





Abstract
A classical prediction from single-locus models is that inbreeding increases the efficiency of selection against partially recessive deleterious alleles (purging), thereby decreasing the mutation load and level of inbreeding depression. However, previous multilocus simulation studies found that increasing the rate of self-fertilization of individuals may not lead to purging and argued that selective interference among loci causes this effect. In this article, I derive simple analytical approximations for the mutation load and inbreeding depression, taking into account the effects of interference between pairs of loci. I consider two classical scenarios of nonrandomly mating populations: a single population undergoing partial selfing and a subdivided population with limited dispersal. In the first case, correlations in homozygosity between loci tend to reduce mean fitness and increase inbreeding depression. These effects are stronger when deleterious alleles are more recessive, but only weakly depend on the strength of selection against deleterious alleles and on recombination rates. In subdivided populations, interference increases inbreeding depression within demes, but decreases heterosis between demes. Comparisons with multilocus, individual-based simulations show that these analytical approximations are accurate as long as the effects of interference stay moderate, but fail for high deleterious mutation rates and low dominance coefficients of deleterious alleles.
ACCORDING to current estimates of spontaneous deleterious mutation rates in multicellular organisms (e.g., Baer et al. 2007; Haag-Liautard et al. 2007; Keightley 2012) and estimated distributions of fitness effects of these mutations (e.g., Eyre-Walker and Keightley 2007; Keightley and Eyre-Walker 2007; Boyko et al. 2008; Haddrill et al. 2010), individuals may typically carry large numbers (possibly up to thousands) of deleterious alleles. Possible consequences of this load of deleterious mutations have been discussed since the early ages of theoretical population genetics (e.g., Haldane 1937). In particular, it may reduce population mean rates of fecundity and viability, thereby increasing vulnerability to extinction (Lynch et al. 1995a,b). It may also affect a number of evolutionary processes, such as the evolution of sex or mating systems: for example, the fact that deleterious alleles are often partially recessive generates inbreeding depression, favoring outcrossing over self-fertilization (e.g., Lande and Schemske 1985; Charlesworth and Charlesworth 1987; Charlesworth 2006).
In very large, panmictic populations and in the absence of epistasis between mutations, genetic associations between deleterious alleles at different loci should remain weak and may be neglected. In diploids, and assuming that the dominance coefficient of deleterious alleles is significantly greater than zero, the mutation load (reduction in mean fitness of the population due to deleterious alleles at mutation–selection balance) is , where U is the deleterious mutation rate per haploid genome (Crow 1970; Agrawal and Whitlock 2012). Furthermore, assuming for simplicity that all deleterious alleles have the same dominance coefficient h, inbreeding depression (defined here as the reduction in fitness of offspring produced by self-fertilization, relative to offspring produced by outcrossing) is (Charlesworth and Charlesworth 2010). Analytical results on the effects of genetic drift and nonrandom mating mainly stem from single-locus models. Inbreeding increases the efficiency of selection against deleterious alleles, lowering the mutation load and inbreeding depression (Lande and Schemske 1985). Genetic drift may also lead to better purging of partially recessive deleterious alleles (Kimura et al. 1963), but this effect causes only a moderate reduction of the mutation load compared to the effect of nonrandom mating and occurs only when the effects of drift and selection are of the same order of magnitude (Glémin 2003). Drift has more noticeable effects when it becomes stronger than selection and allows deleterious alleles to reach fixation, which may increase the load by several orders of magnitude and lowers inbreeding depression (Bataillon and Kirkpatrick 2000). Population subdivision has similar consequences, due to the effects of drift within each local population (Whitlock 2002; Glémin et al. 2003; Roze and Rousset 2004).
