protein cross-link (DPC) repair (Stingele et al. 2014). Wss1 is distantly related to SPRTN, containing a similar N-terminal metalloprotease domain and C-terminal protein–protein interaction domains (Stingele et al. 2014). Most recently, SPRTN’s protease activity was demonstrated for the first time (Lopez-Mosqueda et al. 2016; Stingele et al. 2016; Vaz et al. 2016). Similar to yeast Wss1, SPRTN shows both autocleavage and substrate-cleavage activity, acting on various DNA-binding substrates in a DNA-dependent and replication-coupled manner (Lopez-Mosqueda et al. 2016; Stingele et al. 2016; Vaz et al. 2016). It was suggested that a defect in DPC repair, which imposes replication stress, is the cause of genome instability due to SPRTN dysfunction (Lopez-Mosqueda et al. 2016; Stingele et al. 2016; Vaz et al. 2016); however, a replication-independent function of SPRTN has not been ruled out (Stingele et al. 2016; Vaz et al. 2016).
Upon identifying MH as the SPRTN ortholog, Delabaere et al. (2014) showed that MH is localized transiently in the decondensing sperm nucleus but not in the female nucleus in meiosis II, and that MH is no longer detectable in either the male or the female nuclei at the onset of the first zygotic S phase, just before pronuclear apposition. This pattern of MH localization in early embryos suggests that MH is specifically required for sperm protein removal yet plays a lesser or no role for the ensuing embryonic cycles. Whether MH plays a role in later stages of embryogenesis was not addressed in their study.
Our interest in mh stemmed from our work characterizing the paternal-effect mutation ms(3)k81 (Gao et al. 2011), which leads to the formation of haploid embryos also missing the paternal genome. In this study, we independently defined the mh1 mutant as a point mutation in CG9203 and newly generated deletion alleles of CG9203 were able to recapitulate the original phenotype. Contrary to the results of Delabaere et al. (2014), we showed that MH protein is localized in both parental pronuclei before the first mitosis of the zygote. Moreover, MH forms prominent foci on the satellite-III block on the X chromosome, a known enrichment site of Topoisomerase 2 (Top2) during interphase in early embryogenesis. We found that embryos lacking MH show defects in maternal chromosome condensation and segregation as well as fragmentation at the DNA level during nuclear cycles of the haploid embryos, suggesting that MH has a functional role in later division cycles in addition to its demonstrated role in the integration of the paternal genome into the zygote. Although Top2 localization in embryonic nuclei lacking MH is generally normal, hypomorphic Top2 mutants interact synergistically with mh mutants in affecting oogenesis, suggesting a functional interaction between the two proteins. Our work confirms the conserved role of SPRTN/MH in genome maintenance but opens an exciting avenue of using Drosophila MH to understand the paternal genome remodeling process during early animal development.
Materials and Methods
Fly stocks
Drosophila stocks were raised on cornmeal medium under standard laboratory conditions at 25°. All stocks were obtained from the Bloomington Drosophila stock center unless noted otherwise. All stocks are described in FlyBase (http://flybase.net) unless noted otherwise. The mutant stocks for Top2 were kindly provided by Dr. Pam Geyer of the University of Iowa, and described in Hohl et al. (2012).
Generating mutations and rescuing constructs for mh
