





Abstract
Allergic asthma is a complex disease characterized in part by granulocytic inflammation of the airways. In addition to eosinophils, neutrophils (PMN) are also present, particularly in cases of severe asthma. We sought to identify the genetic determinants of neutrophilic inflammation in a mouse model of house dust mite (HDM)-induced asthma. We applied an HDM model of allergic asthma to the eight founder strains of the Collaborative Cross (CC) and 151 incipient lines of the CC (preCC). Lung lavage fluid was analyzed for PMN count and the concentration of CXCL1, a hallmark PMN chemokine. PMN and CXCL1 were strongly correlated in preCC mice. We used quantitative trait locus (QTL) mapping to identify three variants affecting PMN, one of which colocalized with a QTL for CXCL1 on chromosome (Chr) 7. We used lung eQTL data to implicate a variant in the gene Zfp30 in the CXCL1/PMN response. This genetic variant regulates both CXCL1 and PMN by altering Zfp30 expression, and we model the relationships between the QTL and these three endophenotypes. We show that Zfp30 is expressed in airway epithelia in the normal mouse lung and that altering Zfp30 expression in vitro affects CXCL1 responses to an immune stimulus. Our results provide strong evidence that Zfp30 is a novel regulator of neutrophilic airway inflammation.
VARIATION in gene expression is thought to explain a substantial proportion of complex traits, including disease phenotypes (Nica and Dermitzakis 2008; Cookson et al. 2009; Nicolae et al. 2010). Indeed, one of the main conclusions from genome-wide associations (GWAS) studies of many diseases is that most of the causal variants appear to be regulatory in function, rather than affecting protein-coding gene regions (Hindorff et al. 2009; Nicolae et al. 2010; Maurano et al. 2012). In a simple scenario, a genetic variant causes a change in gene expression (i.e., is an expression quantitative trait locus, eQTL), which in turn influences disease risk (Emilsson et al. 2008). The same types of causal relationships between sequence variation, gene expression, and phenotypes have been documented in model organisms (Schadt et al. 2005; Harbison et al. 2009; Hageman et al. 2011).
We adopted this expression-centric view in an effort to identify new candidate genes for experimental asthma, which is composed of multiple phenotypes including granulocytic (eosinophilic and neutrophilic) inflammation of the airways, mucus metaplasia, and airway hyperresponsiveness. There is ample prior evidence that these traits have a genetic basis (Levitt and Mitzner 1988; Brewer et al. 1999; Whitehead et al. 2003; Kelada et al. 2011) and QTL have been identified for airway hyperresponsiveness on several chromosomes (De Sanctis et al. 1995, 1999; Karp et al. 2000; Ackerman et al. 2005; Camateros et al. 2010; Leme et al. 2010; Himes et al. 2013). However, a systematic evaluation of whether variation in gene expression underlies these traits has not been conducted.
To identify QTL and eQTL that may underlie the phenotypic QTL, we applied a house dust mite (HDM) mouse model of allergic asthma to incipient lines of the Collaborative Cross (CC), a new mouse systems genetics resource (Collaborative Cross Consortium 2012). The CC is composed of a panel of recombinant inbred lines derived from eight-way crosses using five classical inbred strains (C57BL/6J, 129S1/SvImJ, A/J, NOD/ShiLtJ, NZO/HlLtJ) and three wild-derived inbred strains (WSB/EiJ, PWK/PhJ, and CAST/EiJ) (Collaborative Cross Consortium 2012). The genomes of the CC founder strains (Keane et al. 2011; Yalcin et al. 2011) and the features of the CC population have been described recently (Collaborative Cross Consortium 2012). Results obtained using incipient CC lines that were partially inbred (referred to as “preCC” mice) have been notable in terms of phenotypic diversity and identification of new QTL (Aylor et al. 2011; Bottomly et al. 2012; Kelada et al. 2012; Ferris et al. 2013; Kelada et al. 2014).
We measured response to HDM sensitization and challenge in the eight CC founder strains and 151 preCC mice, and the work here is focused primarily on one particular HDM-response phenotype, airway polymorphonuclear neutrophil (PMN) recruitment. PMNs are associated with severe asthma and decrements in lung function in humans (Little et al. 2002; Wenzel 2006). In addition to PMN recruitment, we measured an intermediate phenotype, the concentration of CXCL1 in lung lavage fluid, a hallmark PMN chemokine (Bozic et al. 1995; Frevert et al. 1995). We identified a locus on chromosome (Chr) 7 that controls both phenotypes and integrated lung eQTL data to identify a candidate gene, Zfp30, whose expression is controlled by a cis-regulatory variant in the same region. We propose that this genetic variant regulates both CXCL1 and PMN by altering Zfp30 expression and we tested our model with structural equations and in vitro experiments. Taken together, our results provide strong evidence that Zfp30 is a novel regulator of neutrophilic airway inflammation.