These previous studies are based on single-locus models and therefore do not consider the effects of genetic associations between loci on the mutation load and inbreeding depression. Between-locus associations are generated, however, as soon as population size is finite or mating is nonrandom (even in the absence of epistasis): in particular, correlations in homozygosity, described as “identity disequilibria” (Weir and Cockerham 1973; Vitalis and Couvet 2001), and linkage disequilibria between selected loci (Hill and Robertson 1966; Roze and Lenormand 2005; Kamran-Disfani and Agrawal 2014). Effects of deleterious mutations occurring at many loci have been explored using simulation models of finite or infinite populations (e.g., Charlesworth et al. 1990, 1991, 1992, 1993; Lande et al. 1994; Wang et al. 1999), sometimes showing important deviations from single-locus predictions. In particular, using Kondrashov’s (1985) model to simulate recessive lethal mutations occurring at a very large (effectively infinite) number of unlinked loci in a partially selfing population, Lande et al. (1994) observed that contrary to the predictions of single-locus models, recessive lethals cannot be purged by selfing unless the selfing rate exceeds a threshold value (see also Kelly 2007). Lande et al. (1994) argued that this effect (called “selective interference”) is caused by identity disequilibria. Intuitively, selfing increases homozygosity at each locus and should thus purge recessive lethal mutations; however, if many such mutations segregate in the population, any selfed offspring will almost certainly carry at least one mutation in the homozygous state and will thus not survive. When this is the case, the population is effectively outcrossing, and purging does not occur.
To date, the effects of selective interference in partially inbred populations have been explored only numerically. How these effects scale with the strength of selection against deleterious alleles, dominance coefficients, and recombination rates between loci thus remains unclear. In this article, I derive analytical approximations describing the effect of interference between pairs of loci on the mean frequency of deleterious alleles, the mean and variance in fitness, and the strength of inbreeding depression, assuming weak selection against deleterious alleles. I consider two classical scenarios of nonrandomly mating populations: a single, large population in which individuals self-fertilize at a given rate and a subdivided population with local mating followed by dispersal (island model of population structure). In the first case, interference between loci tends to reduce mean fitness and increase inbreeding depression. These effects are stronger when deleterious alleles are more recessive, but depend only weakly on the strength of selection against deleterious alleles and on recombination rates. In the case of a subdivided population, I first show that combining two different approximations used in previous works (Glémin et al. 2003; Roze and Rousset 2004) yields more accurate expressions for the mutation load, inbreeding depression, and heterosis generated by a single deleterious allele. In a second step, I derive approximations for the effects of interference between loci and show that interference increases inbreeding depression within demes, but decreases heterosis between demes. Comparisons with individual-based, multilocus simulation results show that analytical approximations incorporating the effects of associations between pairs of loci often provide accurate predictions for the mutation load and inbreeding depression as long as the dominance coefficient h of deleterious alleles is not too low. These approximations fail when h becomes close to zero and when the deleterious mutation rate is high, however, probably due to the fact that higher-order interactions (involving three or more loci) become important.
Methods
I consider a diploid population with discrete generations, in which deleterious mutations occur at rate U per haploid genome per generation. For simplicity, I generally assume that all deleterious alleles have the same selection and dominance coefficients (s, h), although distributions of s and h will be considered in the case of a partially selfing population. Deleterious alleles at different loci have multiplicative effects (no epistasis), so that the fitness of an organism carrying j heterozygous and k homozygous mutations is proportional to . In the first model (partial selfing), a parameter α measures the proportion of offspring produced by selfing, while a proportion is produced by random union of gametes. The second model corresponds to the island model of population structure: the population is subdivided into a large number of demes, each containing N adult individuals. These individuals produce large numbers of gametes (in proportion to their fitness), which fuse randomly within each deme to form juveniles. A proportion m of these juveniles disperses, reaching any other deme with the same probability. Finally, N individuals are sampled randomly within each deme to form the next adult generation. I assume soft selection; that is, all demes contribute equally to the migrant pool. In Supporting Information, File S1 and File S2, I derive approximations for the mutation load and inbreeding depression that incorporate effects of pairwise associations between loci, assuming (so that individuals tend to carry many deleterious alleles) and that drift at the whole-population level is negligible relative to selection. In the next sections, these analytical predictions are compared with individual-based, multilocus simulation results. The simulation programs (available from Dryad) are similar to those used in previous work (e.g., Roze and Rousset 2009). Briefly, they represent a finite population of diploids, whose genome consists of a linear chromosome. Each generation, the number of new mutations per chromosome is drawn from a Poisson distribution with parameter U, the position of each mutation along the chromosome being drawn from a uniform distribution (in practice, a chromosome is represented by the positions of the deleterious alleles it carries). To form the next generation, a maternal parent is sampled for each offspring, either among all parents (in the case of a single population undergoing partial selfing) or among all parents from the offspring’s deme of origin (in the case of a subdivided population). In the first case, the parent self-fertilizes with probability α, while with probability a second parent is sampled. In the second case (subdivided population), a second parent is sampled from the same deme as the first. In all cases, the probability that a given parent is sampled is proportional to its fitness. Parents produce gametes by meiosis, a parameter R measuring the genome map length: for each meiosis, the number of crossovers is sampled from a Poisson distribution with parameter R, the position of each crossover being drawn from a uniform distribution. Map length is fixed to 10 M in most simulations, to mimick a whole genome with multiple chromosomes. The program runs for a large number of generations (generally ) and measures the mean number of deleterious alleles per genome, mean fitness, variance in fitness, inbreeding depression, and heterosis (in the case of a subdivided population) every 50 generations.
