





Abstract
With rare exceptions, human tumors arise from single cells that have accumulated the necessary number and types of heritable alterations. Each such cell leads to dysregulated growth and eventually the formation of a tumor. Despite their monoclonal origin, at the time of diagnosis most tumors show a striking amount of intratumor heterogeneity in all measurable phenotypes; such heterogeneity has implications for diagnosis, treatment efficacy, and the identification of drug targets. An understanding of the extent and evolution of intratumor heterogeneity is therefore of direct clinical importance. In this article, we investigate the evolutionary dynamics of heterogeneity arising during exponential expansion of a tumor cell population, in which heritable alterations confer random fitness changes to cells. We obtain analytical estimates for the extent of heterogeneity and quantify the effects of system parameters on this tumor trait. Our work contributes to a mathematical understanding of intratumor heterogeneity and is also applicable to organisms like bacteria, agricultural pests, and other microbes.
HUMAN cancers frequently display substantial intratumor heterogeneity in genotype, gene expression, cellular morphology, metabolic activity, motility, and behaviors such as proliferation rate, antigen expression, drug response, and metastatic potential (Fidler and Hart 1982; Heppner 1984; Nicolson 1984; Campbell and Polyak 2007; Dick 2008). For example, a molecular and phenotypic analysis of breast cancer cells has revealed defined subpopulations with distinct gene expression and (epi)genetic profiles (Shipitsin et al. 2007). Heterogeneity and the existence of subpopulations within single tumors have also been demonstrated via flow cytometry in cervical cancers and lymph node metastases (Nguyen et al. 1993) as well as in leukemias (Wolman 1986). Virtually every major type of human cancer has been shown to contain distinct cell subpopulations with differing heritable alterations (Heppner 1984; Merlo et al. 2006; Campbell and Polyak 2007). Heterogeneity is also present in premalignant lesions; for instance, genetic clonal diversity has been observed in Barrett's esophagus, a condition associated with increased risk of developing esophageal adenocarcinoma (Maley et al. 2006; Lai et al. 2007).
Tumor heterogeneity has direct clinical implications for disease classification and prognosis as well as for treatment efficacy and the identification of drug targets (Merlo et al. 2006; Campbell and Polyak 2007). The degree of clonal diversity in Barrett's esophagus, for instance, is correlated with clinical progression to esophageal adenocarcinoma (Maley et al. 2006). In prostate carcinomas, tumor heterogeneity plays a key role in pretreatment underestimation of tumor aggressiveness and incorrect assessment of DNA ploidy status of tumors (Wolman 1986; Haggarth et al. 2005). Heterogeneity has long been implicated in the development of resistance to cancer therapies after an initial response (Geisler 2002; Merlo et al. 2006) as well as in the development of metastases (Fidler 1978). In addition, tumor heterogeneity hampers the precision of microarray-based analyses of gene expression patterns, which are widely used for the identification of genes associated with specific tumor types (O'Sullivan et al. 2005). These issues underscore the importance of obtaining a more detailed understanding of the origin and temporal evolution of intratumor heterogeneity.
To study the dynamics of intratumor heterogeneity, we construct and analyze a stochastic evolutionary model of an expanding population with random mutational fitness advances. Evolutionary models of populations with random mutational advances have been studied in the context of fixed-size Wright–Fisher processes for both finite and infinite populations (Gerrish and Lenski 1998; Park and Krug 2007; Park et al. 2010); Gerrish and Lenski (1998) studied the speed of evolution in a Wright–Fisher model with random mutational advances in the context of finite but large populations while Park et al. (2010) obtained accurate asymptotic approximations for the evolutionary dynamics of the population, following ideas presented in Park and Krug (2007). The latter work and references therein constitute a substantial exploration of the effects of random mutational advances in fixed-size populations. Our present work complements this research by exploring the effects of random mutational advances in the context of exponentially expanding populations and in particular the implications of these mutational advances on population heterogeneity. Models of exponentially expanding populations are appropriate for the study of situations arising during tumorigenesis, but are also applicable to other organisms undergoing binary replication such as bacteria, agricultural pests, and other microbes and pathogens. Bacterial populations, for instance, are diverging quickly in both genotype and phenotype, as studied by Lenski and colleagues. These investigators examined the dynamics of phenotypic evolution in populations of Escherichia coli that were propagated by daily serial transfer for 1500 days, yielding 10,000 generations of binary fission (Lenski et al. 1991; Lenski and Travisano 1994). The fitness of the bacteria improved on average by 50% relative to the ancestor, and other phenotypic properties, such as cell size, also underwent large changes. Similarly, single malaria isolates have been found to consist of heterogeneous populations of parasites that can have varying characteristics of drug response, from highly resistant to completely sensitive (Foley and Tilley 1997). These findings have implications for treatment strategies, as not all pathogen populations are sensitive to therapeutic interventions, and necessitate the study of diversity dynamics in growing populations of cells.