Materials and Methods
Mice
We obtained 151 male preCC mice (ages 10–14 weeks) from Oak Ridge National Laboratory (Chesler et al. 2008; Kelada et al. 2012; Kelada et al. 2014). Each mouse was from an independent CC line that had undergone 5–14 generations of inbreeding. For each of the eight CC founder strains, we obtained five mice from The Jackson Laboratory. All mice were singly housed, with alpha-dri bedding, in the same facility at NIH under normal 12 hr light/dark cycles. All experiments conducted with mice herein were compliant with an IACUC protocol at an AALAC-approved facility.
Phenotyping protocol
Mice were sensitized with 10 µg of the immunodominant allergen from the Dermatophagoides pteronyssinus species of HDM, Der p 1, administered by intraperitoneal injection on days 0 and 7 of the protocol. On day 14, mice were challenged with 50 µg of Der p 1, administered by orotracheal aspiration (Kelada et al. 2011; Kelada et al. 2014). Mice were phenotyped on day 17 by performing lavage followed by differential cell counts. CXCL1 was measured using a Millipore (Billerica, MA) luminex assay. According to recent sequencing data (Keane et al. 2011), no genetic variation in Cxcl1 would affect detection of CXCL1 by the antibody used. Phenotype data are presented in Supporting Information, Table S1.
Heritability estimates
Narrow sense heritability (h2) was estimated by regression of phenotypes on genotype for the peak locus for the Zfp30 eQTL on chromosome 7. The allele effects for the eQTL indicated three functional alleles and the genotype data were coded accordingly. Genotypes and haplotype frequencies are provided in Table S2.
Genotyping and QTL mapping
Genotypes for 131of preCC mice used in these experiments have been previously reported (Kelada et al. 2012). Twenty additional mice are included in this study. Genotype data are provided in File S1. We genotyped each mouse at the University of North Carolina—Chapel Hill, using one of two Affymetrix SNP arrays (A or B) that were produced in development of the mouse diversity array (MDA) (Yang et al. 2009). After removing uninformative and poorly performing SNPs, these arrays contained 181,752 (A array) and 180,976 (B array) SNP assays, and the set of SNPs on each array did not overlap. Most mice (83%) were genotyped on the B array and the remaining were genotyped on the A array. These training arrays were annotated to NCBI build 36 of the mouse genome, but we mapped QTL boundaries to build 37 positions to integrate with other resources. We report NCBI build 37 positions in our results. We estimated the most probable ancestor for each SNP in each mouse using the GAIN algorithm (Liu et al. 2010) and reconstructed founder haplotypes on the basis of these results. We then merged the nonoverlapping SNP data sets from arrays A and B by imputing unobserved genotypes on the basis of inferred founder haplotype. For QTL mapping, we used HAPPY (Mott et al. 2000) to infer ancestry matrices for an additive genetic model. For efficiency, we then averaged the matrices across SNPs between which GAIN inferred no recombination in the population, and this reduced the mapping data set to 27,059 intervals. We used BAGPIPE (Valdar et al. 2009) to fit a regression model and to report LOD scores. Significance thresholds were determined by permutation. We used the 1.5 LOD drop method to approximate confidence intervals for QTL (Dupuis and Siegmund 1999).
Gene expression analysis
We previously identified eQTL from whole lung RNA using data from a subset of the same mice described here (n = 131) (Kelada et al. 2014). We used these data to identify genes with eQTL in the QTL region on chromosome 7. We included genes whose eQTL peak resided in the confidence interval for the PMN and CXCL1 QTL, spanning 26–30 Mb, and limited the analysis to local eQTL, which we defined as within 10 Mb upstream or downstream of the gene’s position (Kelada et al. 2014).
qRT–PCR and allele-specific PCR
To confirm that expression of Zfp30 measured by microarray was accurate, we performed qRT–PCR. We designed a SYBR green-based qRT–PCR assays for Zfp30 and two genes used for normalization Rps29 and Hprt. We computed the geometric mean of the Ct of the two normalization genes, and this was used to calculate the delta Ct. All primer pairs given below were designed to avoid SNPs in Collaborative Cross founder strains:Zfp30-F 5′-TGTTGGAACAAGGGAAGGAG-3′, Zfp30-R 5′-GCCACGCCTTTGAATTCTT-3′; Rps29-F 5′-GGAGTCACCCACGGAAGT-3′, Rps29-R 5′-TCCATTCAGGTCGCTTAGTC-3′; Hprt-F 5′-GCCCTTGACTATAATGAGTACTTCAGG-3′; and Hprt-R 5′-TTCAACTTGCGCTCATCTTAGG-3′.