Data availability
Dryad DOI: doi:10.5061/dryad.sp01m.
Partial Self-Fertilization
The first effect stems from the fact that the fitnesses of mutant and wild-type homozygotes at locus i are decreased by the same factor from associations with homozygotes at other selected loci; however, the fitness of heterozygotes at locus i is decreased by a smaller factor, since these tend to be associated with heterozygotes at other loci, which have a higher fitness than homozygotes (provided ). Therefore, identity disequilibria have a stronger impact on the fitness of homozygotes than on that of heterozygotes, decreasing the effective dominance coefficient of deleterious alleles and thereby reducing the efficiency of selection against those alleles.
The second effect (deleterious alleles tend to be associated with more heterozygous backgrounds) stems from the fact that because heterozygotes at locus i tend to be heterozygous at locus j (while homozygotes at locus i tend to be homozygous at locus j) and because selection is more efficient among homozygotes than among heterozygotes, selection against the deleterious allele at locus i is less efficient among heterozygotes at locus j than among homozygotes. This effect causes the deleterious allele at locus i to be more frequent among heterozygotes than among homozygotes at locus j, in turn decreasing the efficiency of selection at locus i, since heterozygous backgrounds are fitter than homozygous ones when
In the following, expressions for mean fitness and inbreeding depression δ are obtained by replacing identity disequilibria by their equilibrium values under neutrality. Because allele frequencies are of order (where u is the deleterious mutation rate per locus), this will generate terms of order in the expressions for and δ below. Taking into account the effect of selection acting at loci i and j on would generate terms of order , which should be negligible relative to terms in U and as long as selection is weak (s small). However, is also affected by selection acting at other loci, due to three-locus identity disequilibria. Taking into account the effects of these three-locus associations would introduce terms of order in the expressions for and δ, which may become important when U is sufficiently large. As we will see, some discrepancies are observed between the analytical predictions and the simulation results for high U and low h, probably due to the fact that these higher-order genetic associations (between three or more loci) are not taken into account in the analysis.
Correlations in homozygosity directly increase mean fitness when , because double homozygotes and double heterozygotes have a higher fitness (on average) than genotypes that are homozygous at one locus and heterozygous at the other (e.g., Roze 2009): this effect is represented by the term in in Equation 1 (approximated by Equation 4), corresponding to the factor in Equation 11.
Identity disequilibria tend to decrease the excess of homozygosity at each locus when (Equation 5), increasing mean fitness since homozygotes have a lower fitness than heterozygotes when (term in in Equation 1, which increases as decreases if , as shown by Equation 2). If , is now increased by identity disequilibria, but this again increases mean fitness since homozygotes have a higher fitness than heterozygotes. This second effect corresponds to the term in Equation 11.
Finally, identity disequilibria increase the frequency of deleterious alleles at mutation–selection balance when (as explained above), which decreases mean fitness: this corresponds to the factor in Equation 11.
One can show that effect 3 is stronger than effects 1 and 2 when , causing identity disequilibria to decrease mean fitness (while when , all three effects increase mean fitness). An approximation for the variance in fitness at equilibrium is provided in File S1 (Equation A46); from this expression, it is possible to show that identity disequilibria generally increase the variance in fitness (unless , in which case their effect vanishes).
