





Abstract
In eukaryotes, 40S and 60S ribosomal subunits are assembled in the nucleus and exported to the cytoplasm independently of one another. Nuclear export of the 60S requires the adapter protein Nmd3, but no analogous adapter has been identified for the 40S. Ltv1 is a nonessential, nonribosomal protein that is required for 40S subunit biogenesis in yeast. Cells lacking LTV1 grow slowly, are hypersensitive to inhibitors of protein synthesis, and produce about half as many 40S subunits as do wild-type cells. Ltv1 interacts with Crm1, co-sediments in sucrose gradients with 43S/40S subunits, and copurifies with late 43S particles. Here we show that Ltv1 shuttles between nucleus and cytoplasm in a Crm1-dependent manner and that it contains a functional NES that is sufficient to direct the export of an NLS-containing reporter. Small subunit export is reduced in Δltv1 mutants, as judged by the altered distribution of the 5′-ITS1 rRNA and the 40S ribosomal protein RpS3. Finally, we show a genetic interaction between LTV1 and YRB2, a gene that encodes a Ran-GTP-, Crm1-binding protein that facilitates the small subunit export. We propose that Ltv1 functions as one of several possible adapter proteins that link the nuclear export machinery to the small subunit.
THE synthesis of ribosomes in eukaryotes is a complex and evolutionarily conserved process that involves the processing and folding of four rRNAs and the sequential incorporation of >70 ribosomal proteins into two subunits. Ribosome biogenesis has been studied most extensively in the yeast Saccharomyces cerevisiae, where genetic and biochemical studies have identified >170 nonribosomal factors that are required for, or are members of, large complexes that form as intermediates in the assembly process (for review, see Verma et al. 1995; Kressler et al. 1999; Fromont-Racine et al. 2003; Tschochner and Hurt 2003; Granneman and Baserga 2004; Dlakic 2005). These trans-acting factors include RNA exo- and endonucleases, RNA-modification enzymes, helicases, and export factors, as well as a large number of proteins whose functions are as yet unknown. Current models of ribosome assembly suggest that a number of factors important in 40S biogenesis, as well as a subset of early assembling ribosomal proteins, initially assemble cotranscriptionally on the 35S pre-rRNA to form a 90S preribosomal complex. Cleavage of the 35S rRNA in the 90S complex at sites A0, A1, and A2, which requires the U3 small nucleolar RNA, separates the pre-40S precursor from the pre-60S. Most of the accessory factors are released from the pre-40S at this point, which rapidly exits the nucleus in the company of a much smaller number of new factors necessary for export. Final maturation of the 40S subunit, which includes cleavage of the 20S rRNA to yield the mature 18S rRNA, occurs in the cytoplasm. The 60S-processing machinery is recruited after the release of the 40S precursor. The pre-60S subunit undergoes a series of RNA-processing reactions in the nucleolus before being released to the nucleoplasm where some final maturation events are believed to occur. Pre-60S subunits also acquire factors necessary for subunit export in the nucleoplasm prior to exit through the nuclear pore.
Nuclear export of both the small and large ribosomal subunits requires Crm1, the major export karyopherin in yeast, as well as a RanGTPase system and a subset of nucleoporins (Stage-Zimmermann et al. 2000; Gadal et al. 2001; Moy and Silver 2002). Crm1 exports a broad range of substrates, most of which contain a leucine-rich nuclear export sequence (NES), a short motif with a loosely conserved pattern of three or four hydrophobic residues (Wen et al. 1995; Fornerod and Ohno 2002; la Cour et al. 2004). The intrinsically low binding affinity of Crm1 for NES-bearing cargo is greatly enhanced by binding RanGTP (reviewed in Kutay and Guttinger 2005). In yeast, efficient nuclear export of the small but not the large ribosomal subunit requires the RanGTP-binding protein Yrb2, which interacts with Crm1 (Noguchi et al. 1997; Taura et al. 1998; Moy and Silver 2002). The human homolog of Yrb2, RanBP3, has been shown to stabilize Crm1 in complexes with certain NES-containing cargoes (Englmeier et al. 2001; Lindsay et al. 2001). Finally, nuclear export of the large but not the small ribosomal subunit requires the adapter protein Nmd3, which provides the NES for the subunit (Ho et al. 2000; Gadal et al. 2001; Thomas and Kutay 2003; Trotta et al. 2003). No comparable export adapter protein for the small subunit has yet been identified, despite a screen of >900 temperature-sensitive conditional alleles for 40S export-defective mutants (Moy and Silver 2002).
LTV1 encodes a nonessential, nonribosomal protein that is required for 40S subunit biogenesis. We identified Ltv1 initially due to its interaction with the ankyrin-repeat protein Yar1 (Loar et al. 2004). We showed that cells lacking either YAR1 or LTV1 grow slowly, especially at low temperature, are hypersensitive to inhibitors of protein synthesis, and have aberrant polyribosome profiles. Both Δyar1 and Δltv1 cells produce about half as many 40S subunits as do wild-type cells. Ltv1, but not Yar1, co-sediments in sucrose gradients with 43S/40S subunits, but not with 80S monosomes or with polyribosomes, suggesting a transient interaction with the 40S subunit (Loar et al. 2004). This is consistent with previous data showing that Ltv1 copurifies in substoichiometric amounts with the 40S pool of complexes that contain TAP-tagged Enp1, an early associating ribosome biogenesis factor (Schafer et al. 2003).
Here we further examine the role of Ltv1 in small subunit biogenesis. We show that Ltv1 shuttles between nucleus and cytoplasm in a Crm1-dependent manner and that it contains a functional NES that is sufficient to direct the export of a nuclear localization signal (NLS)-containing reporter. Small subunit export is reduced in cells lacking LTV1, as judged by the reduced export of both the 5′-internal transcribed spacer (ITS1) rRNA fragment and the ribosomal protein RpS3. Finally, we show that LTV1 interacts genetically with the gene for the small subunit export factor Yrb2. We propose that Ltv1 functions as one of several possible adapter proteins that link the nuclear export machinery to the small subunit.
MATERIALS AND METHODS
Yeast strains and culture conditions:
All yeast strains used are listed in Table 1. In experiments involving the GAL1 promoter, the glucose in YPD was replaced with 2% galactose or a mixture of glucose and galactose to a combined total concentration of 2%. Yeast transformations were performed using the lithium acetate method (Gietz et al. 1992). Leptomycin B (LMB) was the generous gift of M. Rosbash.