To validate allele-specific expression of Zfp30, we employed pyrosequencing on lung cDNA samples from CC founder lines (C57BL/6J, WSB/EiJ, A/J, and CAST/EiJ and PWK/PhJ, n = 3/genotype) and F1 heterozygotes from CAST/EiJ × WSB/EiJ, PWK/PhJ × WSB/EiJ, and CAST/EiJ × 129S1/SvImJ crosses (n = 6/cross). We used PCR amplify to a region surrounding a SNP, rs45830063 (C/T), which is located in exon 5 of Zfp30, for which CAST/EiJ and PWK/PhJ have the alternate allele (T). PCR primers were 5′-AAGCCCTACGAATGTGGG-3′ and reverse 5′-TCGAAGGTCCGAGCTACAC-3′, and we used another primer, 5′-ATTCGTAGGGCTTCTCGCC-3′, in the sequencing reaction. Results are presented as percentage allele based on peak heights from the pyrograms.
Conditional correlation analysis
To identify eQTL that may underlie the QTL for PMN and CXCL1, we used conditional correlation. Specifically, we ran Chr 7 QTL scans for PMN, CXCL1, or gene expression, with covariates, either gene expression (GEX) or phenotype (P, i.e., PMN or CXCL1), as follows:
Structural equation models
We used structural equation models (SEMs) (Shipley 2000) to test whether colocalization of the PMN and CXCL1 QTL on Chr 7 reflects a common cause, namely the Zfp30 eQTL, or is the result of chance.
Detection of Zfp30 in the lung
In situ hybridization (ISH) was carried out on 18-μm-thick lung tissue sections from C57BL/6J mice using RNA probes generated from a plasmid containing Zfp30 cDNA sequence corresponding to GenBank accession no. BC049120.
In vitro assays with mouse airway epithelial cells
Primary mouse tracheal epithelial cells (MTECs) were grown under air–liquid interface conditions using a previously described protocol (You et al. 2002). Cells were harvested at selected time points (days 3, 5, 7, 11, 14, and 17) for RNA analysis. For knockdown experiments, we transfected an immortalized airway epithelial cell line, MLE12, with an siRNA for Zfp30 designed by Dharmacon: 5′-GCAAAGAUCUUCACGUGUA-3′. Cells were plated and transfected with 10 pmol of siRNA and then treated with lipopolysaccharide (LPS) 48 hr later to stimulate CXCL1 production. Twenty-four hours after LPS treatment, supernatants were collected and CXCL1 was measured using an ELISA assay (R&D Systems). We verified that siRNA treatment reduced Zfp30 levels using qRT–PCR since no antibody is available to detect ZFP30 protein.
Results
We measured HDM-induced airway PMN recruitment responses among the eight CC founder strains and 151 preCC mice by counting PMN in lung lavage fluid after HDM sensitization and challenge. We also measured concentrations of the well-known PMN chemokine CXCL1 in the same lung lavage fluid. For the sake of simplicity, we refer to these phenotypes as PMN and CXCL1, respectively. CC founder strains exhibited a range of PMN levels (Figure 1A) and the differences among strains provided evidence of heritability (Figure 1A, F-test P < 1.0 × 10−4). Estimates of broad sense heritability for PMN based on interclass correlations (r1) and the coefficient of genetic determination (g2) were 0.56 and 0.39, respectively. CXCL1 also differed significantly among CC founders (Figure 1B, F-test P = 0.01), although the between- vs. within-strain variance was not as great as for PMN, leading lower estimates of broad sense heritability (r1= 0.30 and g2 = 0.18).
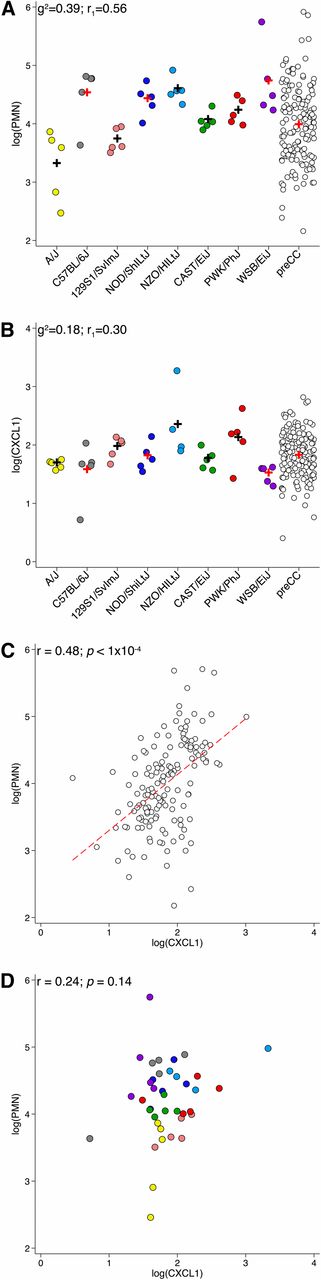
PMN and CXCL1 phenotype distributions and correlations in CC founder strains and preCC mice. (A) Log-transformed PMN counts in lavage fluid. Color coding of strains is as follows: dark gray, C57BL/6J; light blue, NZO/HlLtJ; blue, NOD/ShiLtJ; green, CAST/EiJ; yellow, A/J; salmon, 129S1/SvImJ; red, PWK/PhJ; purple, WSB/EiJ. This color coding is used throughout the manuscript and related papers (Collaborative Cross Consortium 2012). g2 and r1 values denote estimates of broad-sense heritability. (B) Log-transformed CXCL1 levels (pg/ml) in lavage fluid. Plus (+) symbols represent the means. Pairwise correlation of PMN and CXCL1 in (C) preCC mice and (D) CC founders. Dashed line represents linear fit for preCC mice.