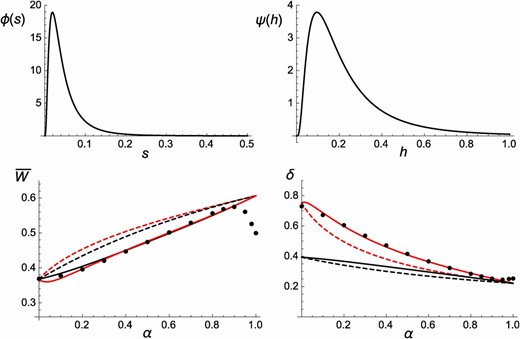
Figure 1 shows that Equation 11 provides accurate predictions for mean fitness when and , while discrepancies are observed for By contrast, ignoring effects of identity disequilibria overestimates mean fitness, in particular when h is low. Figure 1 also shows that is systematically lower than predicted when the selfing rate approaches 1; this effect is likely due to the fact that in the simulations, the effective population size is greatly reduced by background selection effects when outcrossing is very rare, in which case deleterious alleles may increase in frequency due to drift. As shown by Figure S1, reducing the mutation rate from to reduces the effects of identity disequilibria and leads to a better match between predictions from Equation 11 and simulation results for Figure S2 and Figure S3 show that changing the selection coefficient of deleterious alleles to or leads to very similar results (indeed, Equation 11 does not depend on s), except that the effects of drift at high α are stronger for lower values of s. Genomic map length (R) was set to 10 M in these simulations; additional simulations were run for the case of freely recombining loci, but yielded undistinguishable results unless α is close to 1 (in which case free recombination lowers the effects of drift—results not shown). The variance in fitness in the population at equilibrium is shown in Figure 2: when h is low, the variance in fitness is maximized for intermediate values of the selfing rate α, mainly due to the effects of identity disequilibria (which are maximized for intermediate values of α).
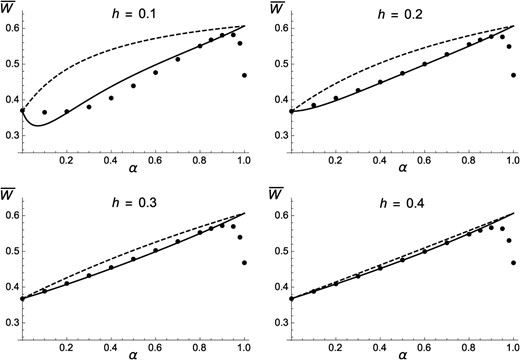
Average fitness at equilibrium as a function of the rate of self-fertilization α, for different values of the dominance coefficient of deleterious alleles (h), and deleterious mutation rate per haploid genome . Solid curves, analytical approximation including effects of identity disequilibria (Equation 11); dashed curves, neglecting effects of identity disequilibria (obtained by setting in Equation 11); solid circles, simulation results (in this and the following figures, error bars are smaller than the size of circles). In the simulations, , , and .
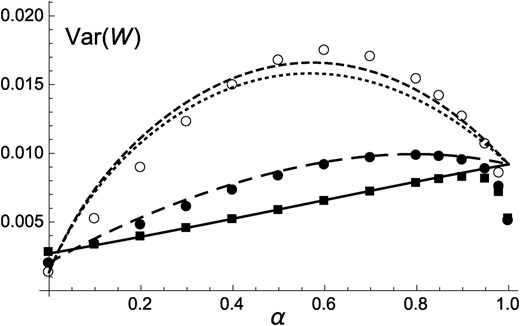
Variance in fitness in the population at equilibrium, as a function of the rate of self-fertilization α and for different values of the dominance coefficient of deleterious alleles. Curves correspond to predictions from Equation A46 in File S1 (dotted, ; long dashed, ; solid, ). Short-dashed curve, adding the term given in Equation A47 in File S1 for ; symbols, simulation results for (open circles), (solid circles), and (solid squares). Parameter values are the same as in Figure 1.
Figure 3 compares the value of inbreeding depression measured in simulations with predictions from Equation 14, also showing that taking into account the effects of identity disequilibria leads to more accurate predictions (although discrepancies appear for ). Results for the case of fully recessive mutations () are shown in Figure 4: in agreement with Lande et al. (1994), for high mutation rates ( or 0.5) purging occurs only when the selfing rate exceeds a threshold value. Below this threshold, the population is effectively outcrossing, which is confirmed by the fact that mean fitness stays very close to the average fitness of a panmictic population ( when ) multiplied by the outcrossing rate (see Figure S4). Figure 4 also shows that while Equation 14 provides better predictions than the equivalent expression ignoring identity disequilibria, it does not fully capture the effect of selective interference for intermediate selfing rates and high values of U, indicating that higher-order genetic associations (in particular, joint homozygosity at multiple loci) must have important effects for these parameter values.