Yeast strains
Strain | Genotype | Source |
|---|---|---|
| LY134 | MATα his3D1 leu2D0 lys2D0 ura3D0 (BY4742) | Open Biosystems |
| LY135 | MATa his3D1 leu2D0 met15D0 ura3D0 Δyar1∷kanR | Open Biosystems |
| LY136 | MATα his3D1 leu2D0 lys2D0 ura3D0 Δltv1∷kanR | Open Biosystems |
| BY4743 | MATa/MATα his3D1/his3D1, leu2D0/leu2D0, lys2D0/LYS2, MET15/met15D0, ura3D0/ura3D0 | Open Biosystems |
| LY139 | MATa/MATα his3D1/his3D1 leu2D0/leu2D0 lys2D0/LYS2 MET15/met15D0 ura3D0/ura3D0 Δltv1∷kanR/Δltv1∷kanR | Open Biosystems |
| PSY2070 | MATa Δyrb2∷HIS3 ura3-52 leu2Δ1 trp1 his3Δ200 | P. Silver |
| MNY8 | MATa leu2-3,112 trp1-1 ura3-1 ade2-1 his3-11,15 can1-100 Δcrm1∷kanR pDC-CRM1T539C (LEU2/CEN) | M. Rosbash |
| LY181 | MATa Δyrb2∷HIS3, Δltv1∷kanR, his3- leu2- ura3- met? trp? | This study |
Strain | Genotype | Source |
|---|---|---|
| LY134 | MATα his3D1 leu2D0 lys2D0 ura3D0 (BY4742) | Open Biosystems |
| LY135 | MATa his3D1 leu2D0 met15D0 ura3D0 Δyar1∷kanR | Open Biosystems |
| LY136 | MATα his3D1 leu2D0 lys2D0 ura3D0 Δltv1∷kanR | Open Biosystems |
| BY4743 | MATa/MATα his3D1/his3D1, leu2D0/leu2D0, lys2D0/LYS2, MET15/met15D0, ura3D0/ura3D0 | Open Biosystems |
| LY139 | MATa/MATα his3D1/his3D1 leu2D0/leu2D0 lys2D0/LYS2 MET15/met15D0 ura3D0/ura3D0 Δltv1∷kanR/Δltv1∷kanR | Open Biosystems |
| PSY2070 | MATa Δyrb2∷HIS3 ura3-52 leu2Δ1 trp1 his3Δ200 | P. Silver |
| MNY8 | MATa leu2-3,112 trp1-1 ura3-1 ade2-1 his3-11,15 can1-100 Δcrm1∷kanR pDC-CRM1T539C (LEU2/CEN) | M. Rosbash |
| LY181 | MATa Δyrb2∷HIS3, Δltv1∷kanR, his3- leu2- ura3- met? trp? | This study |
Yeast strains
Strain | Genotype | Source |
|---|---|---|
| LY134 | MATα his3D1 leu2D0 lys2D0 ura3D0 (BY4742) | Open Biosystems |
| LY135 | MATa his3D1 leu2D0 met15D0 ura3D0 Δyar1∷kanR | Open Biosystems |
| LY136 | MATα his3D1 leu2D0 lys2D0 ura3D0 Δltv1∷kanR | Open Biosystems |
| BY4743 | MATa/MATα his3D1/his3D1, leu2D0/leu2D0, lys2D0/LYS2, MET15/met15D0, ura3D0/ura3D0 | Open Biosystems |
| LY139 | MATa/MATα his3D1/his3D1 leu2D0/leu2D0 lys2D0/LYS2 MET15/met15D0 ura3D0/ura3D0 Δltv1∷kanR/Δltv1∷kanR | Open Biosystems |
| PSY2070 | MATa Δyrb2∷HIS3 ura3-52 leu2Δ1 trp1 his3Δ200 | P. Silver |
| MNY8 | MATa leu2-3,112 trp1-1 ura3-1 ade2-1 his3-11,15 can1-100 Δcrm1∷kanR pDC-CRM1T539C (LEU2/CEN) | M. Rosbash |
| LY181 | MATa Δyrb2∷HIS3, Δltv1∷kanR, his3- leu2- ura3- met? trp? | This study |
Strain | Genotype | Source |
|---|---|---|
| LY134 | MATα his3D1 leu2D0 lys2D0 ura3D0 (BY4742) | Open Biosystems |
| LY135 | MATa his3D1 leu2D0 met15D0 ura3D0 Δyar1∷kanR | Open Biosystems |
| LY136 | MATα his3D1 leu2D0 lys2D0 ura3D0 Δltv1∷kanR | Open Biosystems |
| BY4743 | MATa/MATα his3D1/his3D1, leu2D0/leu2D0, lys2D0/LYS2, MET15/met15D0, ura3D0/ura3D0 | Open Biosystems |
| LY139 | MATa/MATα his3D1/his3D1 leu2D0/leu2D0 lys2D0/LYS2 MET15/met15D0 ura3D0/ura3D0 Δltv1∷kanR/Δltv1∷kanR | Open Biosystems |
| PSY2070 | MATa Δyrb2∷HIS3 ura3-52 leu2Δ1 trp1 his3Δ200 | P. Silver |
| MNY8 | MATa leu2-3,112 trp1-1 ura3-1 ade2-1 his3-11,15 can1-100 Δcrm1∷kanR pDC-CRM1T539C (LEU2/CEN) | M. Rosbash |
| LY181 | MATa Δyrb2∷HIS3, Δltv1∷kanR, his3- leu2- ura3- met? trp? | This study |
Plasmid construction:
pNLSSV40-NESPKI-GFP2 (pPS1372) and pNLSSV40-GFP-lacZ (pPS815) were both gifts of P. Silver. pYAR1-GFP (Ld46), pLTV1-GFP (Ld47), and pRPS3-GFP (Ld50) were each constructed by PCR amplification of the yeast ORF from genomic DNA using gene-specific primers with 5′ tails that included specific restriction enzyme recognition sites. Each PCR product was cleaved with the appropriate restriction enzymes and ligated into the BamHI and XbaI sites for YAR1, the BamHI and SmaI sites for LTV1, and the BamHI and ClaI sites for RPS3 in the MCS of pUG23 (generous gift of J. H. Hegemann). pNLSSV40NESLTV1-GFP2 (Ld61) was constructed by gapped plasmid repair using in vivo homologous recombination (Ma et al. 1987). pPS1372 was linearized with EcoRI and transformed into yeast along with a 200-bp PCR product containing the 12 aa putative LTV1 NES. The LTV1 NES was amplified using a forward primer that contained 39 bp homologous to the NLS of pPS1372 plus 21 bp of LTV1 sequence just upstream of the NES and a reverse primer that contained 40 bp of sequence homologous to the green fluorescent protein (GFP) gene in pPS1372 and 21 bp of LTV1 sequence just downstream of the NES. Plasmid DNA was rescued from yeast transformants and amplified in bacteria, and the correct structure of the construct was confirmed by DNA sequencing.
Northern blotting:
Wild-type and isogenic deletion strains were grown to a density of 0.4 OD660/ml in YPD medium. Total RNA was extracted using glass bead homogenization in the presence of TRIZOL reagent (Invitrogen), and then precipitated overnight in isopropanol at −20° before drying and resuspending in nuclease-free H2O. A total of 10 μg of total RNA from each strain was subjected to electrophoresis in a 6% formaldehyde/1% agarose gel prepared in MOPS buffer and then transferred by capillary blotting to nylon membranes (Amersham Biosciences). Blots were probed with [32P]ATP end-labeled oligonucleotide DNA probes complementary to select rRNA sequences (described in Kressler et al. 1997), and hybridization signals were detected and analyzed using a Typhoon imaging system (Amersham Biosciences).
Ribosome sedimentation and protein analysis:
Yeast strains were grown in YPD at 30° to a density of 0.5 OD660/ml. At time of harvest, cultures were incubated with 100 μm cycloheximide for 15 min to interrupt protein translation. Cell homogenates were prepared and ribosomal complexes were isolated on sucrose gradients as previously described (Loar et al. 2004). The protein distribution in sucrose gradient fractions was assessed by immunoblot using antibodies directed against green fluorescent protein (Invitrogen) or the yeast ribosomal proteins RpS2 and RpL3 (gift of J. Warner). After transfer to nitrocellulose membranes, individual proteins were detected using the above antibodies at 1:2000 dilution, HRP-coupled secondary antibodies at 1:20,000 dilution, and chemiluminescence detection reagent (Perkin-Elmer Life Sciences).
Fluorescent microscopy of living cells:
To visualize GFP fluorescence in living cells, strains carrying GFP vectors were grown in the appropriate selective media to 0.05–0.5 OD660/ml. Two milliliters of culture was harvested by centrifugation, washed twice in water, and resuspended in 50 μl of water. To stain living cells with DAPI, ∼2 ml of cells was harvested by centrifugation, washed in 1× phosphate-buffered saline (PBS) (13.7 mm NaCl, 0.27 mm Kcl, 0.43 mm Na2HPO4 · 7H2O, 0.4 mm KH2PO4), and resuspended in 200 μl 1× PBS. DAPI was added to a final concentration of 2.5 μg/ml and cells were incubated at room temperature for 60 min. Cells were again harvested by centrifugation, washed twice in water, and resuspended in 50 μl of water. Cells were then mounted on a polylysine-coated microscope slide and viewed with the ×100 objective under oil immersion using either the FITC or the DAPI channels of a Zeiss Axioskop fluorescence microscope. Images were recorded using either a SPOT CCD camera and image capture software (Insight) or a CoolSnap camera and software (RS Photometrics). Digital images were processed using Photoshop (Adobe).