Among preCC mice, there was substantial variation in both PMN and CXCL1 (Figure 1, A and B). The distributions of both phenotypes exceeded and interpolated the mean values of the eight founder strains, indicative of a polygenic basis of inheritance. Importantly, PMN and CXCL1 were strongly correlated among preCC mice (Figure 1C, Pearson r = 0.48, P < 1.0 × 10−4), whereas there was only weak evidence of correlation among CC founder strains (Figure 1D).
We then mapped QTL for PMN and CXCL1 (Figure 2) and identified one QTL for PMN on Chr 2 (Der p 1-induced pulmonary neutrophils, Dpn1; peak LOD = 7.54, padj < 0.05; 79.8–98.0 Mb) and two suggestive peaks on Chr 4 (LOD = 6.02, padj < 0.2; 3.3–10.0 Mb), and Chr 7 (LOD = 6.04, padj < 0.2; 24.6–29.8 Mb). For CXCL1, we identified a single QTL on Chr 7 (Dpc1; LOD = 7.66, padj < 0.05; 26.4–30.5 Mb), overlapping the PMN peak. Since Cxcl1 is located on Chr 5, Dpc1 is trans-regulator of CXCL1. The phenotypic correlation and colocalization of Chr 7 QTL for PMN and CXCL1 (Dpc1) suggested a variant linked to both phenotypes, and we focused our attention on this region for the rest of investigation.
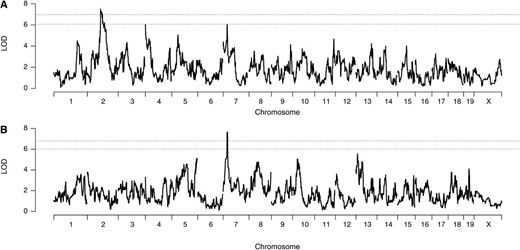
Genome scans of (A) PMN and (B) CXCL1. Dashed lines represent 95 and 80% significance thresholds. Note the colocalized QTL for both traits on chromosome 7.
We asked whether we could prioritize candidate genes for Dpc1 using gene expression data. We queried a lung eQTL data set we recently generated (Kelada et al. 2014) and focused our search for candidate genes on genes with a local eQTL in Dpc1 region on Chr 7 (24.6–30.5 Mb). Fifty-two genes corresponding to 70 array probes met this criterion (Table S3). We then used a conditional correlation approach to evaluate whether any of these 70 eQTL could explain the PMN and CXCL1 QTL. We evaluated whether inclusion of GEX into the QTL model for PMN or CXCL1 caused a substantial drop in the LOD score at the peak locus on Chr 7 (models 1 and 2 in Materials and Methods), which is indicative of a causal relationship (i.e., QTL → GEX → P). We also tested if the gene expression trait is reactive to the phenotype (QTL → P → GEX) by comparing LOD scores obtained by from QTL models in which GEX is the phenotype and the covariate is either CXCL1 or PMN (models 3 and 4 in Materials and Methods). Zfp30 was the only gene that had a local eQTL, showed a large drop in the LOD score for both PMN and CXCL1 (Figure 3) in the conditioned model, and did not show evidence of being reactive to PMN or CXCL1 (Table S3). Additionally, out of all 52 genes, the pairwise correlations between gene expression and CXCL1 and PMN were strongest for Zfp30 (r = −0.36, P < 1.0 × 10−4 and r = −0.39, P < 1.0 × 10−4, respectively; Table S3).