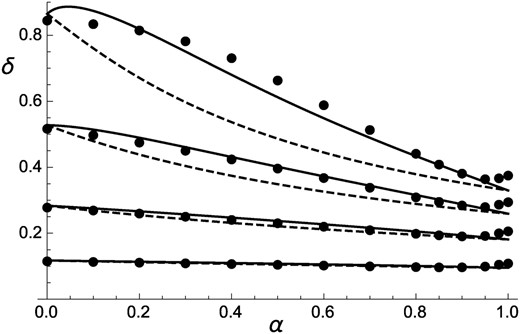
Inbreeding depression as a function of the rate of self-fertilization α, for different values of the dominance coefficient of deleterious alleles (, 0.2, 0.3, and 0.4 from top to bottom), and deleterious mutation rate per haploid genome Solid curves, analytical approximation including effects of identity disequilibria (Equation 14); dashed curve, neglecting effects of identity disequilibria (setting in Equation 14); solid circles, simulation results (same parameter values as in Figure 1).
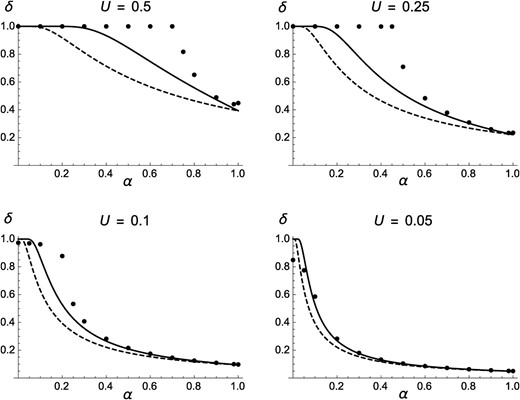
Inbreeding depression as a function of the selfing rate α: same as Figure 3 with fully recessive deleterious alleles () and different values of the deleterious mutation rate U.
The previous results assume that all deleterious alleles have the same selection and dominance coefficients. However, File S1 shows that they are easily extended to the more realistic situation where s and h vary among loci, as long as we can assume that selection is much stronger than drift at most loci. In that case, mean fitness and inbreeding depression at equilibrium do not depend on the strength of selection against deleterious alleles and can be obtained by integrating terms appearing in the equations above over the distribution of dominance coefficients of these alleles (see Equations A56 and A57 in File S1). To test these results, I modified the simulation program so that the distribution of selection coefficients of deleterious alleles is log-normal, with density function (where μ and are the mean and variance of ), truncated at (this has a negligible effect for the parameter values considered here). Available data on fitness effects of deleterious alleles point to an absence of correlation between homozygous and heterozygous effects of deleterious mutations (at least for mutations having sufficiently large homozygous effect, e.g., Manna et al. 2012), the distribution of heterozygous effects () being much less variable than the distribution of homozygous effects (s). Here, I assume for simplicity that all deleterious alleles have the same heterozygous effect θ: as a consequence, s and h are negatively correlated, and the distribution of dominance coefficients () is given by . Figure 5 shows the distributions of s and h for , setting μ and θ so that and (that is, and ); Figure S5 shows h as a function of s for these parameter values. As shown by Figure 5, Equations A56 and A57 in File S1 provide accurate predictions for mean fitness and inbreeding depression when s and h vary across loci (as before, discrepancies appear when α approaches one, due to finite population size effects). It also shows that introducing a variance in h has little effect on mean fitness (its value being well predicted by the expression assuming fixed h), while it strongly increases inbreeding depression, in particular when the selfing rate is small. This may be understood from single-locus results: inbreeding depression increases faster than linearly as h decreases (the effect of h on δ being more marked when α is small), causing inbreeding depression to increase as the variance of h increases. By contrast, the effect of h on mean fitness is weaker and vanishes when Finally, Figure S6 shows that when , the variance of h generates positive inbreeding depression, which is slightly increased by identity disequilibria.