By mobilizing a P element inserted at the 5′ region of mh, we recovered 16 mutant lines that failed to rescue maternal-effect lethality when placed over the original mh1 allele or the mh deficiency Df(1)shtdEPΔ. Diagnostic PCR analyses suggest that all 16 alleles have molecular lesions in the mh region. We chose four alleles to perform detailed molecular characterization by PCR amplifying the affected region, followed by sequencing. Figure 1A provides a schematic representation of the alleles of mh. For the mh6 allele, nts 15,474,352–15,474,733 were deleted (nt designations are based on FlyBase release 5) thus affecting the HEXXH active site. For mh9, nts 15,473,021–15,475,059 were deleted with an addition of 51 nt of filler sequences. This deletion affects almost the entire mh gene. For mh31, nts 15,474,231–15,475,059 were deleted with an addition of 10 nt of filler sequences, also affecting HEXXH. For mh18, nts 15,473,451–15,476,522 were deleted with an addition of 2 nt of filler sequences. Although data used to compile the figures were taken from examining mutants of mh6 and mh9, the other two alleles were similarly examined and displayed similar cellular phenotypes.
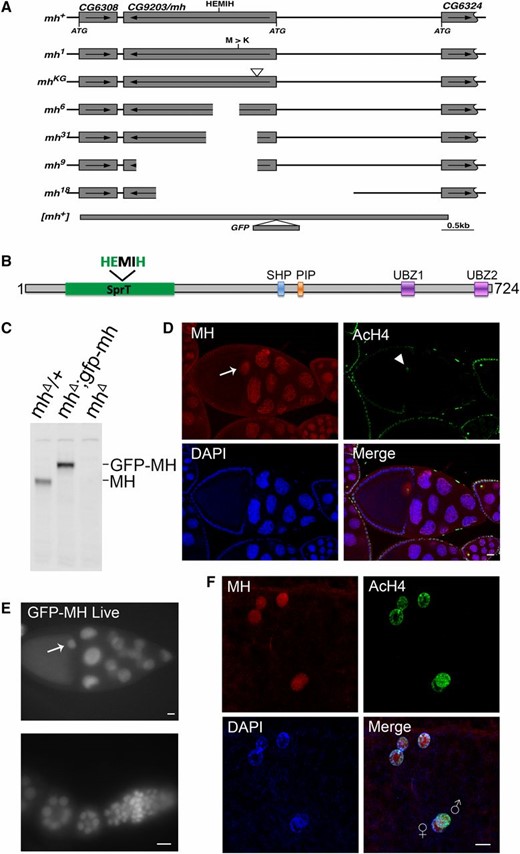
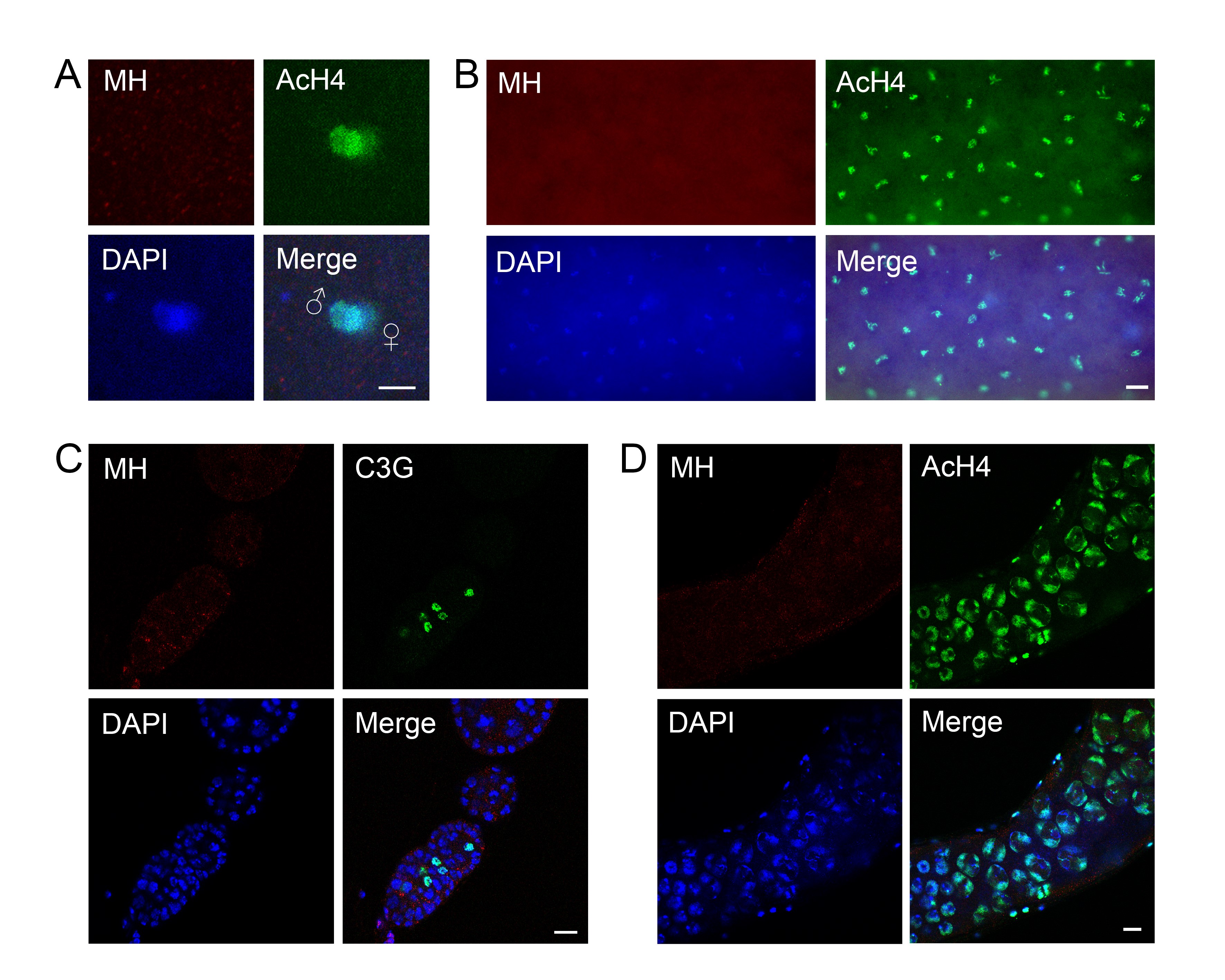
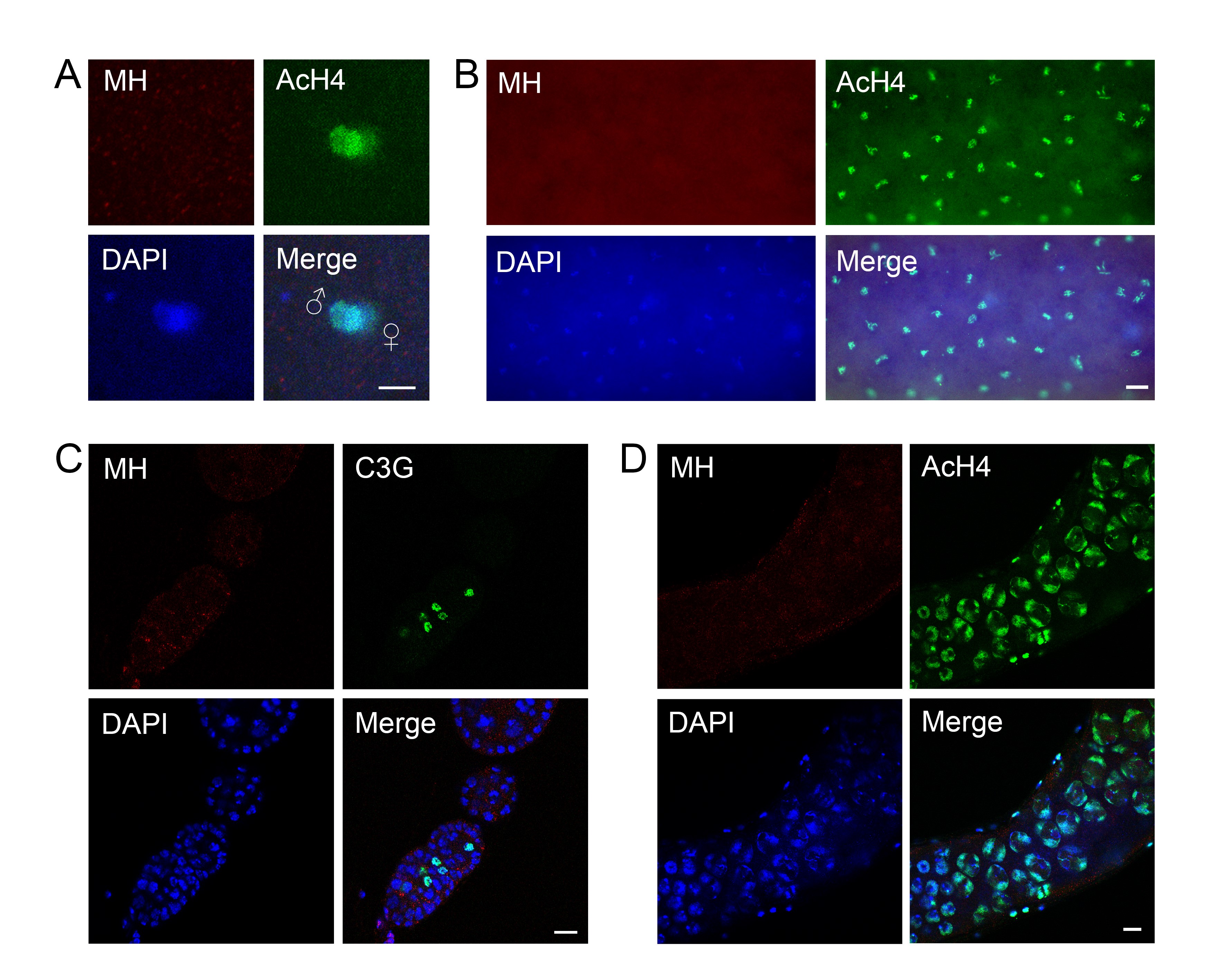
The mh genomic locus and MH protein localization. (A) The genomic structure of the mh locus and mh alleles. The names of the different mh alleles are given to the left of the diagrams. The neighboring genes are also included in the diagram, with the coding region (from the ATG start codon to the translational stop) denoted as rectangular boxes, and the direction of transcription denoted with an arrow. The approximate position of the HEMIH active site is also indicated. In the original mh1 allele, the approximate position of the Met to Arg change is indicated. The approximated insertional position of the P element (KG) used to generate new mh mutations is indicated by an inverted triangle. For the new mh alleles (6, 9, 18, and 31), the approximate extent of the deleted segment is indicated by an empty space. The approximate extent of the rescuing genomic fragment of mh+ is denoted with a thick line and the insertional position of the GFP tag is also indicated. (B) A diagram showing annotated domains of MH based on homology to SPRTN proteins identified in various organisms. (C) Western blot results showing that an anti-MH antibody specifically recognizes the MH protein. Ovary extracts from females with the indicated genotypes were used. The increase in molecular weight of the protein band in the middle lane, when compared with that in the left lane, was due to the fusion of GFP to the MH protein. (D) Protein localization by immunostaining of egg chambers in wild-type ovaries. MH protein is produced in germline cells and deposited in the oocyte nucleus (arrow). The AcH4 is used as another marker for chromatin in addition to DAPI signals. Note that chromatin of the oocyte nucleus is visible with anti-AcH4 staining (arrowhead ). (E) Live GFP signals from egg chambers and a germarium expressing a GFP-tagged MH fusion protein. The GFP signals show essentially identical patterns with those from staining with anti-MH. The oocyte nucleus is marked with an arrow. (F) MH localization in embryos before the first zygotic cycle. MH is present in the nuclei of the polar body (the three nuclei on the top), and in the juxtaposing parental pronuclei in the middle. The male pronucleus is marked with a higher level of AcH4. Bars, 10 μm.
To construct a rescuing construct for mh mutants, an ∼6-kb fragment (nts 15,472,231–15,478,078) was amplified from wild-type genomic DNA followed by sequencing to confirm the lack of mutations created by PCR. This fragment was cloned into the pTV2gw vector (Gao et al. 2009a) and transformed into flies using standard P-element-mediated transformation. This mh fragment was also modified to include an egfp gene fused in frame with, and at the N terminus of, mh (Figure 1A) using the method of recombineering previously described (Gao et al. 2009a). At least two independent lines were used to rescue mh mutants for each of the two constructs. We discovered that maternal lethality caused by the following mutant combinations can all be rescued with the mh genomic fragment: four new mh alleles when homozygous or trans-heterozygous with Df(1)shtdEPΔ, and the original mh1 allele when trans-heterozygous with Df(1)shtdEPΔ. Active site mutations of mh were constructed based on the original mh genomic fragment using site-directed mutagenesis, and introduced similarly to the mh mutant background for rescuing experiments.