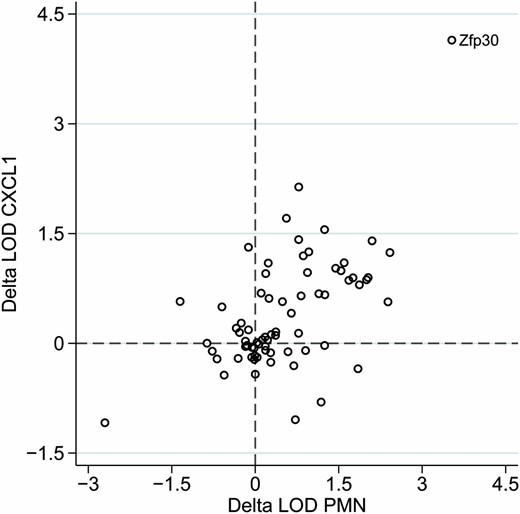
Conditioned QTL scans of CXCL1 and PMN with gene expression identify Zfp30 as a strong candidate gene. x- and y-axes represent the change in the LOD score (delta LOD) at the peak locus on Chr 7 when expression of genes with local eQTL is included in the QTL model. Zfp30 stands out because of the large delta LOD for both CXCL1 and PMN.
To characterize the Zfp30 eQTL, we examined the pattern of gene expression among CC founders and preCC mice (Figure 4). CC founders and preCC mice of the same haplotype had equivalent levels of Zfp30 gene expression, indicative of a limited effect of genetic background, and were distributed in three groups composed of PWK/PhJ or CAST/EiJ alleles (group 1), C57BL/6J or WSB/EiJ alleles (group 2), and A/J, 129S1/SvImJ, NOD/ShiLtJ, or NZO/HlLtJ alleles (group 3). PreCC mice homozygous for group 3 alleles had 2.8 and 1.8 times higher Zfp30 expression compared to preCC mice homozygous for group 1 and group 2 alleles, respectively (P < 1 × 10−4 for both comparisons). Likewise, preCC mice homozygous for C57BL/6J or WSB/EiJ alleles had 1.6 times higher expression than preCC mice homozygous for PWK/PhJ or CAST/EiJ alleles had (P < 1.0 × 10−4). Gene expression among preCC mice that were heterozygous at the eQTL peak (n = 41; far right side of Figure 4A) was consistent with a semidominant mode of inheritance.
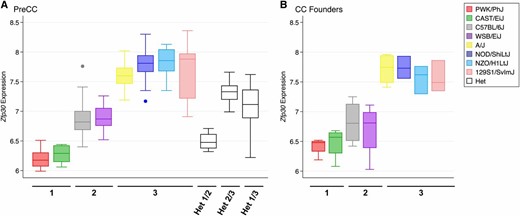
Zfp30 gene expression among CC founder strains and preCC mice. Array-based measurements of Zfp30 expression from 131 preCC mice partitioned by their founder haplotype at the peak locus for the eQTL on Chr 7 (A) and three to four mice per CC founder strain (B) are shown. Heterozygous preCC are shown at the far right in A.
We compared array-based measurements of Zfp30 expression to qRT–PCR-based measurements among CC founder strains (n = 3–4 mice/strain) and found a strong correlation overall (R2 = 0.85, P = 0.001; Figure S1). Further, we performed an analysis of allele-specific gene expression using pyrosequencing (Figure S2). We measured the amount of expressed reference allele (C) for rs45830063 among F1 mice (n = 6–7/cross) from crosses between CC founder lines from each of the three expression groups. As expected on the basis of array data, F1 heterozygotes of group 2 and 3 mice expressed more of the C allele than the T allele (P < 0.05 for both t-test comparisons), and we also observed higher C allele expression among group 1/3 heterozygotes compared with group 2/3 heterozygotes (P < 0.05).
Collectively, our results indicate that there are three functional alleles in CC founder strains and preCC mice and that cis-regulatory variation in Zfp30 affects gene expression in a semidominant manner. The allele effects pattern is consistent with CC founder haplotypes spanning the majority of the Zfp30 locus. We examined sequence of CC founder strains (Keane et al. 2011) for Zfp30, including 3 kb of upstream sequence and 1.5 kb of downstream sequence (30,566,017–30,581,221 bp, containing 288 SNPs in total) and found that except for regions downstream of exon 4, the phylogeny of the CC founders supports three distinct alleles (Figure S3). More specifically, there are 140 SNPs in this delimited region and 99.3% of them (n = 139) are consistent with three alleles and therefore are potential causal variants (Table S4).
These three alleles account for 78% of variation in Zfp30 expression (i.e., h2 = 0.78), which is consistent with the broad sense heritability we estimated (H2 = 0.58–0.73) based on array data from the CC founder strains. Thus, the eQTL is the source of most variation in Zfp30 expression. The Zfp30 allele effects were consistent with those for CXCL1 (h2 = 0.14) and PMN (h2 = 0.08) (Figure 5). The narrow sense heritability estimate for CXCL1 approaches the lower bound of the broad sense heritability estimates (0.18–0.30), indicating that Dpc1 is the major source of heritable variation of CXCL1 in the context of allergen challenge. PMN (H2 = 0.39–0.56) has a more complex genetic architecture, with contributions from two other loci. This accumulated evidence led us to conclude that Dpc1 is the Zfp30 eQTL.