In situ hybridization:
Wild-type and mutant yeast strains were grown to stationary phase at 30° in YPD and then diluted 10-fold into fresh YPD and incubated for 3 hr. Cells were fixed in 4% formaldehyde for 30 min and then harvested and washed in buffer B (0.1 m KPO4, 1.2 m sorbitol). Spheroplasts were generated by incubating the cells with 2 mg/ml lyticase (Sigma) in the presence of ribonuclease inhibitors (Sigma) in a volume of 500 μl buffer B. Permeabilized cells were adhered to polylysine-coated glass coverslips. In situ hy-bridization of the 5′-ITS1 probe was conducted after the method of Amberg et al. (1992). A Cy3-labeled oligonucleotide complementary to a portion of the 5′-ITS1 rRNA was constructed and used in the hybridization as previously described (Moy and Silver 2002). Following hybridization and washing, the coverslips were counterstained with 5 μg/ml DAPI and mounted onto slides. Individual cells were viewed under the ×100 objective of an Olympus BX51 epifluorescence microscope using the DAPI and rhodamine fluorescence channels. Images were collected with a Qcolor camera system (Olympus) and postprocessed using Photoshop (Adobe).
RESULTS
Mutants lacking YAR1 or LTV1 exhibit aberrant rRNA processing:
We have previously shown that mutants lacking LTV1 or YAR1 are both stress sensitive and defective in ribosome biogenesis; these mutants produce about half as many 40S subunits as do wild-type cells (Loar et al. 2004). As ribosome biogenesis mutants often exhibit defects in rRNA processing, we asked whether rRNA processing was altered in either Δyar1 or Δltv1 cells. Northern blot analysis of the steady-state levels of rRNA in wild-type or mutant cells reveals several aberrant rRNA precursors in both mutants (Figure 1). In wild-type cells, the 18S rRNA is generated by cleavage of the 35S precursor first at sites A0 and A1, followed by cleavage at site A2. The 20S rRNA thus generated is converted to the mature 18S in the cytoplasm by cleavage at site D. In Δyar1 mutants, there is a significant accumulation of both 20S and 21S rRNA precursors, as well as a reduction in the 27SA2 precursor, indicative of a reduction in the rate of cleavage at sites A2 and D in this mutant (Figure 1). In Δltv1 mutants, there is somewhat less 20S than in Δyar1, but the 21S and 23S precursors are strikingly abundant, 35S and 32S precursors are apparent, and there is a reduction in the 27SA2 intermediate. Both the accumulation of the 21S, 23S, and 32S rRNAs detected by probe 4 and the reduction in the 27SA2 species can be explained by an inhibition of the A2 cleavage (Figure 1). In addition, accumulation of the 23S and the 35S precursors suggests that cleavage at sites A0 and A1 are also inhibited in Δltv1. We note here that an aberrant RNA species previously identified in Δltv1 cells as 20S, on the basis of size, is likely identical to the 21S band that we describe here (Ihmels et al. 2002). In summary, cleavage at site A2 appears to be slowed in both Δyar1 and Δltv1 cells, and cleavage at sites A0–A1 is additionally reduced in Δltv1 cells.
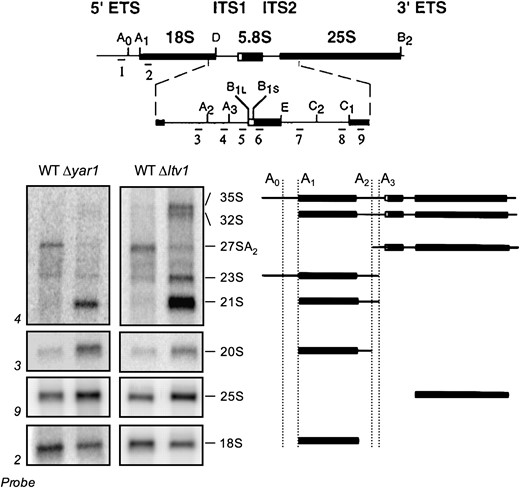
Ribosomal RNA-processing defects in Δyar1 and Δltv1 strains. Wild-type parental strain (LY134) and isogenic deletion strains Δyar1 (LY135) and Δltv1 (LY136) were grown at 30° in YPD medium. Following total RNA extraction, 10 μg of RNA from each strain was subjected to formaldehyde–agarose gel electrophoresis and processed for Northern blotting. Blots were probed with the 32P-labeled oligonucleotide probes depicted at the top of the figure (adapted from Kressler et al. 1997). The sizes of the various rRNA intermediate species are indicated. A schematic of the structure of these intermediates is shown at the right.
Neither Yar1 nor Ltv1 are nucleolar-localized proteins:
Since cells lacking Ltv1 or Yar1 exhibit an rRNA-processing defect, we tested whether either of these proteins were nucleolar localized. The SSU processome, which is required for processing the pre-rRNA at sites A0, A1, and A2 is found in the nucleolus (Dragon et al. 2002; Bernstein et al. 2004), as are other nonribosomal proteins that are necessary for the A0–A2 cleavages (Bousquet-Antonelli et al. 2000). To determine the cellular location of Yar1 and Ltv1 in vivo, we tagged each gene with GFP and expressed the fusion proteins from a low-copy vector under the control of a Met-repressible promoter. We confirmed that each of the GFP-tagged proteins is functional by expressing the GFP fusion protein in strains lacking the corresponding wild-type gene. Even in growth conditions that permit only low levels of expression (Mumberg et al. 1994), we found that pLTV1-GFP was able to completely suppress the slow-growth phenotype of Δltv1 cells at both 30° and 18° (Figure 2). This experiment also shows that ectopic expression of Ltv1-GFP is not deleterious in wild-type cells expressing the endogenous LTV1 gene. Yar1-GFP is also able to complement the slow-growth phenotype of Δyar1 cells (data not shown). Microscopic analysis of cells (both LTV1 and Δltv1) expressing low levels of Ltv1-GFP revealed a primarily cytosolic distribution of GFP (Figure 3 and data not shown). We observed GFP fluorescence over both the nucleus and the cytoplasm, the vacuole being the only organelle from which fluorescence was excluded (Figure 3A, top right; see also Huh et al. 2003). A similar distribution of Yar1-GFP fluorescence is observed (Figure 3A, top center). Thus neither Yar1 nor Ltv1 exhibit the nucleolar-localized phenotype expected of a protein with a direct role in early pre-rRNA processing.
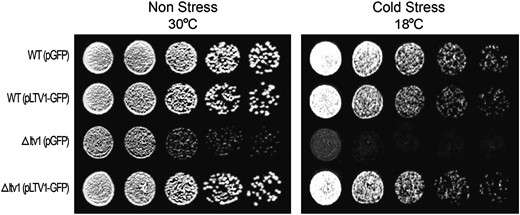
pLTV1-GFP rescues the Δltv1 cold-sensitive mutant phenotype. Wild-type (WT) (LY134) and Δltv1 (LY136) strains transformed with either pGFP (pUG23) or pLTV1-GFP (Ld47) were grown in selective media to midlog phase. Cell density was determined by hemocytometer and cells were diluted to 3.2 × 106 cells/ml. Serial 1:4 dilutions were made and 5-μl aliquots of each dilution were spotted onto plates lacking histidine and containing 500 μm methionine. Plates were incubated at 30° and photographed after 37 hr or at 18° and photographed after 64 hr.