To produce MH protein in flies using the Gal4–upstream activation sequence (UAS) system, mh complementary DNA (cDNA) clones with or without the active site mutations were cloned into the pUASp vector and transformed into flies as P elements. These elements were combined with a nos-Gal4 driver and the mh mutations. While expression of the wild-type protein under this setting rescued maternal lethality of mh homozygous mutants, expression of the active site mutants did not.
Mitotic chromosome squash on embryos
Embryos were collected every 2 hr, dechorionated with 50% bleach, washed with embryo wash buffer (0.7% NaCl, 0.05% Triton X-100), permeabilized with octane for 5 min, and rehydrated in embryo wash buffer for 1 min. Rehydrated embryos were treated with 0.05 mM colchicine in 0.7% NaCl for 20 min; followed by 10 min in a hypotonic solution (0.5% sodium citrate); then fixed with 11:11:2 of methanol, acetic acid, and water mix for at least 5 min; and squashed to spread mitotic chromosomes (Gao et al. 2009b). Spread mitotic chromosomes were visualized by DAPI staining.
Antibodies and Western blot
Guinea pig (GP) anti-MH antibody was raised against a His-tagged antigen consisting of residues 387–724 of MH (GenBank sequence AAF48462.1) and affinity purified using the same antigen. The affinity-purified MH antibody was used at 1:2000 on Western blots and 1:1000 in immunostaining experiments. For the preparation of Drosophila ovary extracts, ovaries were dissected in PBS and homogenized in 1× Laemmli buffer (Bio-Rad, Hercules, CA), boiled for 5 min, separated by SDS-PAGE, and subjected to Western blot.
Immunofluorescent staining and FISH
Ovaries were dissected in fresh PBS and fixed with freshly diluted 3.7% formaldehyde in PBS for 20 min at room temperature, and immunofluorescent labeling of proteins was performed following standard protocols (Sullivan et al. 2000). Embryos for immunofluorescent staining and FISH were collected within the desired time point, dechorionated with 50% bleach, washed with embryo wash buffer (0.7% NaCl, 0.05% Triton X-100), fixed with 1:1 freshly diluted 3.7% formaldehyde in PBS and heptane, and devitellinized in 1:1 methanol and heptane following the slow formaldehyde fix method (Sullivan et al. 2000). FISH was performed as previously described (Zhang et al. 2014). The probe for the ribosomal DNA (rDNA) locus was made from plasmid DNA carrying one unit of the rDNA repeats kindly provided by Dr. Igor Dawid of the National Institutes of Health. Following FISH, immunofluorescent staining of MH was performed by following standard protocols (Sullivan et al. 2000). Primers for generating DNA probes are available upon request. The following primary antibodies were used: GP anti-MH (1:1000), rabbit anti-acetylated histone H4 (anti-AcH4) (1:1000, Abcam), rabbit anti-Top2 (1:1000, kindly provided by Dr. Paul Fisher from the State University of New York at Stony Brook), anti-C(3)G (kindly provided by Dr. Mary Lilly of the National Institutes of Health). Fluorescent images were taken with a Carl Zeiss (Thornwood, NY) Axioplan 2 compound microscope or a Carl Zeiss 780 confocal microscope.
Southern blotting
Genomic DNA was purified from embryos with standard phenol extraction and the isopropanol precipitation method. The concentrations of DNA were quantified using Qubit dsDNA HS Assay Kit (Q32854, Invitrogen, Carlsbad, CA) and Qubit Fluorometer. Genomic DNA was treated with or without HaeIII, separated on 1.2% agarose gel, and transferred to positively charged nylon membrane (Amersham Hybond-N+, RPN119B, GE Healthcare Life Sciences) for Southern blot following the manufacturer’s protocol. The probe used for hybridization was labeled and detected using a AlkPhos Direct Labeling and Detection System with CDP-Star (RPN3691GE, Healthcare Life Sciences) following the manufacturer’s protocol. The signal intensity was quantified by the ImageQuant software. The marker used was an Invitrogen 1 Kb Plus DNA Ladder. Primers for generating DNA probes are available upon request.
Data availability
The genetic strains and antibodies generated in this study are available upon request. The authors state that all data necessary for confirming the conclusions presented in the article are represented fully within the article.
Results
mh is a mutation in the CG9203 gene
We mapped the original mh1 mutation within the region uncovered by the chromosomal deficiency Df(1)shtdEPΔ (Tanaka-Matakatsu et al. 2007), which includes 11 annotated genes. Targeted sequencing revealed an ATG to AAG change in the coding region of CG9203, converting a Met to an Arg, along with other base changes that might represent SNPs. Since the mutated Met is highly conserved among CG9203 homologs, we decided to pursue the hypothesis that the mh1 phenotype is due to the loss of CG9203. The Met to Arg change in the mh1 mutant was independently identified by Delabaere et al. (2014).
Using a transposable P element inserted at the 5′ region of the CG9203 locus (P{SUPor-P}CG9203KG05829), we conducted transposase-mediated deletion of the CG9203 locus. We recovered 16 alleles that failed to complement the original mh1 allele. Molecular characterization of four alleles revealed that they have different parts of the CG9203 gene deleted, including the active site motif of HEMIH, thus generating likely null alleles of the gene (mhΔ, Figure 1, A and C, also see Materials and Methods for a detailed description of each of the alleles). Importantly, all of the alleles led to maternal-effect lethality when placed over the chromosomal deficiency Df(1)shtdEPΔ. These results strongly suggest that maternal lethality of mh1 is due to the loss of CG9203.
We also constructed transgenes carrying a genomic fragment encompassing the CG9203 locus (Figure 1A, also see Materials and Methods). When this transgene was introduced into the mh mutant backgrounds (homozygous combinations of mh1 and mhΔ alleles; all trans-heterozygous combinations of mh1, mhΔ, and Df(1)shtdEPΔ), the maternal-effect lethality was prevented; further confirming that the mh phenotype is due to the loss of CG9203. Using the same genomic fragment, we constructed an mh locus N-terminally tagged with an egfp gene (Figure 1A) so that we can monitor the GFP-MH fusion protein inside Drosophila cells. This new construct also rescued the mh phenotype.