(Top) Distributions of s and h assuming a log-normal distribution of s with and (so that ) and fixed heterozygous effects of deleterious alleles (so that ). See text for more explanations. (Bottom) Mean fitness and inbreeding depression as a function of the selfing rate α. Black circles, simulations results, using the distributions of s and h shown at the top; black curves, analytical predictions for fixed h, set to (from Equations 11 and 14); red curves, analytical predictions for varying h (from Equations A56 and A57 in File S1); dashed/solid curves, neglecting/including the effects of identity disequilibria. The mutation rate is set to ; in the simulations, and .
Population Structure
File S2 shows how approximations for L, δ, and H can be derived, assuming that deme size N is large, while the migration rate m and strength of selection s are small. As in the previous section, the total population size is supposed very large (large number of demes), so that the effects of drift at the whole population level can be neglected. In a first step, I show that improved approximations for L, δ, and H generated by mutation at a single locus can be obtained by combining previous results (Glémin et al. 2003; Roze and Rousset 2004). Then, I extend these results to the case of deleterious alleles occurring at a large number of loci, incorporating effects of pairwise associations among loci.
Single-locus results
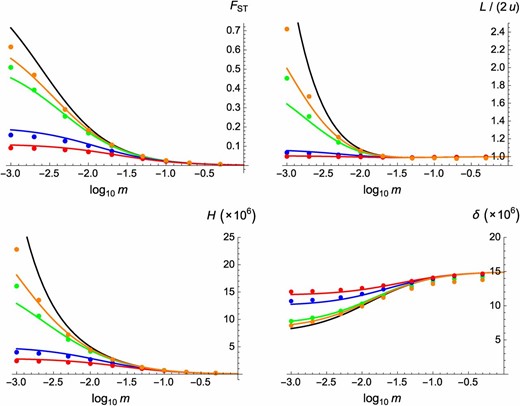
Equilibrium values of , mutation load L (divided by its value in a panmictic population, ), heterosis, and inbreeding depression in a subdivided population, when selection acts at a single locus. The x-axes show the migration rate between demes (on a log scale), and the different colors correspond to different values of s: 0.005 (orange), 0.01 (green), 0.05 (blue), and 0.1 (red). Colored curves, predictions from Equations 20 and 22–24; circles, one-locus simulation results (30 replicates of generations; error bars are smaller than the size of circles); black curves, predictions from Roze and Rousset (2004) (obtained by replacing Γ by in Equations 20 and 22–24). Other parameter values: , , ; in the simulations the number of demes is set to 200, and back mutations occur at rate .
Finally, we can note that when the migration rate m is set to zero, the model represents an infinite number of replicates of a single population of size N. The above results thus predict that the variance in frequency of a deleterious allele due to drift in a single finite population should be as long as the average frequency of the deleterious allele remains small (from Equation 20, with ). Furthermore, expressions for the average allele frequency, mutation load, and inbreeding depression are obtained by setting in Equations 21–23. Figure 7 shows that these approximations are indeed accurate as long as N is not too small (so that the deleterious allele stays rare in the population).
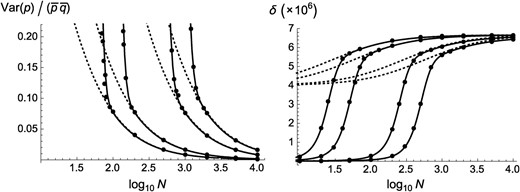
Variance of deleterious allele frequency (scaled by ) and inbreeding depression in a single finite population, as a function of population size N (on a log scale). Solid curves correspond to predictions obtained from numerical integration over the standard diffusion result for the distribution of allele frequency (e.g., equation 9.3.4 in Crow and Kimura 1970; see also Bataillon and Kirkpatrick 2000), while dashed curves correspond to (left) and to the expression obtained by replacing by in Equation 23 (right). Circles, one-locus simulation results (averages over 30 replicates of – generations). Parameter values: , 0.01, 0.05, 0.1 (from right to left); ; ; back mutation rate, .