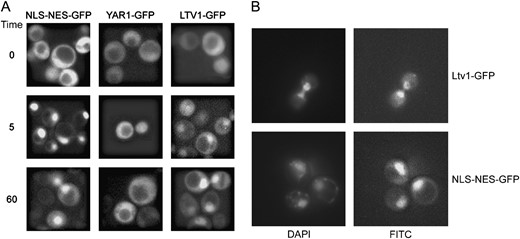
Nuclear export of Ltv1-GFP is Crm1 dependent. (A) pLTV1-GFP (Ld47), pYAR1-GFP (Ld46), and NLSSV40-NESPKI-GFP (pPS1372) plasmids were transformed in the LMB-sensitive crm1(T539C) (MNY8) strain. Strains expressing these GFP-tagged proteins were grown overnight at 30° in appropriate selective media to early to midlog phase, harvested, and photographed as described in materials and methods. LMB was added to an aliquot of each culture to a final concentration of 100 ng/ml and images of living cells captured beginning 5 and 60 min after addition of LMB. Time, in the left-most column, is the time since addition of LMB to cells. (B) Living cells expressing Ltv1-GFP (Ld47) or NLSSV40-NESPKI-GFP (pPS1372) protein were stained with DAPI and then treated with LMB. Cells were photographed 5 min after the addition of LMB.
Ltv1 is a shuttle protein whose export is Crm1 dependent:
The subcellular distribution seen for Yar1-GFP and Ltv1-GFP has also been described for proteins that shuttle between the nucleus and the cytoplasm. For example, the adaptor protein Nmd3, which is necessary for the export of the 60S subunit, gives a fluorescent signal over the nucleus and cytoplasm in exponentially growing cells that becomes nuclear localized if Crm1 export is inhibited (Ho et al. 2000; Gadal et al. 2001). Since Ltv1 interacts with Crm1 in a two-hybrid assay (Ito et al. 2001), we were especially interested in whether Ltv1 might partition between the nucleus and cytoplasm in a Crm1-dependent manner. LMB is a potent and very specific inhibitor of eukaryotic Crm1 proteins; it binds covalently to a conserved cysteine in the cargo-binding site, interfering with the formation of Crm1-RanGTP-NES export cargo complexes and inhibiting export (Kudo et al. 1999). Because the S. cerevisiae Crm1 lacks this conserved cysteine, it is normally resistant to LMB. However, a single amino acid substitution in the S. cerevisiae protein (T539C) renders cells LMB sensitive (Neville and Rosbash 1999).
To determine if either Yar1 or Ltv1 might be shuttle proteins, we visualized the subcellular distribution of the GFP-tagged proteins in cells expressing the LMB-sensitive Crm1T539Cp in the presence and absence of the drug. Cells were grown to early log phase and viewed prior to LMB addition and then 5 and 60 min after addition of LMB. Fluorescence microscopy revealed that LMB causes a rapid nuclear accumulation of Ltv1-GFP, comparable to that of the previously characterized shuttle reporter NLSSV40-NESPKI-GFP (Taura et al. 1998; Neville and Rosbash 1999) (Figure 3A). In contrast, Yar1-GFP showed little or no nuclear accumulation upon LMB treatment. Either the detected Yar1 fluorescence is over, but not within nuclei, or Yar1 can leave nuclei in a Crm1-independent manner. In LMB-treated cells, the GFP fluorescence of Ltv1-GFP and NLSSV40-NESPKI-GFP colocalizes with DAPI-stained nuclei (Figure 3B). These data indicate that Ltv1, but not Yar1, is a shuttle protein whose export from the nucleus is Crm1 dependent.
LTV1 has a functional NES that drives export of NLS-GFP in a Crm1-dependent manner:
We next searched for sequences within the Ltv1 protein that might support a direct interaction between Ltv1 and Crm1. Crm1 binds almost all of its substrates through a leucine-rich NES located in the substrate. Numerous studies have contributed to a definition of the following consensus sequence for the leucine-rich NES: ϕ-x(2–3)-ϕ-x(2–3)-ϕ-x-ϕ (where ϕ is L, I, V, F, M and x is any amino acid) (Wen et al. 1995; Bogerd et al. 1996; Henderson and Eleftheriou 2000; la Cour et al. 2004). The presence of leucines is not essential, although leucine and isoleucine are the most common hydrophobic residues, and many NESs that diverge from this postulated consensus have been identified (for review, see Fornerod and Ohno 2002). In addition to the pattern of hydrophobic residues, an analysis of 67 functional NESs reveals that NES regions are rich in glutamate, aspartate, and serine and that the majority are negatively charged (la Cour et al. 2004). Analysis of the known structures of a small number of NES-containing proteins indicates that the side chains of the first three N-terminal hydrophobic residues in the NES align to form a hydrophobic stripe along an α-helix (Rittinger et al. 1999; la Cour et al. 2004). Most NES sequences are bound weakly by Crm1, and the binding affinity of Crm1 for the NES is greatly increased by RanGTP binding. A weak NES may be biologically important, as artificially selected strong NES peptides seem to block the efficient unloading of cargo at the nuclear pore (Engelsma et al. 2004; Kutay and Guttinger 2005).
We identified a single potential NES sequence in Ltv1 at amino acid 169 (Figure 4). This NES is conserved in other fungal Ltv1 homologs and is negatively charged, and the first three N-terminal hydrophobic residues in the NES would align to form a hydrophobic stripe in a helical wheel projection of the sequence. However, this putative NES differs from the consensus at the fourth conserved hydrophobic position: the usual L/I is changed to a charged residue in Ltv1. The change at the fourth position is intriguing because a charged residue is also present at this position in both of the proposed NES elements in the yeast 60S adaptor protein Nmd3 (Gadal et al. 2001).

Sequence alignment identifies a potential NES in Ltv1. Ten experimentally validated NES sequences were aligned in Clustal X. Profile alignment was then used to align this NES profile to the S. cerevisiae Ltv1 sequence. Only one sequence was identified in Ltv1, which was then compared to the aligned fungal Ltv1 proteins to determine whether this sequence was conserved between distantly related fungal species. The conserved sequence is shown here.
To test whether the putative NES in Ltv1 is a functional in vivo target for Crm1, we asked whether the sequence was sufficient to drive export of a heterologous fusion protein in a Crm1-dependent manner. We replaced the NES from protein kinase inhibitor (PKI) in the NLSSV40-NESPKI-GFP reporter construct (Taura et al. 1998) with a 40-amino-acid segment of Ltv1 containing the putative NES by in vivo gap repair via homologous recombination. The structure of our construct was confirmed by DNA sequencing. We then compared the intracellular location of GFP in cells expressing Crm1T539Cp (MNY8) transformed with our pNLSSV40-NESLTV1-GFP, with a pNLSSV40-GFP reporter, or with the pNLSSV40-NESPKI-GFP reporter in the presence and absence of the Crm1 inhibitor LMB.
If the Ltv1 sequence is a Crm1 target, addition of this putative NES to the strong SV40 NLS sequence in these vectors should result in the detection of GFP fluorescence in both the cytoplasm and the nucleus. As illustrated in Figure 5 and quantified in Table 2, cells transformed with the pNLS-GFP reporter exhibit nearly complete nuclear localization of GFP. In contrast, most cells with the PKI NES added to the NLS (pNLS-NESPKI-GFP) exhibit a distribution of fluorescence over both the nucleus and the cytoplasm. Cells transformed with the pNLS-NESLTV1-GFP reporter, with the Ltv1 NES, exhibited a nearly identical distribution of fluorescence to that observed in cells with the PKI NES reporter. That some cells exhibit nuclear accumulation of GFP indicates that addition of the Ltv1 sequence did not destroy the function of the NLS sequence—thus the reporter protein is not cytosolic simply because the nuclear localization signal is defective. Furthermore, since addition of LMB results in the nuclear accumulation of both fusion proteins, it demonstrates that the cytoplasmic GFP signal observed in the absence of LMB is Crm1 dependent.