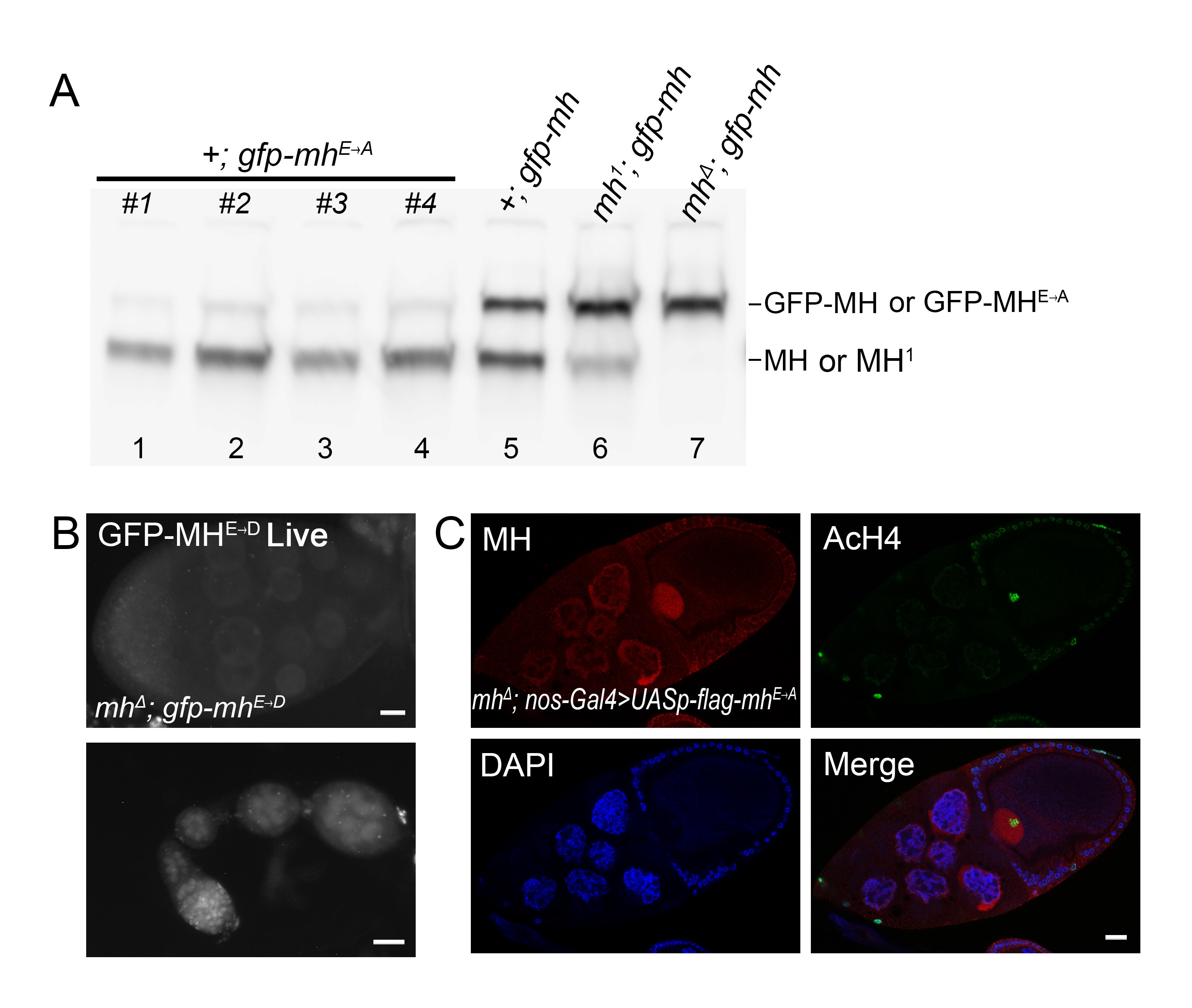
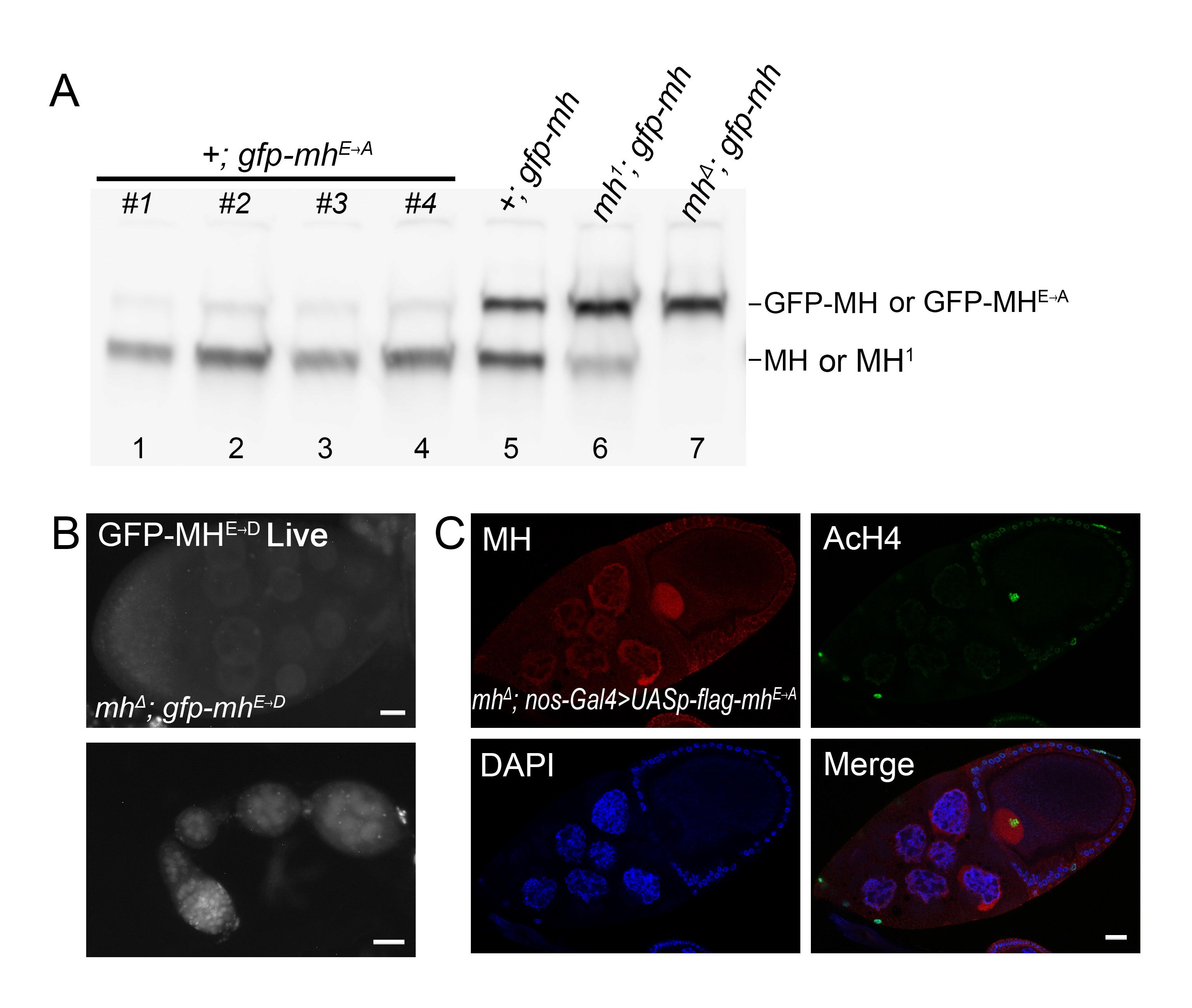
As its mammalian homolog SPRTN, MH has a SprT domain that contains a highly conserved HEXXH metalloprotease motif, in which the glutamic acid serves as the active site for the protease (Figure 1B). We constructed genomic-rescuing fragments with the Glu (E) residue mutated individually to Ala (A), Asp (D), or Gln (Q), and discovered that none of them was able to rescue, suggesting that protease activity is essential for MH function. However, as shown in Supplemental Material, Figure S1A, the E to A mutant protein was produced at a lower level compared with that produced from a wild-type mh transgene (lanes 1–5). By observing GFP fluorescence directly, we also found that the fluorescence of the E to D mutation was also weaker than that of wild-type GFP-MH (compare Figure 1E and Figure S1B). Therefore, the inability of active site mutations to rescue could be at least partly due to insufficient protein expression. A similar protein instability associated with MH active-site mutations was reported by Delabaere et al. (2014). We also attempted to test the effect of overexpressing the mutant protein in the female germline. We used nanos-Gal4 to drive the expression of an mh cDNA either with or without the E to A mutation. While expressing wild-type MH protein was able to rescue female sterility, expressing the mutant protein was not, even though the mutant protein was detectable by immunostaining (Figure S1C).