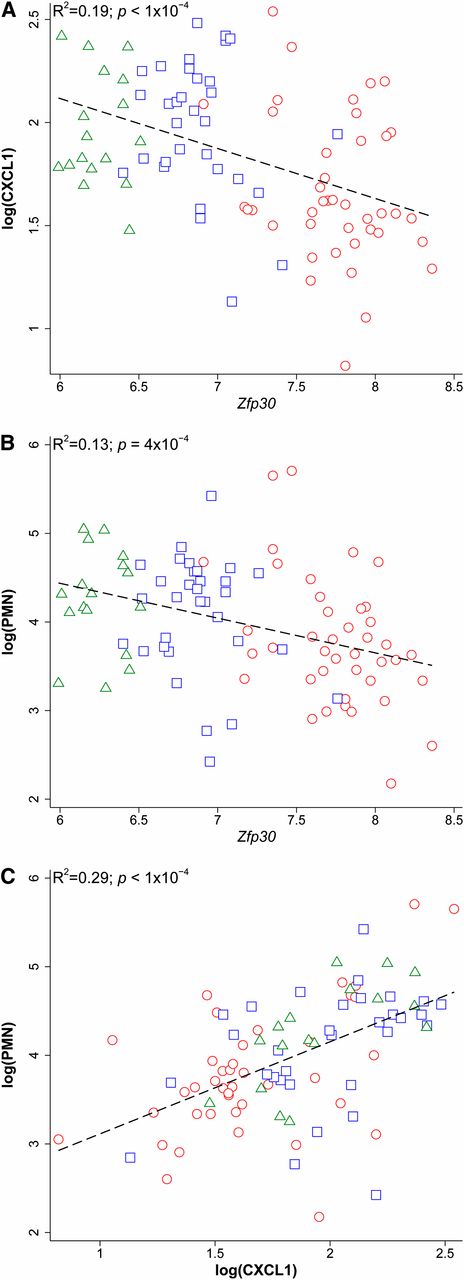
Pairwise correlations of Zfp30 gene expression, CXCL1, and PMN as a function of Chr 7 locus CC founder genotype. Correlations between traits among homozygous preCC mice are shown, and mice are coded according to eQTL allele effect groups. Red circles represent group 3 homozygotes, blue squares represent group 2 homozygotes, and green triangles represent group 1 homozygotes. (A) CXCL1 vs. Zfp30 gene expression. (B) PMN vs. Zfp30 gene expression. (C) PMN vs. CXCL1. Dashed lines represent linear fit.
We used SEMs to test if the Dpc1 effect on CXCL and PMN was independent of Zfp30 expression. We evaluated the fit of causal (QTL → CXCL1 → PMN), reactive (QTL → PMN → CXCL1), and independent (CXCL1 ← QTL → PMN) models that included Zfp30 gene expression (Figure 6). We found that two models (Figure 6, E and F) fit the data equally well, and both indicate that (1) the localization of the CXCL1 and PMN QTL are not independent, (2) Zfp30 expression underlies both CXCL1 and PMN, and (3) Zfp30 has an effect on PMN independent of CXCL1 (curved arrows). We cannot resolve the directionality of the causal relationship between CXCL1 and PMN. Nonetheless, we conclude that the association between Dpc1 and both CXCL1 and PMN is secondary to Dpc1’s effect on Zfp30 expression.
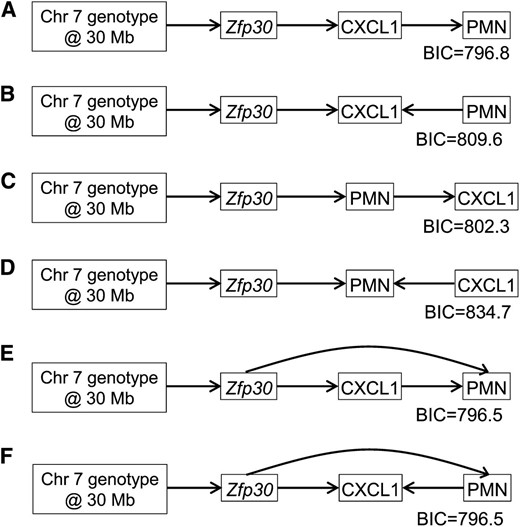
Structural equation models of Chr 7 QTL, Zfp30 gene expression, CXCL1, and PMN. A series of linear models were built and the results are depicted graphically by nodes, which represent the phenotypes, and directed edges that indicate causal relationships. Models E and F are the best-fit models because they have the lowest Bayesian information criterion (BIC) values. Both models E and F place Zfp30 upstream of CXCL1 and PMN and also indicate that Zfp30 may have an effect on PMN independent of CXCL1. Model F also indicates that PMN may feedback on CXCL1.