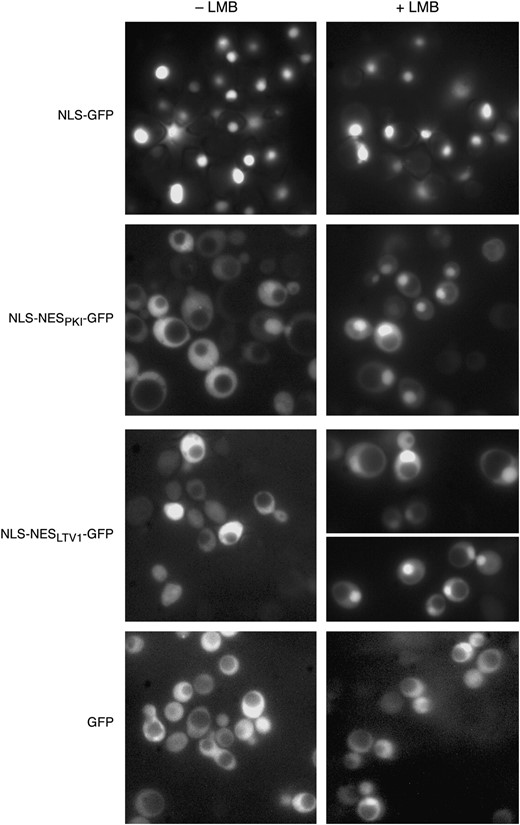
Ltv1 NES is sufficient to drive nuclear export of NLS-GFP reporter. MNY8 cells, transformed with pNLS-GFP (pPS815), pNLS-NESPKI-GFP (pPS1372), pNLS-NES LTV1-GFP (Ld61), or pGFP (pUG23), were grown in selective media to early log phase, collected, and washed in sterile water. Images of cells were captured using fluorescent microscopy and then LMB was added to an aliquot of cells to a final concentration of 100 ng/ml and photographed at 15 min after addition.
Ltv1 NES drives GFP export in a Crm1-dependent manner
Plasmid | Time after LMB (min) | No. of cells | % with nuclear localized GFP |
|---|---|---|---|
| pNLS-GFP-lacZ | 0 | 67 | 100 |
| 15 | 34 | 100 | |
| pNLS-NESPKI-GFP2 | 0 | 105 | 39 |
| 15 | 110 | 62 | |
| pNLS-NESLTV1-GFP2 | 0 | 101 | 40 |
| 15 | 120 | 53 | |
| 30 | 118 | 75 | |
| pGFP | 0 | 84 | 5 |
| 15 | 43 | 0 |
Plasmid | Time after LMB (min) | No. of cells | % with nuclear localized GFP |
|---|---|---|---|
| pNLS-GFP-lacZ | 0 | 67 | 100 |
| 15 | 34 | 100 | |
| pNLS-NESPKI-GFP2 | 0 | 105 | 39 |
| 15 | 110 | 62 | |
| pNLS-NESLTV1-GFP2 | 0 | 101 | 40 |
| 15 | 120 | 53 | |
| 30 | 118 | 75 | |
| pGFP | 0 | 84 | 5 |
| 15 | 43 | 0 |
Ltv1 NES drives GFP export in a Crm1-dependent manner
Plasmid | Time after LMB (min) | No. of cells | % with nuclear localized GFP |
|---|---|---|---|
| pNLS-GFP-lacZ | 0 | 67 | 100 |
| 15 | 34 | 100 | |
| pNLS-NESPKI-GFP2 | 0 | 105 | 39 |
| 15 | 110 | 62 | |
| pNLS-NESLTV1-GFP2 | 0 | 101 | 40 |
| 15 | 120 | 53 | |
| 30 | 118 | 75 | |
| pGFP | 0 | 84 | 5 |
| 15 | 43 | 0 |
Plasmid | Time after LMB (min) | No. of cells | % with nuclear localized GFP |
|---|---|---|---|
| pNLS-GFP-lacZ | 0 | 67 | 100 |
| 15 | 34 | 100 | |
| pNLS-NESPKI-GFP2 | 0 | 105 | 39 |
| 15 | 110 | 62 | |
| pNLS-NESLTV1-GFP2 | 0 | 101 | 40 |
| 15 | 120 | 53 | |
| 30 | 118 | 75 | |
| pGFP | 0 | 84 | 5 |
| 15 | 43 | 0 |
RpS3-GFP export is inhibited in Δltv1 cells:
Given that Ltv1p shuttles between nucleus and cytoplasm in a Crm1-dependent manner, that it contains a functional NES sequence capable of driving reporter export, and that it interacts in a two-hybrid assay with Crm1 and with Nup116, a nucleoporin implicated in small subunit export (Moy and Silver 1999; Ito et al. 2001), we hypothesized that Ltv1's role in small subunit biogenesis might be at the level of pre-40S export rather than at the level of rRNA processing per se. To test this hypothesis, we constructed an RpS3-GFP reporter plasmid that we could use to measure small subunit export. Large subunit ribosomal proteins tagged with GFP have been used very effectively to identify mutants in 60S export (Hurt et al. 1999; Gadal et al. 2001). For example, Rpl25p-GFP is incorporated into 60S ribosomal subunits and rapidly exported out of the nucleus in wild-type cells. As a result, GFP is distinctly nuclear excluded, with both the nucleus and the vacuole appearing dark in an otherwise fluorescing cell. In contrast, mutants impaired in 60S subunit export accumulate Rpl25p-GFP fluorescence in the nucleus.
We constructed a C-terminal RPS3-GFP fusion in a low-copy, methionine-repressible vector. The function of this fusion protein was tested in two ways. First, we determined that RpS3-GFP is assembled into 40S subunits by fractionating wild-type cell extracts expressing the fusion protein. RpS3-GFP was identified in the same sucrose gradient fractions as the endogenous protein RpS2, namely in the 40S region and with the 80S peak and larger polyribosomes (Figure 6). No free RpS3-GFP was detected in the soluble protein fractions, which suggests that most of the exogenous RpS3-GFP is incorporated into ribosomes and any excess is likely degraded (Hendrick et al. 2001). Second, we found that expression of RPS3-GFP can complement the lethality of Δrps3 mutants. We transformed the pRPS3-GFP expression vector into a heterozygous diploid Δrps3∷kanR/+ strain and sporulated the diploid, selecting for the plasmid. Spores were germinated and tested for mating type, and for the ΔrpS3∷kanR allele by replica printing onto plates containing G418. We were able to isolate both G418-sensitive and G418-resistant haploid segregants covered by the pRPS3-GFP vector (data not shown). That expression of RpS3-GFP protein is able to complement the lethality of the Δrps3 allele confirms that the fusion protein is functional and that the GFP tag does not interfere with either the assembly of RpS3 into ribosomes or the function of these ribosomes in translation.
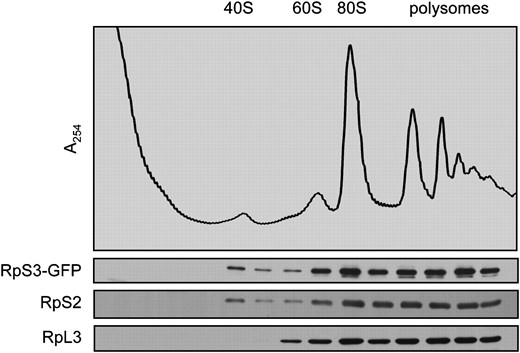
A fluorescent-tagged RpS3 fusion protein is incorporated into functional ribosomes. Wild-type (LY134) cells transformed with pRPS3-GFP (Ld50) were cultured to early log phase in media lacking histidine and methionine and then fractionated to yield a postnuclear homogenate. Ribosomal complexes were further separated on a 15–50% sucrose gradient. Individual gradient fractions were processed for Western blotting with antibodies to GFP and to small (S2) and large (L3) ribosomal subunit proteins.