MH protein is localized in both parental pronuclei before the first mitosis
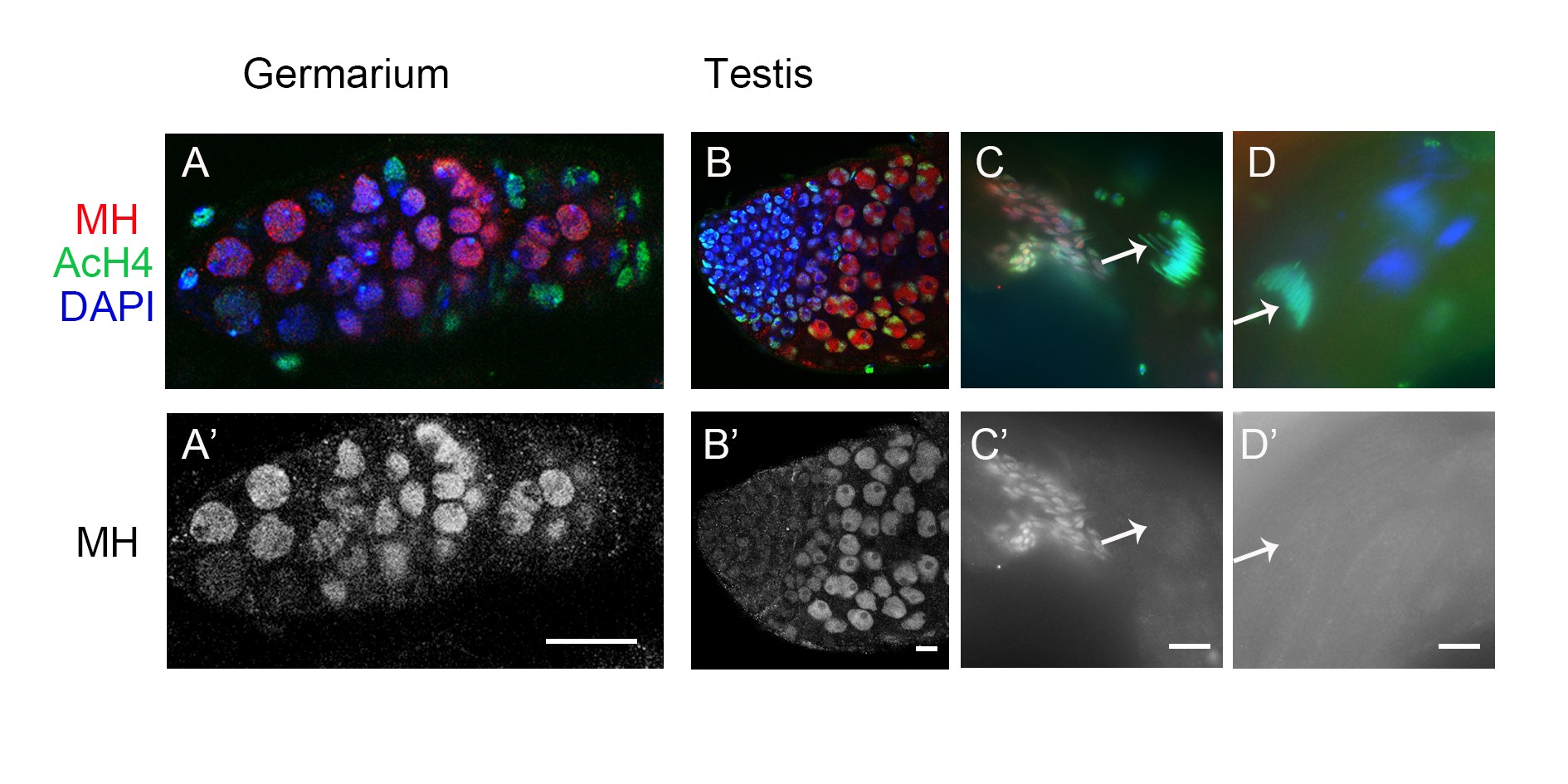
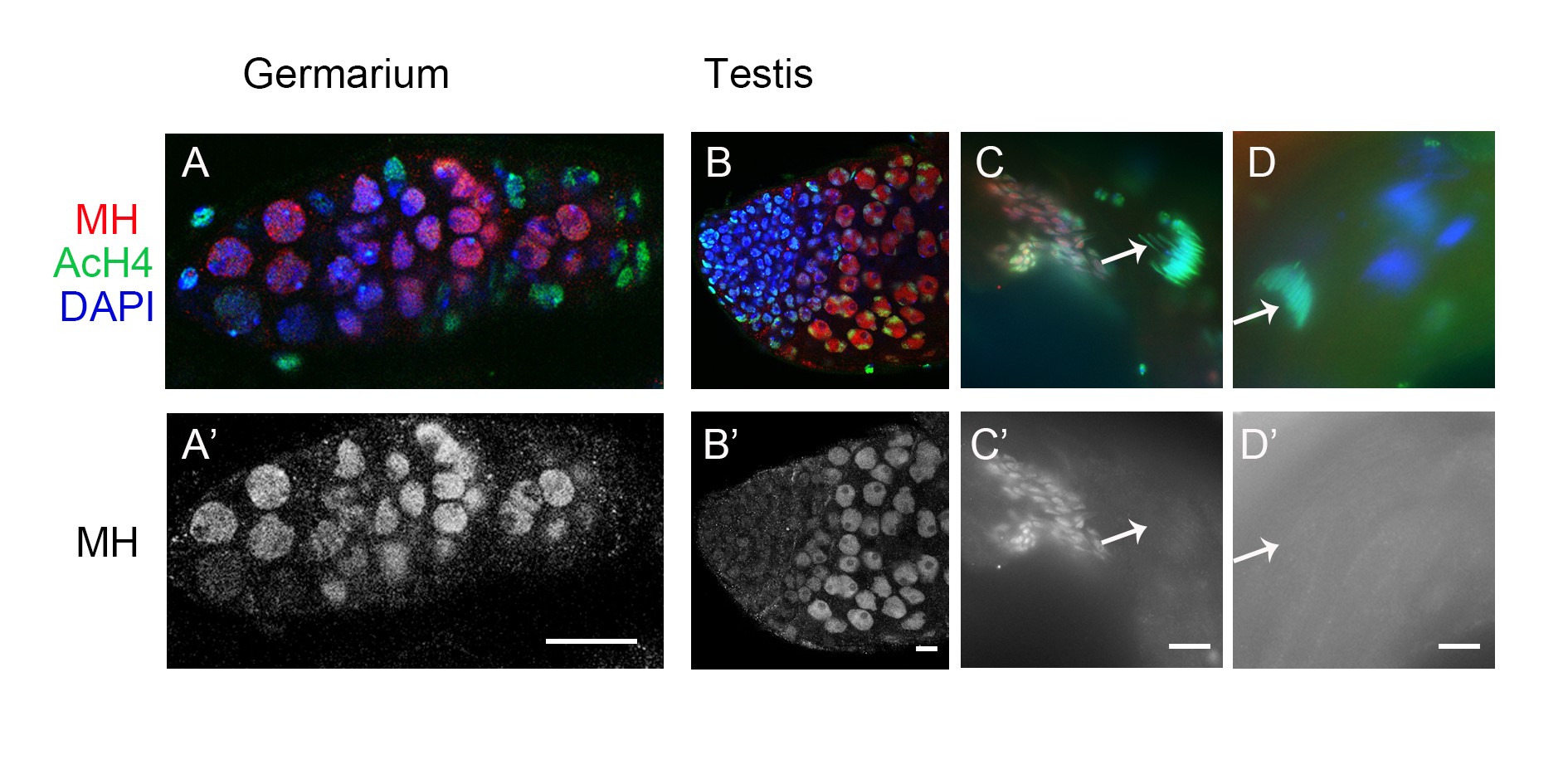
To help elucidate MH function, we generated a polyclonal antibody against the C-terminal part of the MH protein to examine its distribution in different tissues. To verify the specificity of this antibody, we performed Western blot analyses on ovary extracts from wild-type or gfp-mh rescued mh females, with extracts from mh mutant females as a negative control. As expected, the antibody specifically recognized MH and GFP-MH fusion protein (Figure 1C). Immunostaining of wild-type ovaries using this antibody showed that MH is expressed in germline cells but not in somatic follicle cells (Figure 1D and Figure S2). This pattern of MH localization was also confirmed by live GFP signals in the ovaries of gfp-mh females (Figure 1E). In addition, our antibody produced negative staining patterns in various tissues taken from mh mutants as shown in Figure S3, again confirming the specificity of our MH antibody.
The loss of paternal chromosomes during and after the first zygotic mitosis in embryos from mhΔ-homozygous females (here after referred to as mhΔ embryos) suggests that maternally derived MH might be specifically required for the proper behavior of only the paternal genome. Interestingly, staining of embryos collected every 15 min revealed that MH is present in both the maternal and paternal pronuclei before the first zygotic mitosis (Figure 1F, n = 35). When we used AcH4 as a marker to distinguish between the two parental pronuclei, we did not observe an overt difference in the amount of MH in either nucleus. The presence of MH in the maternal pronucleus is not unexpected considering that MH is deposited into the nuclei of oocytes during oogenesis (Figure 1, D and E). Since we did not observe MH in the nuclei of mature sperm, even though it is present in earlier stages of spermatogenesis (Figure S2), maternal MH proteins must have been recruited to paternal chromosomes before the first division.