Given this preponderance of evidence for Zfp30 as a novel factor in the airway inflammation, we sought to validate its role using ex vivo and in vitro approaches. We first confirmed its expression in mouse lung using ISH and found that Zfp30 was expressed in airway epithelia in the lung and trachea (Figure 7A). Expression outside of the epithelia is also apparent, but expression appears to be most abundant in the epithelia. We examined Zfp30 gene expression in airway epithelia in more detail using MTECs. Zfp30 expression increased over the time course of MTEC growth and differentiation in air–liquid interface cultures, peaking at day 17, where expression was 24 times greater than in whole lung tissue (Figure 7B). These results demonstrate that Zfp30 is expressed in well-differentiated airway epithelia, a cell type of the lung that is known to have an important role in innate and acquired immune responses. Finally, we tested whether altering Zfp30 expression in immortalized mouse airway epithelial cells (MLE12) in vitro affected immune response to LPS, a known inducer of CXCL1. As predicted on the basis of our in vivo preCC data showing a negative correlation between Zfp30 expression and CXCL1, knockdown of Zfp30 led to heightened CXCL1 in supernatants of cells exposed to LPS (Figure 7C, 43% change, P < 0.05). These results corroborate our findings from preCC mice and indicate that Zfp30 is a novel regulator of immune responses in the lung.
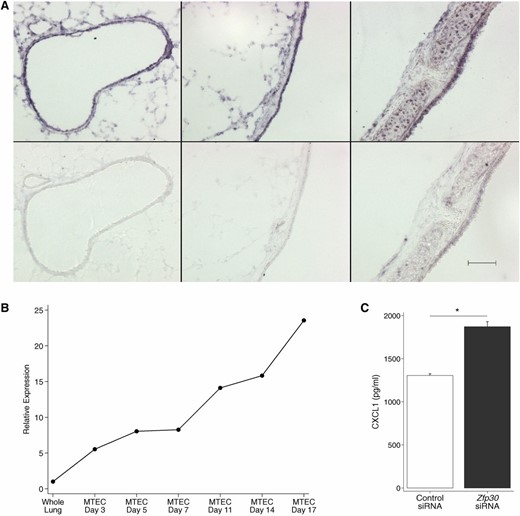
Zfp30 expression in mouse airways and its effect on CXCL1. (A) In situ hybridization using a Zfp30 RNA probe in the lung and trachea of a C57BL/6J mouse. Representative images are shown. Top: sections probed with anti-sense probe. Bottom: sections probed with the sense (control) probe. Left, medium airway; middle, large airway; right, trachea. Scale bar, 100 μm. (B) Time course of Zfp30 gene expression in mouse tracheal epithelial cells (MTEC) grown in culture shows a correlation between MTEC differentiation and Zfp30 expression. Cells are subjected to air–liquid interface on day 14. (C) siRNA-based knockdown of Zfp30 enhances CXCL1 secretion in response to LPS treatment. MLE12 cells were stimulated with 100 ng/ml LPS after transfection with a control siRNA or siRNA designed for Zfp30. (*) P < 0.05. Data are representative of three experiments with similar results.
Discussion
We used an inbred strain survey, QTL mapping, conditional correlation analysis, and causal inference to identify Zfp30 as a candidate gene for neutrophilic response to HDM sensitization and challenge. The use of preCC mice in QTL mapping experiments afforded us some notable strengths. First, mapping resolution was on the order of 5 Mb for the phenotype QTL on Chr 7, a small size in comparison to typical F2-intercross studies. Additionally, by employing the hypothesis that functionally similar Zfp30 alleles are similar in sequence and are descended from a common ancestor (Kelada et al. 2012), we identified a set of putative causal variants.
Our SEMs integrated four levels of information [genetic variation, Zfp30 expression, CXCL1 (protein) expression, and PMN] and identified a novel regulator of a classical chemokine response. The best-fit models predict that decreasing Zfp30 expression will increase CXCL1 and hence PMN. Subsequent in vitro assays validated these predictions. The SEMs also indicate that the relationship between CXCL1 and PMN may involve feedback. This is consistent with observations that PMN can secrete CXCL1 in a feed-forward loop (Scapini et al. 2000). These results provide a foundation for future experiments that will characterize the functional role of Zfp30 in neutrophilic airway disease.