Effects of interference between selected loci
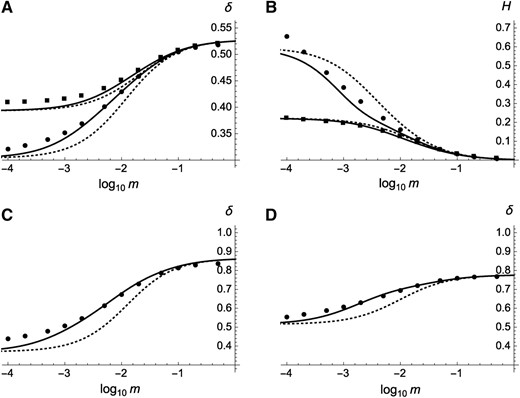
Inbreeding depression (A, C, and D) and heterosis (B) when deleterious mutations occur at a large number of loci, as a function of the migration rate between demes (on a log scale). Circles, multilocus simulation results; solid curves, predictions from Equations 31 and 33; dotted curves, predictions ignoring effects of interactions between loci (setting and to zero in Equations 31 and 33). Parameter values: (A and B) , , (squares, top curves in A, bottom curves in B), (circles, bottom curves in A, top curves in B); (C) , , ; and (D) , , Deme size: In the simulations the number of demes is set to 200 and genome map length to M.
Discussion
Theoretical predictions regarding the effect of the mating system of organisms on the mutation load and inbreeding depression are often based on single-locus models. However, as previously shown by Lande et al. (1994), some of these predictions may not hold when considering more realistic situations involving multiple selected loci. In particular, when the genomic mutation rate toward recessive deleterious alleles is sufficiently high, inbreeding depression is maintained at high levels irrespective of the selfing rate of individuals (contrary to the predictions of single-locus models), unless selfing exceeds a threshold value. This selective interference effect has been invoked by Scofield and Schultz (2006) and by Winn et al. (2011) to explain the lack of evidence of purging in meta-analyses comparing species with intermediate selfing rates to species with a low selfing rate (while species with high selfing rates show reduced inbreeding depression): for example, Winn et al. (2011) observed that species with intermediate selfing rates (between 0.2 and 0.8) present similar levels of inbreeding depression to those of species with lower selfing rates (<0.2). Furthermore, it has been proposed that this effect may allow the stable maintenance of mixed mating systems (involving both selfing and outcrossing), since the classical prediction that only complete selfing or complete outcrossing should be evolutionarily stable (Lande and Schemske 1985) is based on the assumption that inbreeding depression is a decreasing function of the selfing rate.
Most previous studies of selective interference were based on Kondrashov’s (1985) simulation model, representing deleterious alleles occurring at an infinite number of unlinked loci, in an infinite population. Lande et al. (1994) considered the case of fully (or almost fully, i.e., ) recessive lethal mutations () and found that selective interference becomes important when the genomic deleterious mutation rate is sufficiently high (0.2–1). Kelly (2007) showed that strong homozygous effects of deleterious alleles are not necessarily needed for interference to occur (the effect being actually stronger with than with ), while h has to be sufficiently low to observe interference. Winn et al. (2011) modeled transitions from outcrossing to partial selfing and showed that increased selfing leads to lower levels of inbreeding depression (purging) when and and when and , but not when and (for a genomic mutation rate of 1), inbreeding depression staying close to 1 in the last situation.
To date, no analytical model has explored the mechanisms of selective interference. In this article, I showed that analytical approximations can be obtained in regimes where interference stays moderate, by considering the effects of pairwise interactions between selected loci and assuming weak selection. As we have seen, the mechanisms underlying interference in partially inbred populations depend on the form of inbreeding considered. In a single, large population undergoing partial selfing, interference between loci is mainly driven by identity disequilibria between those loci (as long as the fitness of heterozygotes departs from the average of both homozygotes a each locus, i.e., ). However, identity disequilibria affect inbreeding depression through several mechanisms: correlations in homozygosity directly reduce δ, but also indirectly decrease homozygosity at each locus (which also reduces δ) and decrease the efficiency of selection against deleterious alleles, allowing them to be maintained at higher frequencies (thereby increasing δ). This last effect (which predominates over the first two) corresponds to the verbal explanation proposed previously to explain selective interference (purging is prevented by identity disequilibria, e.g., Lande et al. 1994; Winn et al. 2011). However, we have seen that this effect itself involves three different mechanisms: reduction of the effective dominance coefficient of deleterious alleles, decrease in homozygosity at each locus, and positive correlations between the presence of a deleterious allele at a given locus and heterozygosity at other loci. The results presented here also show that interference is affected little by the strength of selection against deleterious alleles (at least as long as selection is weak to moderate) or by linkage, as long as genome map length is sufficiently high—in agreement with the simulation results obtained by Charlesworth et al. (1992), showing that the effect of linkage on mean fitness and inbreeding depression in partially selfing populations often remains slight.