Our results contradict those of Delabaere et al. (2014), which stated that MH is only present in the decondensing male nucleus but absent in both parental pronuclei before their juxtaposition. One possible cause for the difference in MH-staining results from the two studies is that our antibody was raised against a bacterially expressed fusion protein while Delabaere et al. (2014) used antibodies against peptides, either from MH or the V5 tag. Perhaps our antibodies detected MH epitopes on parental pronuclei that the antibodies used by Delabaere et al. (2014) were not able to recognize. We noted that our antibodies were able to recognize the MH active-site mutant proteins, which are expressed at a lower level than the wild-type protein, on Western blots and in immunostaining experiments (Figure S1); yet the antibodies from Delabaere et al. (2014) could not. MH distribution in later embryonic nuclei was not investigated by Delabaere et al. (2014), so it is not clear whether this discrepancy in staining results was limited to the earliest embryonic cycle.
Loss of MH leads to chromosomal defects of the maternal genome during mitosis
Our results from MH staining of early embryos suggest that differential distribution of MH between the male vs. the female pronuclei cannot explain the specific loss of the paternal genome in mh mutant embryos. In addition, we observed abundant nuclear MH in later stages of embryogenesis (see below), suggesting that MH’s function in maintaining chromosome stability is not limited to the very early divisions. If our hypothesis were correct, we would observe chromosome instability in later cycling nuclei even though they are haploid. Indeed, we observe chromosome bridges in later embryonic divisions (Figure 2A). To further characterize haploid mitosis in mhΔ mutants, we performed mitotic squash on 0–2 hr embryos (syncytium embryos in nuclear division cycles). While some of the mhΔ embryos showed normally condensed and segregated haploid chromosomes (Figure 2C), various degrees of aneuploidy, condensation, and segregation defects of maternal chromosomes were frequently observed (92%, n = 153, examples are shown in Figure 2, D–F), which is suggestive of MH’s function in later division cycles of embryogenesis. As a control for chromosome integrity of haploid embryos, we used those produced by wild-type females fertilized with males harboring the paternal-effect mutation in ms(3)k81. In these ms(3)k81-fathered embryos, the paternal chromosomes are lost due to telomere fusions involving only the paternal genome (Gao et al. 2011). Over 90% of the nuclei (165 out of 182) display chromosomes with normal morphology (examples are shown in Figure 2G). Therefore, chromosome instability of mhΔ embryos is not due to haploidy but the loss of MH functions. This supports our proposition that MH also has somatic functions in genome maintenance during embryonic development.
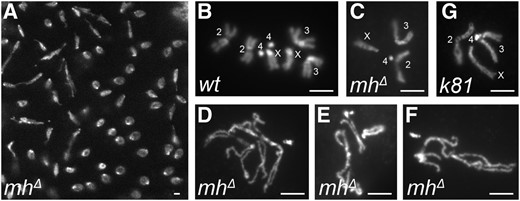
Mitotic chromosome defects in mh-mutant embryos. (A) Asynchronous mitosis and chromosome bridges in an mhΔ embryo. (B) Diploid mitotic chromosomes from a wild-type embryo. (C–F) Mitotic chromosomes from mh mutant embryos. A rare case of a normal-looking haploid genome is shown in (C) with chromosomes labeled. Chromosomes in (D–F) show various degrees of condensation defects and/or aneuploidy. (G) Mitotic chromosomes of a haploid nucleus from an embryo fathered by a ms(3)k81 mutant male. Chromosomes are labeled and appear normal. Bars, 2 μm.
MH protein localizes in the nucleus and forms prominent foci during interphase
To better understand MH’s role in early zygotic cycles, we collected 0- to 2-hr-old embryos for immunostaining to investigate the dynamics of MH localization. Interestingly, MH forms prominent foci in the nuclei during interphase (Figure 3A). In some embryos, there is only one MH focus in each nucleus, while in others there are one or two spots per nucleus. We suspected that these foci are associated with the X chromosome, thereby XY embryos would have one spot in each nucleus, while nuclei in XX embryos would show one or two spots depending on the focal plane and the status of chromosome or chromatid pairing. Interphase MH foci were also observed with live GFP fluorescence from embryos carrying the gfp-mh construct (Figure 3B), confirming results from antibody staining of fixed samples. MH foci disappear as the chromosomes condense in mitotic prophase (Figure 3A). After telophase, MH is recruited back into the nuclei as the embryo enters the next cell cycle. In summary, the localization of MH appears to be spatially and temporally regulated.
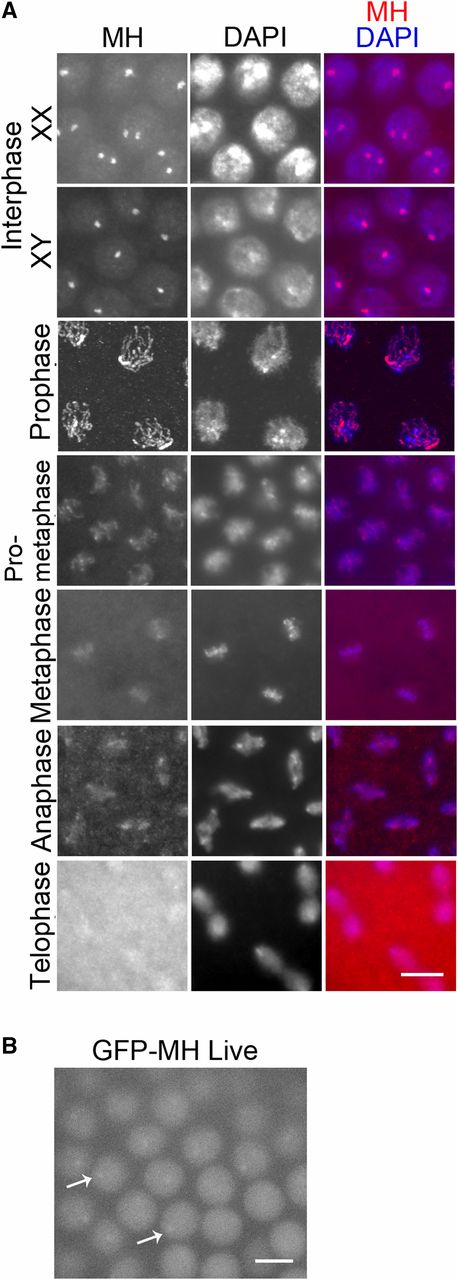
MH localization in wild-type syncytial embryos. (A) Localization of MH by immunostaining. MH protein distributes evenly in the nucleus but forms prominent foci during interphase, with one or two foci in each nucleus in XX embryos and one focus in each nucleus in XY embryos. During the mitotic program, MH is distributed on the condensed chromosomes, except in telophase. (B) Live GFP signal from an embryo expressing GFP-MH. GFP signal appears evenly nuclear but displays one to two foci per nucleus. Examples are indicated by arrows. This is consistent with the appearance of interphase MH foci revealed by immunostaining in (A). Bars, 5 μm.
Interphase MH foci colocalize with the 359-bp satellite
To identify the genomic location of MH foci in interphase, we combined FISH with probes to candidate loci and immunostaining (immuno-FISH) of MH. Based on the large size of these foci, we hypothesized that they represent a large and possibly repetitive locus on the X chromosome. A well-known repetitive locus on the X is the rDNA locus, which also has a copy on the Y chromosome (Figure 4A). In our immuno-FISH experiment using an rDNA probe, we observed two types of staining patterns that we interpreted as being examples for the XY and the XX embryos. In XY embryos, MH foci were adjacent to one of the two rDNA loci; in XX nuclei, MH foci and rDNA loci were always next to each other (Figure 4B). This supports the hypothesis that MH foci are on the X chromosome, but more importantly are in close proximity to the rDNA locus. In the genome of D. melanogaster, adjacent to the rDNA locus on X there is an ∼11-Mbp region called satellite III (Figure 4A), which is comprised of 359-bp, AT-rich repeats (Hsieh and Brutlag 1979b). To test whether MH foci colocalize with satellite III, we used a probe for the 359 repeats in MH immuno-FISH (Shermoen et al. 2010). As shown in Figure 4C, satellite III and MH foci colocalize very well.