To determine if Ltv1 functions in 40S export, we transformed the pRPS3-GFP vector into both wild-type and Δltv1 cells. Cells were grown to early log phase on selective media and then living cells were observed for GFP fluorescence. Wild-type yeast expressing RpS3-GFP protein export it so efficiently that a nuclear exclusion phenotype is observed; most cells exhibit cytoplasmic GFP staining with nuclear and vacuolar exclusion (Figure 7, Table 3). This phenotype is identical to that observed in cells expressing Rpl25-GFP (Gadal et al. 2001; Schafer et al. 2003) or in cells expressing Rpl11b-GFP (Stage-Zimmermann et al. 2000). As protein localization can be influenced by overexpression, we varied the expression levels of the RpS3-GFP construct by growing cells in increasing concentrations of methionine to repress the Met promoter. The percentage of wild-type cells exhibiting nuclear exclusion of GFP fluorescence was roughly the same over a range of methionine concentrations (∼60%). In the Δltv1 strain, only one-third to one-half as many cells exhibit nuclear exclusion of RpS3-GFP (Table 3). This result indicates that export of RpS3-GFP is inhibited substantially in Δltv1 cells relative to wild-type cells. The extent of inhibition for small subunit export is consistent both with the fact that Δltv1 mutants are viable and with the observation that the total number of 40S subunits in Δltv1 cells is reduced by ∼50% relative to wild-type cells (Loar et al. 2004).
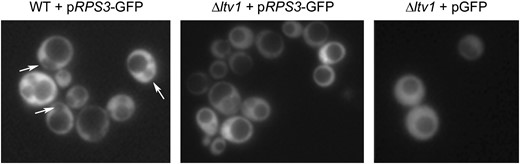
RpS3-GFP export is retarded in Δltv1 cells. Wild-type (WT) (LY134) and Δltv1 (LY136) cells transformed with either pRPS3-GFP (Ld50) or pGFP (pUG23) were grown in media lacking histidine and methionine to early log phase, harvested, and visualized by fluorescence microscopy as described in materials and methods. Arrows identify the nuclear-excluded phenotype scored in Table 3.
RpS3-GFP is retained in the nucleus in Δltv1 cells
0 μm Met | 300 μm Met | 600 μm Met | ||||
|---|---|---|---|---|---|---|
| Strain | % with nuclear exclusion | N | % with nuclear exclusion | N | % with nuclear exclusion | N |
| Wild type + pRPS3-GFP | 66 | 100 | 54 | 108 | 60 | 126 |
| Δltv1 + pRPS3-GFP | 36 | 205 | 17 | 101 | 17 | 24 |
| Δltv1 + pGFP | 0 | 41 | 1 | 96 | 0 | 8 |
0 μm Met | 300 μm Met | 600 μm Met | ||||
|---|---|---|---|---|---|---|
| Strain | % with nuclear exclusion | N | % with nuclear exclusion | N | % with nuclear exclusion | N |
| Wild type + pRPS3-GFP | 66 | 100 | 54 | 108 | 60 | 126 |
| Δltv1 + pRPS3-GFP | 36 | 205 | 17 | 101 | 17 | 24 |
| Δltv1 + pGFP | 0 | 41 | 1 | 96 | 0 | 8 |
RpS3-GFP is retained in the nucleus in Δltv1 cells
0 μm Met | 300 μm Met | 600 μm Met | ||||
|---|---|---|---|---|---|---|
| Strain | % with nuclear exclusion | N | % with nuclear exclusion | N | % with nuclear exclusion | N |
| Wild type + pRPS3-GFP | 66 | 100 | 54 | 108 | 60 | 126 |
| Δltv1 + pRPS3-GFP | 36 | 205 | 17 | 101 | 17 | 24 |
| Δltv1 + pGFP | 0 | 41 | 1 | 96 | 0 | 8 |
0 μm Met | 300 μm Met | 600 μm Met | ||||
|---|---|---|---|---|---|---|
| Strain | % with nuclear exclusion | N | % with nuclear exclusion | N | % with nuclear exclusion | N |
| Wild type + pRPS3-GFP | 66 | 100 | 54 | 108 | 60 | 126 |
| Δltv1 + pRPS3-GFP | 36 | 205 | 17 | 101 | 17 | 24 |
| Δltv1 + pGFP | 0 | 41 | 1 | 96 | 0 | 8 |
ITS1 has a distinct localization in Δltv1 cells:
If Ltv1 is required for the efficient nuclear export of small ribosomal subunits, then we might also expect to detect mislocalization of the 5′-ITS1 rRNA in Δltv1 mutants. The ITS1 is a 210-nt RNA fragment that is normally cleaved from the 20S pre-rRNA in the cytoplasm as the final step in generating the mature 40S subunit from the 43S precursor (Figure 1); it is then degraded in the cytoplasm by an exonuclease. The ITS1 in situ hybridization assay monitors the cellular localization of this RNA and was developed to screen for defects in small ribosomal subunit export (Moy and Silver 1999, 2002; Leger-Silvestre et al. 2004). In wild-type cells, ITS1 staining is concentrated in the nucleolus, with some staining of the cytoplasm depending on the rate of exonuclease degradation of the RNA fragment. In some nuclear-export-defective mutants, ITS1 staining is reduced in the cytoplasm and increases in the nucleoplasm, consistent with release of pre-40S subunits from the nucleolus but blockage of nuclear exit. This assay was used to establish the small subunit export function of Yrb2, the RanGTP-binding protein that is required for the export of several NES-containing proteins (Taura et al. 1998; Moy and Silver 2002).
We grew wild-type, Δltv1 and Δyrb2 mutants to early log phase at 30° and then fixed cells and hybridized with a Cy3-labeled oligonucleotide probe specific for the ITS1 sequence. Fixed cells were also stained with DAPI to identify the nucleoplasm. In wild-type cells, we observed ITS1 staining (usually in a small crescent shape) that was distinct and separate from the DAPI staining of chromatin (Figure 8). Quantification of the number of wild-type cells in which the ITS1 signal colocalizes with the DAPI signal revealed that only 16% of wild-type cells exhibited an overlapping stain pattern (Table 4). In the Δyrb2 strain, the ITS1 signal is more diffuse and the ITS1 and DAPI signals overlap in ∼50% of the cells, consistent with the ITS1 being released from the nucleolus and accumulating in the nucleoplasm, as previously reported (Moy and Silver 2002). The nucleolar vs. nuclear distribution of the ITS1 signal in Δltv1 cells is intermediate between that of wild-type and Δyrb2 strains. Quantitative analysis shows that 29% of Δltv1 cells, roughly twice the number of wild-type cells, have overlapping ITS1 and DAPI staining, indicative of nuclear retention of the ITS1 after release from the nucleolus (Table 4).
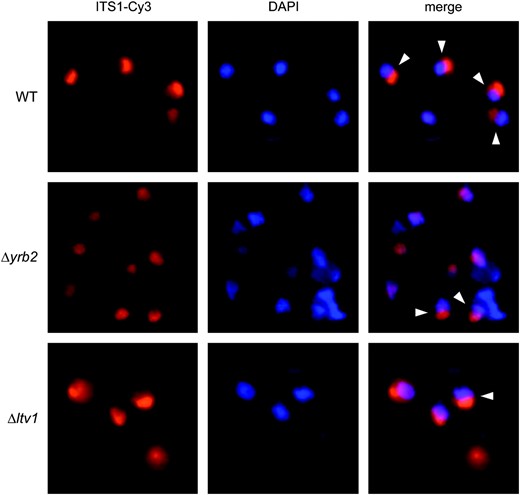
Deletion of YRB2 or LTV1 results in altered localization of an ITS1 rRNA fragment. A Cy3-labeled DNA probe complementary to the 5′-ITS1 sequence was hybridized with fixed and permeabilized wild-type (WT) (BY4743), Δyrb2 (PSY2070), or Δltv1 (LY139) mutant cells. The nuclear DNA was counterstained with DAPI prior to viewing in an epifluorescence microscope. Overlaid images show the colocalization or apposition of the ITS1 and DAPI signals. Cells exhibiting distinct, nonoverlapping DAPI/ITS1 signals, as scored in Table 4, are indicated with arrowheads.