When inbreeding results from limited dispersal (population structure), interference effects are more complicated as they involve associations between loci as well as between different individuals from the same spatial location. However, we have seen that when selection and migration are weak while deme size is large, the main effect of interference between loci (assuming partially recessive deleterious alleles) is to increase the effective migration rate at each locus (Ingvarsson and Whitlock 2000), thereby reducing probabilities of identity between alleles present in different individuals from the same deme. This may either increase or decrease the strength of selection against deleterious alleles, depending on parameter values, but it always increases inbreeding depression within demes, while reducing heterosis between demes. In contrast to the case of partial selfing in a single population, this effect does not involve identity disequilibria (correlations in homozygosity across loci), but does involve other types of associations between alleles present in different individuals from the same deme (moments of linkage disequilibrium and allele frequencies, see Equations B44 and B45 in File S2). Furthermore, an important difference between partial selfing and population structure is that the mutation load and inbreeding depression in a structured population may be affected by the strength of selection against deleterious alleles (in particular when migration is weak, see Figure 6). The effects of interference between loci also depend on the strength of selection, being more marked for lower values of s.
Is selective interference likely to have important consequences in natural populations? Confirming previous results, we have seen that interference leads to substantial deviations from single-locus results for parameter values leading to strong inbreeding depression (high U, low h), independently of the strength of selection against deleterious alleles. In particular, the total absence of purging as the selfing rate increases (up to a threshold value) is observed only when inbreeding depression is close to 1 (while for lower values of δ, interference only dampens the decline of inbreeding depression with selfing). As observed by Winn et al. (2011), this condition may be fulfilled in gymnosperms, which show very high levels of inbreeding depression. In contrast, angiosperms show lower values of inbreeding depression (on average), for which selective interference may not be sufficiently strong to prevent purging. According to the results shown here, interference between deleterious alleles may thus not represent a sufficient explanation for the lack of evidence for purging in angiosperms in Winn et al.’s (2011) meta-analysis (for selfing rates between 0 and 0.8). Other possible explanations may be a lack a sufficient power to detect purging or synergistic epistasis between deleterious alleles, which tends to flatten the relationship between inbreeding depression and the selfing rate (Charlesworth et al. 1991). Note also that, as discussed by Winn et al. (2011), most estimates of inbreeding depression compiled in their data set were obtained under greenhouse conditions and may thus be biased downward if inbreeding depression tends to be stronger in harsher environments (Armbruster and Reed 2005). More empirical studies of inbreeding depression in different sets of conditions are thus needed to assess the potential importance of interactions between loci on selection against deleterious alleles.
Finally, because the suppression of purging due to interference occurs only when inbreeding depression is maximal, this mechanism does not seem a likely explanation for the evolutionary maintenance of mixed mating systems (as proposed in previous articles), since selfing should be strongly disfavored when δ is close to 1. Nevertheless, the effects of associations between loci on the evolution of mating systems remain little explored (but see Kamran-Disfani and Agrawal 2014). Besides affecting inbreeding depression, between-locus associations may modulate the advantage of selfers due to more efficient purging (e.g., Uyenoyama and Waller 1991; Epinat and Lenormand 2009) and possibly generate additional selective forces acting on a modifier locus affecting the selfing rate. These effects are still waiting for analytical exploration.
Acknowledgments
I thank the bioinformatics and computing service of the Roscoff Biological Station for computing time and Joachim Hermisson and two anonymous reviewers for helpful suggestions and comments. This work was supported by the French Agence Nationale de la Recherche (ANR-11-BSV7-013).
Footnotes
Communicating editor: J. Hermisson
Supporting information is available online at www.genetics.org/lookup/suppl/doi:10.1534/genetics.115.178533/-/DC1.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}