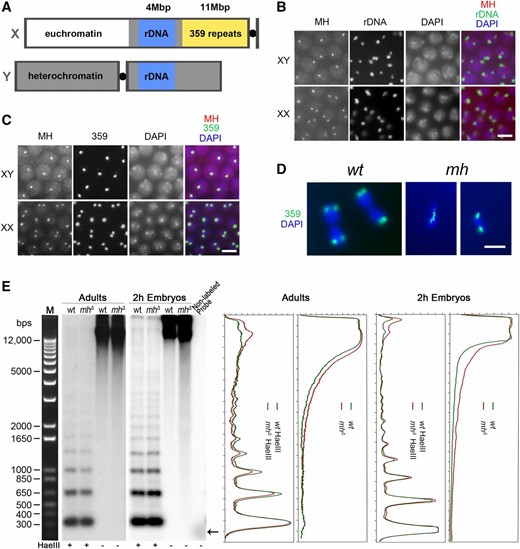
MH maintains the stability of the 359-bp repeat satellite on the X chromosome. (A) A diagram showing the relative position of various genetic elements on the X and Y chromosomes. (B) Immuno-FISH with anti-MH and a probe for the rDNA locus showing that interphase MH foci are always adjacent to the rDNA locus. (C) Immuno-FISH with anti-MH and a probe for the 359-bp repeat locus showing that the two colocalize. (D) DAPI-stained images of embryonic nuclei in anaphase hybridized with a probe to the 359 repeats. In the wild-type nuclei, the sister copies of the 359 repeats have been separated to the two daughter cells. In the two nuclei from mhΔ embryos, one displays a chromosomal bridge with 359 repeats localized on the bridge. The other had the two sister copies separated to the daughter nuclei. (B–D) Bars, 5 μm. (E) Southern blot analysis of genomic DNA using a probe for the 359-bp repeat. The left panels show signals from the Southern blot membranes. The last lane contained the template DNA used to generate the probe for 359 repeats with → denoting the running position of DNA for probe production. The right panels show signal quantification of the membranes. Genomic DNA was taken either from adults or 0–2 hr embryos of the indicated genotypes. HaeIII digestion of genomic DNA is used to show the 359-bp ladders and equal loading of DNA samples. Nondigested genomic DNA was used to detect the presence of fragmented DNA, which appears as lower molecular weight smears on the membrane. wt, wild type.
The 359-bp repeats have been previously shown to play an important role in proper chromosome segregation (Dernburg et al. 1996). In particular, hybrid incompatibility involving the D. melanogaster 359 repeats with the D. simulans genome has been implicated as the cause for embryonic lethality of hybrids between the two species (Ferree and Barbash 2009). We therefore were interested in whether the 359 repeats also experience instability in mh mutant embryos. We performed FISH for the 359 repeats in mh mutant embryos and observed chromosome segregation abnormalities, such as chromosome bridges, in ∼24% of anaphase/telophase nuclei (n = 536) involving the satellite region; suggesting that the repeats experience instability (examples are shown in Figure 4D). We also observed chromosome abnormality that does not seem to involve the 359 repeats (Figure 4D).
To provide additional evidence that the 359 repeats are indeed experiencing instability under the mh mutant background, we used Southern-blot analysis to assay the integrity of the satellite by using genomic DNA purified from embryos and female adults and a fragment from the 359-bp repeat as the probe (Figure 4E). When digested with the HaeIII restriction endonuclease, the satellite III block can be cleaved into a ladder of monomer and multimers (due to sequence variation at the HaeIII cut site) of the 359-bp repeats (Hsieh and Brutlag 1979a). We used HaeIII-digested DNA to control DNA loading (Figure 4E). Genomic DNA (without HaeIII digestion) from 0 to 2 hr mhΔ embryos, when compared with DNA from similarly staged wild-type embryos, shows a significantly more smeary appearance on the membrane (Figure 4E), which indicates the presence of fragmented genomic DNA involving the 359 repeats in mhΔ embryos. Adult genomic DNA without HaeIII digestion has a similar level of fragmented satellite-III sequences in wild type and mhΔ mutants (Figure 4E), consistent with the proposition that the strongest phenotype of loss of MH manifests during embryonic development.
Similar nuclear patterns of Top2 and MH during embryonic mitosis
The striking enrichment of MH on the 359 repeats implies that it is a major site of MH action. Considering that MH potentially acts as a protease, investigating the state of other proteins similarly enriched at the same region could reveal potential MH substrates. Top2, an enzyme essential for releasing DNA topological stress, is known to be enriched at the 359-bp satellite in embryonic interphase (Käs and Laemmli 1992; Ferree and Barbash 2009). In addition, when the transient covalent Top2-DNA intermediates were stabilized in Drosophila cells by chemicals poisonous to Top2 function, regularly spaced Top2 cleavage sites were detectable in the repeated region, generating a cleavage ladder with a periodicity of ∼359 bp (Käs and Laemmli 1992).
As expected, double staining of syncytium embryos with MH and Top2 antibodies showed that interphase MH foci colocalize with Top2 foci (Figure 5A). According to a previous report on live imaging of rhodamine-labeled Top2 injected into embryos (Swedlow et al. 1993), nuclear Top2 increases throughout interphase and reaches a maximum in late interphase, then decreases throughout prophase and anaphase, and reaches a minimum in late telophase. Correspondingly, the cytoplasmic concentration decreases in interphase and increases in mitosis (Swedlow et al. 1993). Our immunostaining results support this report and indicate that the distribution dynamics of Top2 is similar to that of MH throughout the mitotic cycles in embryos. Interestingly, also similar to MH, Top2 is present in both parental pronuclei before the first zygotic division (Figure 5B), suggesting that Top2 may also be involved in formatting the paternal genome during fertilization.
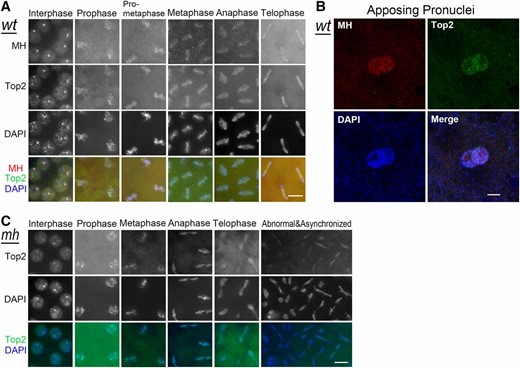
MH and Top2 localization in syncytial embryos. (A) Colocalization of MH and Top2. Immunostaining of wild-type embryos shows that Top2 forms interphase foci that colocalize with MH foci. MH and Top2 also colocalize with each other on chromosomes throughout mitosis. (B) Immunostaining of Top2 in an early embryo, showing that Top2 is present on both parental pronuclei. (C) Top2 localization in mhΔ embryos appears normal. Bars, 5 μm. wt, wild type.
This MH and Top2 colocalization suggests that MH might functionally interact with Top2. To test the simple hypothesis that MH is needed for normal Top2 localization at the 359-bp repeats (either promoting or inhibiting Top2 localization), we performed Top2 immunostaining in mhΔ embryos. However, in these embryos, Top2 localization seems normal (Figure 5C). Notably, Top2 was consistently observed at the lagging chromosomes in anaphase and telophase of both wild-type and mhΔ embryos (Figure 5, A and C), especially in mhΔ embryos when mitosis had become abnormal (Figure 5C). Therefore, MH is not responsible for recruiting Top2 onto chromosomes and Top2 might function upstream of MH, which might not be surprising given the possibility that Top2 could be a substrate of MH (see Discussion).
Mh and Top2 interact genetically to effect oogenesis
It is currently unfeasible to investigate whether Top2 and MH genetically interact in embryos to regulate chromosome stability as Top2 mutant embryos do not initiate zygotic divisions (Hohl et al. 2012; Hughes and Hawley 2014). To provide additional evidence supporting a functional interaction between MH and Top2, we turned our attention to oogenesis where MH is abundantly present and during which Top2’s function has been demonstrated (Hohl et al. 2012; Hughes and Hawley 2014; Mengoli et al. 2014). As reported by Hohl et al. (2012), certain combinations of hypomorphic Top2 mutants are viable and support limited oogenesis. We combined mh null mutations and Top2 hypomorphic ones by genetic crosses, and used daily egg production as a functional readout. As shown in Figure 6A, when female fecundity was followed from day 2 to day 58, the mh single mutation does not affect egg production significantly when compared with the wild-type background. On the other hand, a Top2 single mutation (trans-heterozygous for Top235-5 and Top217-3 alleles) drastically reduces female fecundity. This effect of Top2 mutations on oogenesis was exacerbated by an mh mutation, and the double mutants ceased to produce eggs by day 20. In addition, Top2 and MH act synergistically to control ovary development so that ovaries from the double mutants are visibly smaller than those from either of the single mutants (Figure 6B). These results suggest that the mh mutation dramatically affects oogenesis in a Top2 mutant background but not in the wild-type background, indicative of a functional interaction between the two proteins. However, we cannot be certain that the genetic interaction in the ovary is related to the embryonic colocalization of the two proteins as we did not analyze the ovaries of the double mutants to determine the cellular basis for this genetic interaction.