ITS1 accumulates in Δltv1 cells
Strain | % DAPI and ITS1 separate | % DAPI and ITS1 overlapping | No. of cells |
|---|---|---|---|
| Wild type | 84 | 16 | 91 |
| Δltv1 | 71 | 29 | 104 |
| Δyrb2 | 48 | 52 | 66 |
Strain | % DAPI and ITS1 separate | % DAPI and ITS1 overlapping | No. of cells |
|---|---|---|---|
| Wild type | 84 | 16 | 91 |
| Δltv1 | 71 | 29 | 104 |
| Δyrb2 | 48 | 52 | 66 |
ITS1 accumulates in Δltv1 cells
Strain | % DAPI and ITS1 separate | % DAPI and ITS1 overlapping | No. of cells |
|---|---|---|---|
| Wild type | 84 | 16 | 91 |
| Δltv1 | 71 | 29 | 104 |
| Δyrb2 | 48 | 52 | 66 |
Strain | % DAPI and ITS1 separate | % DAPI and ITS1 overlapping | No. of cells |
|---|---|---|---|
| Wild type | 84 | 16 | 91 |
| Δltv1 | 71 | 29 | 104 |
| Δyrb2 | 48 | 52 | 66 |
LTV1 genetically interacts with YRB2:
If Ltv1 does function in Crm1-mediated export of the small subunit, then we might expect a functional interaction between Ltv1 and Yrb2. Efficient nuclear export of the small subunit in yeast requires Yrb2 as well as Crm1 (Moy and Silver 2002). Δyrb2 cells are viable but slow growing at 15°, accumulate the 20S rRNA precursor, and mislocalize the ITS1 to the nucleoplasm, phenotypes similar to those observed in Δltv1 cells (Moy and Silver 2002). In addition, polyribosome profiles of Δyrb2 cells reveal that these cells have fewer 40S subunits than wild-type cells (Moy and Silver 2002).
To test for a genetic interaction between LTV1 and YRB2, we constructed the heterozygous diploid, Δyrb2∷HIS3/+, Δltv1∷kanR/+, transformed it with a URA3-containing Gal-regulated YRB2 expression vector, and sporulated the resultant diploids, selecting for the vector. We tested random spores for genotype and mating type. Haploid segregants bearing both the Δyrb2 and the Δltv1 allele were then tested for their ability to lose the wild-type YRB2 URA3-containing plasmid on 5-fluoroorotic acid (5-FOA) plates. Both Δyrb2 and Δyrb2Δltv1 double mutants were able to grow on 5-FOA, indicating that the combination of knockout alleles is not lethal (data not shown). We then tested the growth rate of the double mutant relative to both single mutants at both 30° and 18°, after curing the cells of the pGAL-YRB2 vector. As shown in Figure 9, Δyrb2Δltv1 mutants exhibit a synthetic slow-growth phenotype relative to both the Δltv1 and the Δyrb2 mutants at both 30° and 18°. In liquid culture, the doubling time at 30° of the double mutant is at least twice that of either single mutant (6.2 hr. vs. 2.3 hr for Δltv1 and 3.1 hr for Δyrb2). This synthetic phenotype indicates a genetic interaction between LTV1 and YRB2 and suggests that their gene products are functionally related. Synthetic relationships can occur between genes acting in a single biochemical pathway or between genes within two distinct pathways if one process functionally compensates for the other (Hartman et al. 2001).
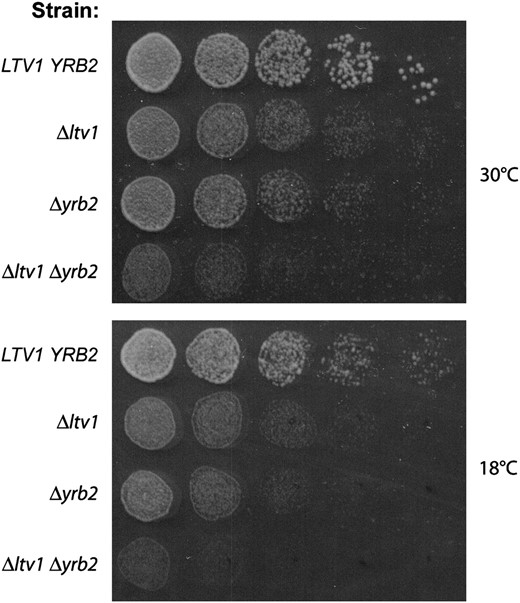
Genetic interaction between LTV1 and YRB2. (A) Wild-type strain LY134 (BY4742), its isogenic derivative LY136 (Δltv1∷KanR), PSY2070 (Δyrb2∷HIS3), and the double-mutant LY181 (Δyrb2∷HIS3 Δltv1∷KanR) were grown to midlog phase in YPD liquid media. Cell density was determined by hemocytometer and each culture was diluted to 3.2 × 106 cells/ml and then serially diluted 1:4 four times. We spotted 5-μl aliquots of each dilution from left to right on two YPD plates, which were incubated at 18° (photographed after 4 days) or at 30° (photographed after 1.5 days).
Overexpression of YRB2 from a high-copy vector is toxic in wild-type cells; it causes a slow-growth phenotype, results in nuclear retention of Crm1-GFP, and inhibits Crm1-dependent nuclear export and 40S export (Taura et al. 1998; Moy and Silver 2002). We tested whether overexpression of YRB2 was also toxic in cells lacking LTV1 by growing wild-type and Δltv1 mutant strains transformed with a pGAL-YRB2 vector on plates that allow either low-level expression or overexpression of YRB2. We confirmed that overexpression of YRB2 inhibits the growth of wild-type cells, but find that in the Δltv1 mutant, overexpression of YRB2 has no further deleterious effect on the growth of Δltv1 cells at either 30° or 18° (Figure 10). This suggests that one or more of the deleterious consequences of high levels of Yrb2 require the presence of Ltv1.
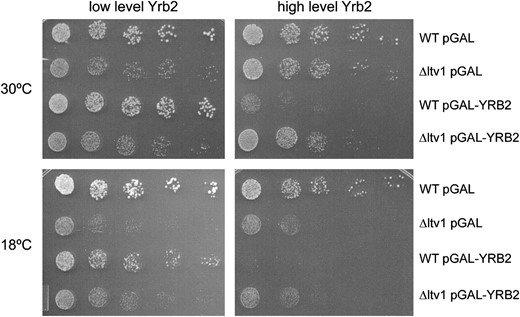
Toxicity of Yrb2 overexpression requires Ltv1. Wild-type (LY134) and Δltv1 (LY136) strains transformed either with the empty vector (pGAL) or with pPS1082 (pGAL-YRB2) were grown in selective media containing 2% glucose or 2% galactose to midlog phase. Cells were diluted and spotted onto selective plates containing the same galactose/glucose concentrations as the liquid media and incubated as described in Figure 2.
DISCUSSION
Ribosome adapters and nuclear export:
Both large and small ribosomal subunits are thought to be accompanied through the nuclear pore by export factors that couple them to the Crm1 karyopherin. Nuclear export of the 60S subunit requires the essential protein Nmd3 (Ho et al. 2000; Gadal et al. 2001). Nmd3 is a predominantly cytoplasmic protein in exponentially growing cells, but becomes localized to the nucleus if Crm1 is inhibited with LMB (Ho et al. 2000; Gadal et al. 2001; Johnson et al. 2001). Nmd3 binds to 60S subunits, but is released in the cytoplasm prior to subunit joining (Ho and Johnson 1999; Hedges et al. 2005). Nmd3 has a consensus NES signal in the C terminus that is sufficient to direct the export of an NLS-GFP reporter (Ho et al. 2000), and the nuclear export of Nmd3 itself is Crm1 dependent (Ho et al. 2000; Gadal et al. 2001; Johnson et al. 2001). C-terminal deletion mutants of NMD3 lacking the NES are dominant-negative inhibitors of cell growth and block 60S subunit export, as measured by the localization of RpL25-GFP (Belk et al. 1999; Ho et al. 2000). If the PKI NES is supplied to NES-deficient Nmd3 protein, 60S biogenesis is restored, suggesting that export of Nmd3 is necessary for export of the 60S subunit (Ho et al. 2000). These data have been used to argue that Nmd3 is an adapter protein responsible for providing an NES to the large ribosomal subunit in trans, thus linking it to Crm1 and providing for its nuclear export.