PMN are one of the first responders to innate immune stimuli. Their trafficking to the site of injury is controlled by the local gradient of CXC cytokines including CXCL1 and CXCL2 (MIP-2), as well as GM–CSF, which controls PMN release from the bone marrow. PMN contribute to the clearing of infection through a variety of mechanisms including the release of antibacterial proteins (Kolaczkowska and Kubes 2013). In humans, PMN are associated with asthma severity (Wenzel et al. 1997; Jatakanon et al. 1999) and some have suggested that the addition of PMN to eosinophilic inflammation may represent a distinct asthma subtype, one that is resistant to corticosteroids (Wenzel et al. 1999). Thus PMN are associated with both acute and chronic immune responses. At the present, it is not clear from our results whether the PMN response we observed was due to an acquired immune response to HDM per se or are the result of an innate immune response, but the fact that Zfp30 expression was primarily localized to airway epithelia (not an immune cell) suggests the latter. Innate immune responses are known to prime acquired immune responses through activation of pattern recognition receptors (Schenten and Medzhitov 2011); hence the role of Zfp30 in both innate and acquired immune responses should be investigated further. It is worthwhile noting that two previous studies identified QTL for innate immune response phenotypes at approximately the same location on Chr 7. Using A/J × C57BL/6J (AxB) recombinant inbred strains, Matesic et al. (1999) found that variation at this locus modifies LPS-induced splenocyte proliferation responses. Likewise, Denny et al. (2003) found that this region controls survival and bacterial load after pulmonary infection with Streptococcus pneumoniae in an F2 intercross between BALB/cO1aHsd and CBA/CaO1aHsd strains. In both of these studies, the strains used had contrasting alleles at the Zfp30 locus [according to Sanger data (Keane et al. 2011), BALB/c has the same allele as A/J and CBA has the same allele as C57BL/6J]. Hence these results are consistent with the hypothesis that the variation in Zfp30 affects responses to a variety of immune stimuli.
We do not yet know how Zfp30 controls CXCL1 and PMN influx. We examined gene expression data we previously collected and found that Zfp30 expression does not change as a function of allergen challenge in whole lung or isolated airway epithelia (unpublished results). Thus baseline variation in Zfp30 expression appears to be a determinant of the response to house dust mite allergen. It is somewhat surprising that a gene whose expression is not modulated by allergen challenge can play such an important role in neutrophil recruitment responses to allergen. How baseline variation in Zfp30 expression modifies these responses remains to be determined.
Little is known about the function of ZFP30 (the protein product of Zfp30), except from predictions based on amino acid sequence motifs. ZFP30 is a C2H2-type zinc finger that contains a Krupple-associated box (KRAB) domain, which are known to repress gene expression (Witzgall et al. 1994), perhaps through the induction of heterochromatin (Groner et al. 2010). Zfp30 expression was negatively associated with CXCL1 protein (and PMN) but was not associated with lung Cxcl1 RNA in preCC mice (not shown), suggesting that Cxcl1 RNA is not a direct target of ZFP30 (in contrast to ZFP36, tristetraprolin, which is well-known regulator of Cxcl1 RNA levels; Datta et al. 2008). The targets and sequence specificity of ZFP30 are clearly important questions to address in the future and will be necessary to understanding how variation in this gene affects immune response.
Allergic asthma is a complex trait that is influenced by multiple genetic loci and environmental exposures. Neutrophilic inflammation is one feature of the disease and is itself a complex trait. Our results suggest that the PMN response to HDM has contributions from at least three loci. We used a systems genetics approach to show that PMN is mediated by CXCL1 and Zfp30 expression, and Zfp30 expression is under local genetic control by Dpc1. This biological cascade is undoubtedly a small piece of a larger puzzle. Nonetheless, we have tied a novel genetic variant to a complex biological response through its most proximal endophenotype, Zfp30 expression.
Acknowledgments
We thank Drs. Francis S. Collins (National Institutes of Health, NIH) and David A. Schwartz (University of Colorado School of Medicine) for support and advice during the course of this project, Dr. Darla R. Miller (University of North Carolina, UNC) for providing research support that facilitated the mouse experiments conducted herein, John Didion (UNC) for his assistance with array probe SNP mapping, Kim Burns of the Marsico Lung Institute’s Histology Core for assistance with mouse lung tissue sections, and Amanda Raimer for technical assistance with the pyro-sequencing assays. This work was supported in part by the intramural program of the National Human Genome Research Institute, National Institutes of Health (NIH) (ZIA-HG200361), the U.S. Department of Energy, Office of Biological and Environmental Research, and by NIH grants U01CA134240, U01CA105417, F32GM090667, K99ES021535, GM070683, an National Institutes of General Medical Sciences Centers of Excellence in Systems Biology program grant GM076468, and the Expression Localization Core Facility funded by National Institute of Neurological Disorders and Stroke Center grant P30 NS045892.
Footnotes
Supporting information is available online at http://www.genetics.org/lookup/suppl/doi:10.1534/genetics.114.168138/-/DC1.
Sequence data from this article have been deposited with the GenBank Data Libraries under accession no. BC049120.
Communicating editor: D. W. Threadgill
Literature Cited
Collaborative Cross Consortium, 2012 The genome architecture of the Collaborative Cross mouse genetic reference population. Genetics 190: 389–401.
Nica, A. C., and E. T. Dermitzakis, 2008 Using gene expression to investigate the genetic basis of complex disorders. Hum. Mol. Genet. 285: 17: R129–R134.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}