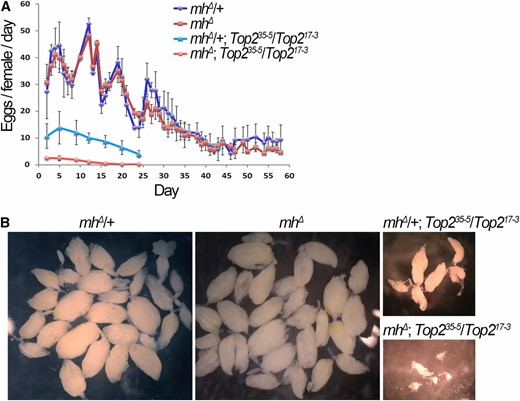
MH and Top2 interact to control ovarian development. (A) mh interacts with Top2 to control female fecundity. Egg production from individual females with the indicated genotypes were quantified until egg laying was exhausted. Double mutants of mh and Top2 have a stronger egg-laying defect than either single mutants. (B) Ovarian sizes in mh and Top2 single and double mutants. Light field images taken at the same magnification show a synergistic effect of mh and Top2 mutations on ovary size. Wild-type and mh single mutant ovaries were dissected from 50-day-old females, while Top2 single mutant and mh, Top2 double mutant ovaries were dissected from 20-day-old females.
Discussion
The mh mutation was recovered as the first Drosophila mutation that produces gynohaploid embryos >40 years ago (Gans et al. 1975). The identification by Delabaere et al. (2014), as well as by us independently in this study, that the mh gene encodes a conserved protease for the resolution of DNA–protein cross-links has shed light on the fascinating process of paternal genome remodeling which MH helps regulate. Embryos lacking MH protein lose their paternal genome during the first zygotic mitosis, hence the paternal chromosomes appear to be more sensitive to the loss of MH than the maternal chromosomes. A thorough understanding of MH function has to provide an explanation for the precipitous loss of the paternal genome. In mhΔ embryos, the paternal pronucleus is formed and appears normally decondensed (Figure S3 and Delabaere et al. 2014), and the replication of paternal DNA is initiated. Therefore, MH is not essential for paternal pronucleus decondensation, a role previously assigned to chromatin remodeling factors such as CHD1 and ISWI (Konev et al. 2007; Doyen et al. 2015). Our immunostaining results clearly showed that MH is localized to both the maternal and paternal pronuclei before the first zygotic division takes place, suggesting that differential distribution of MH proteins between the two genomes must not be the underlying mechanism for the different requirements of MH during zygote formation, as previously suggested (Delabaere et al. 2014). In addition, MH is abundantly present in zygotic nuclei during subsequent divisions. Therefore, we favor the hypothesis that at the molecular level MH carries a similar function during the first and the subsequent embryonic divisions, and that its function is more stringently required for the proper behavior of the paternal genome during fertilization. This could be because the paternal genome, enriched with sperm-specific chromatin proteins, requires more extensive remodeling in which MH participates. This differential requirement, even in the presence of similar levels of MH proteins on the two parental genomes in the wild-type background, results in the precipitous loss of the paternal genome when maternal MH is depleted.
To better elucidate MH’s role in genome maintenance, we focused most of our efforts on characterizing MH during later divisions of embryonic development based on the reasoning that later divisions are more accessible to molecular and cytological analyses. Indeed, we discovered that MH is required for proper chromosomal integrity in later divisions. MH appears to be associated to chromatin through most of the cell cycle. Strikingly, in interphase nuclei, MH is enriched at the largest block of repetitive sequences in the fly genome: the 359 satellite. A better understanding of how MH is recruited to the satellite would improve our understanding of MH’s biological function. We do not favor the hypothesis that this recruitment is based on a specific DNA sequence at the repeats. MH appears on chromatin throughout the genome and its distribution on mitotic chromosomes does not show a specific enrichment (Figure 3, Figure 4, and Figure 5). In addition, in other cells where MH is present, such as those in the ovary, we did not observe a specific enrichment of MH (Figure 1). Instead, we suggest that MH’s enrichment at the satellite is caused by molecular events happening at the satellite during early embryogenesis. We envision that the large block of repetitive sequences creates a challenge for the rapid genome replication during these zygotic divisions, each lasting <20 min. Perhaps replication slowing and/or replication-fork collapse are more frequent at the satellite, which could result in chromatin intermediates that are recognized by MH, and the resolution of these intermediates requires MH. Indeed, in the absence of MH, we observed DNA configurations indicative of breakages at the satellite (Figure 4).
MH is predicted to be a protease associated with chromatin. Recently, this class of proteins has garnered much attention. The yeast WSS1 protein is a DNA-activated protease required for the removal of protein molecules cross-linked with DNA, such as Topoisomerase 1 (Stingele et al. 2014). Most recently, the MH homolog SPRTN has been shown to function similarly to WSS1 in mammalian somatic cells (Lopez-Mosqueda et al. 2016; Stingele et al. 2016; Vaz et al. 2016). Strikingly, we discovered that Top2 colocalizes with MH at the 359 satellite. In fact, the 359 satellite represents a major Top2 cleavage site in vivo. In addition, the prolonged presence of Top2 at the 359 satellite has been proposed as the underlying cause for hybrid lethality involving D. simulans and D. melanogaster (Käs and Laemmli 1992; Ferree and Barbash 2009). Top2 forms a covalent intermediate with its DNA substrate (Nitiss 2009). Therefore, we speculate that Top2 is an MH substrate in Drosophila and that MH’s recruitment to the satellite is to resolve Top2-DNA intermediates. If our model is correct, we would expect to see more abundant Top2 at the satellite in mh mutant embryos, but this was not observed in our immunostaining experiments. It is possible that the increased presence of Top2 at the satellite is too subtle to be detected by immunostaining. We attempted to detect Top2-DNA adducts using anti-Top2 Western blot by purifying genomic DNA from embryos. Unfortunately, the current reagents available to us have not yielded convincing results. Nevertheless, mammalian SPRTN was shown to proteolytically cleave Top2 molecules (Lopez-Mosqueda et al. 2016; Vaz et al. 2016), which lends support to our proposition that Top2 is an MH substrate in Drosophila as well.
We speculate that the primary role of MH when dealing with paternal chromatin remodeling during fertilization is to eliminate proteins that have been specifically attached to paternal DNA. These proteins could be expressed and deposited during spermatogenesis similarly to the MST-HMG-Box proteins. However, one must not overlook the possibility that MH’s substrate(s) on the paternal genome actually comes from the maternal pool of factors that are specifically recruited to the paternal genome to facilitate its unpacking. Some of these maternal factors have to be removed once their mission is complete. Regardless of the source of MH substrates, paternal or maternal, they are likely conserved given that the MH protease is highly conserved and that many of the processes that it regulates are also conserved. Since the loss of SPRTN is lethal in mammals, the unique phenotype of the Drosophila mh mutants ensures that future studies of MH will likely yield exciting insights into the process that guarantees the incorporation of the paternal genome into the zygote.
Acknowledgments
We thank Igor Dawid, Pam Geyer, Mary Lilly, and Paul Fisher for sharing reagents. We thank members of the Rong laboratory for comments on the manuscript. This work was supported by the Intramural Research Program of the National Cancer Institute, and by grants from the National Natural Science Foundation of China (31660331 and 31460299 to J.C. and 31371364 to Y.S.R.).
Footnotes
Supplemental material is available online at www.genetics.org/lookup/suppl/doi:10.1534/genetics.117.200949/-/DC1.
Communicating editor: B. R. Calvi
Literature Cited
Author notes
These authors contributed equally to this work.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}