No analogous adapter protein for the pre-40S subunit has been identified in any cell type. This is despite a screen of 900 temperature-sensitive alleles for yeast genes defective in small subunit export (Moy and Silver 2002). One possible explanation for this lack of candidates is that export of the small subunit, unlike that of the large subunit, may involve multiple adapter proteins whose export functions are partially redundant. Several kinds of evidence now suggest that the nonribosomal protein Ltv1 may be one such export adapter. We showed previously that cells lacking LTV1 have aberrant polyribosome profiles and produce about half as many 40S subunits as do wild-type cells (Loar et al. 2004). Δltv1 cells are viable, but slow growing, especially at low temperature, consistent with the reduced rate of ribosome biogenesis (Loar et al. 2004). In addition, Ltv1 co-sediments with 43S/40S subunits, but not with 80S ribosomes, indicating that Ltv1 is released from the small subunit prior to subunit joining. This is consistent with previous studies that identified Ltv1 as a nonstoichiometric member of 40S but not 90S preribosomal complexes purified using the TAP-tagged ribosomal biogenesis factor Enp1 (Schafer et al. 2003). We provide evidence here that Ltv1 is a nuclear-cytoplasmic shuttle protein whose export is Crm1 dependent. Furthermore, Ltv1 contains an NES consensus sequence that is sufficient to drive the nuclear export of an NLS-GFP reporter protein, suggesting that Ltv1 is a direct Crm1 cargo. We show that in Δltv1 mutants the rate of nuclear export of the small subunit reporter, RpS3-GFP, is reduced relative to wild-type cells. Since RpS3-GFP is normally incorporated into 40S subunits very efficiently, this result implies that Ltv1 functions in small subunit export. RpS3 export is not completely blocked when Ltv1 is absent, and thus it is likely that the pre-40S subunit can be exported via other mechanisms, albeit less efficiently. Any alternative export pathway, however, is likely to involve Crm1 as inhibition of Crm1 with LMB blocks all 40S export. Finally, we show that LTV1 genetically interacts with YRB2, which encodes a Ran-binding protein that also interacts with Crm1 and is required for efficient 40S export. On the basis of these data, we propose that Ltv1 functions as a nonessential adapter for the small subunit, providing an NES in trans and thus linking the 40S subunit to the Crm1 nuclear export machinery.
Role of Yrb2:
The genetic interaction between LTV1 and YRB2 is intriguing, given the involvement of Yrb2 in ribosome biogenesis. Yrb2 is necessary for the efficient export of 40S subunits, which accumulate in the nucleoplasm of Δyrb2 cells (Moy and Silver 2002; Figure 8), and YRB2 overexpression results in an accumulation of Crm1-GFP in the nucleus and an inhibition of Crm1-mediated export (Taura et al. 1998). As RanBP3, the human homolog of Yrb2, stabilizes the association between Crm1 with certain NES-containing cargos (Englmeier et al. 2001; Lindsay et al. 2001), one hypothesis is that Yrb2 functions similarly in yeast to stabilize the interaction between Crm1 and the small subunit, and perhaps some other NES-containing cargos as well. Overexpression of Yrb2 might be deleterious because it monopolizes Crm1 in tight complexes, perhaps restricting its release of cargo and its recycling. As Ltv1 is necessary for the deleterious effects of Yrb2 overexpression on cell growth (Figure 10), Ltv1 may be necessary for the interaction among Crm1, Yrb2, and the small subunit.
rRNA-processing defects: Direct or indirect?
Deletion of either YAR1 or LTV1 results in the accumulation of specific pre-rRNA precursors (Figure 1). In a Δyar1 strain, 20S and 21S rRNA species are more abundant than in the corresponding wild-type strain, while in Δltv1 mutants, the 21S rRNA species is especially abundant, as are the 23S, 32S, and 35S rRNAs. The 20S rRNA accumulates if cleavage at site D is inhibited, a step that is executed by Nob1 in the cytoplasm after export (Fatica et al. 2003). The 21S rRNA persists if cleavage at site A2 is inhibited and cleavage at the A3 site occurs first, and the 23S and the 35S precursors accumulate if cleavage at sites A0 and A1 are also inhibited. All three of these early cleavages occur in the nucleolus and require the U3 snRNP. We think it is unlikely that either Yar1 or Ltv1 has any direct role in rRNA cleavage. Rather, we believe that these rRNA precursors accumulate as an indirect consequence of inhibiting other steps in ribosome biogenesis. If ribosome assembly can be thought of as a series of linked biochemical reactions, then the failure to remove products at any step would likely lead to the accumulation of intermediates. Alternatively, 43S preribosomes trapped in the nucleus might be unable to recycle associated factors required for earlier steps in assembly/pre-rRNA processing. Such a link between ribosome assembly and export has been noted previously by others. For example, excess hNMD3 in the nucleus alters the kinetics of rRNA processing in oocytes (Trotta et al. 2003), and normal rRNA processing is disrupted in strains lacking both Rai1p and Kap123p, a phenotype that is suppressed by overexpressing Nmd3 (Sydorskyy et al. 2003). We suggest that the rRNA processing defects in Δyar1 and Δltv1 mutants are indirect effects, which may reflect a coupling between ribosome assembly and nuclear export that deserves further exploration.
Role of RpS3 in ribosome biogenesis:
The ribosome export adapter hypothesis presumes that ribosomal proteins on the surface of subunits do not themselves directly interact with Crm1, but must do so through an adapter. Consistent with this hypothesis, Crm1 has not been reported to interact in a two-hybrid assay with any ribosomal proteins. Ltv1, on the other hand, interacts with both Crm1 and with one small subunit protein, RpS3 (Ito et al. 2001). RpS3 has recently been implicated in small subunit export (Ferreira-Cerca et al. 2005). Cells depleted for RpS3 accumulate 35S, 23S, and 21S rRNA precursors, similar to Δltv1 cells. Pulse-labeling experiments indicate that RpS3-depleted cells accumulate 20S rRNA to wild-type levels in the nucleus but fail to export it, leading the authors to propose that RpS3, along with five other ribosomal proteins that exhibit this phenotype, is an integral part of the export machinery for 20S-containing subunits. More recently, RpS3, Ltv1, and Enp1 have been shown to form a salt-resistant complex that can be released from pre-40S subunits, and all three proteins are phosphorylated, probably all by Hrr25 kinase. These authors confirm that RpS3 depletion affects 40S export and suggest that RpS3, Enp1, and Ltv1 may function in recruiting nuclear export factors and in initiating nuclear export (Schafer et al. 2006).
We previously showed that overexpression of RPS3 rescues the slow-growth and stress-sensitive phenotypes of Δyar1 but not of Δltv1 mutants and proposed that Yar1 might function as an RpS3 chaperone but that Ltv1 likely acts downstream of RpS3 (Loar et al. 2004). The exposed location of RpS3 on the beak of the small subunit (Spahn et al. 2001), the export phenotype of RpS3-depleted cells, and an interaction with Ltv1 all suggest that RpS3 could be a target for Ltv1 binding. Further work will be necessary to determine whether the RpS3:Ltv1 interaction is necessary for nuclear export of the small subunit.
Footnotes
Communicating editor: M. Johnston
Acknowledgement
We thank the students of the Molecular Biology 312 class of 2003, especially David Woessner for construction of the pRPS3-GFP vector. We are grateful to Jim Posakony for his careful reading of the manuscript and for many useful discussions. This work was supported by National Institutes of Health grants GM-061643 and GM-061643-02 to D.E.L. and GM-068208 to R.M.S.
References
Ito, T., T. Chiba, R. Ozawa, M. Yoshida, M. Hattori et al.,
Johnson, A. W., J. H. Ho, G. Kallstrom, C. Trotta, E. Lund et al.,
Taura, T., H. Krebber and P. A. Silver,

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}