In this article, we consider an exponentially expanding population of tumor cells in which (epi)genetic alterations confer random fitness changes to cells. This model is used to investigate the extent of genetic diversity in tumor subpopulations as well as its evolution over time. The mathematical framework is based on the clonal evolution model of carcinogenesis, which postulates that tumors are monoclonal (i.e., originating from a single abnormal cell) and that over time the descendants of this ancestral cell acquire various combinations of mutations (Merlo et al. 2006; Campbell and Polyak 2007). According to this model, genetic drift and natural selection drive the progression and diversity of tumors. Our work complements studies of the effects of random mutational fitness distributions on the growth kinetics of tumors (Durrett et al. 2010) and contributes to the mathematical investigation of intratumor heterogeneity (Coldman and Goldie 1985, 1986; Michelson et al. 1989; Kansal et al. 2000; Komarova 2006; Haeno et al. 2007; Schweinsberg 2008; Bozic et al. 2010; Durrett and Moseley 2010).
MATERIALS AND METHODS
We consider a multitype branching process model of tumorigenesis in which (epi)genetic alterations confer an additive change to the birth rate of the cell. This additive change is drawn according to a probability distribution v, which is referred to as the mutational fitness distribution. Cells that have accumulated i ≥ 0 mutations are denoted as type-i cells. Initially, the population consists entirely of type-0 cells, which divide at rate a0, die at rate b0, and produce type-1 cells at rate u. The initial population, whose size is given by V0, is considered to be sufficiently large so that its growth can be approximated by , where λ0 = a0 − b0 and time t is measured in units of cell division. Type-1 cells divide at rate a0 + X, where X ≥ 0 is drawn according to the distribution v, and give rise to type-2 cells at rate u. All cell types die at rate b0 < a0. In general, a type-(k − 1) cell with birth rate ak−1 produces a new type-k cell at rate u, and the new type-k cell type divides at rate ak−1 +X, where is drawn according to v. Each type-k cell produced by a type-(k − 1) cell initiates a genetically distinct lineage of cells, and the set of all of its type-k descendants is referred to as its family. The total number of type-k cells in the population at time t is given by and the set of all type-k cells is called the kth wave or generation k. The total population size at time t is given by .
Note that in our model, mutations are not coupled to cell divisions but instead occur at a fixed rate per unit of time. This model can be modified to exclusively consider (epi)genetic alterations that occur during cell division by assuming that type-0 individuals divide at rate α0 and during each division, there is a chance μ that a mutation occurs, producing a type-1 individual. These two model versions are equivalent for wave-1 cells if a0 = α0(1 − μ) and u = α0μ. For later waves, the model must be altered so that the mutation rate is dependent upon the genetic constitution of the cells, since accumulation of alterations modifies the fitness of the cell and hence the rate at which further changes are accumulated. Analysis of this model would then require the mutation rate term to be inside the integral over the support of the fitness distribution; however, this modification does not alter the limiting results significantly. Our model and results can also be modified to account for generation-dependent mutation rates (Coldman and Goldie 1985; Park et al. 2010).
The mutational fitness distribution v determines the effects of each (epi)genetic change that is accumulated in the population of cells. We consider fitness distributions concentrated on [0, b] for some b > 0. We discuss two distinct classes of distributions: (i) v is discrete and assigns mass gi to a finite number of values ; (ii) v is continuous with a bounded density that is continuous and positive at b. Figure 1 shows a snapshot of the population decomposition in a sample simulation for case ii and illustrates the complex genotypic composition of the population of cells generated by our model.
![A sample cross-section of tumor heterogeneity. (A) The composition of a tumor cell population at t=150 time units after tumor initiation. Each “wave” of cells, defined as the set of cells harboring the same number of (epi)genetic alterations, is represented by a different color. Individual clones of cells with identical genotype are shown as circles, positioned on the horizontal axis according to fitness (i.e., birth rate) and on the vertical axis according to clone size. Note that later waves tend to have larger fitness values. (B) The time at which individual clones of cells were created during tumor progression. The color scale depicts the time of emergence of each clone. In A and B, parameters are a0=0.2, b0=0.1, v∼U([0,0.05]), and u=0.001.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/genetics/188/2/10.1534_genetics.110.125724/6/m_461fig1.jpeg?Expires=1716554805&Signature=HBbx-aiqZWgo5l9ROvNM5dlmiIlJgDlpoA1bGGaVTGTk65c1oSEkxSHDP8WAnNFZlGSSNE9tO2KBYNiZYpMbgPi3ic0BZmQ0dbH~P7k4VchWfjeFpUhP2WS9Wq98ztk63pS87qr2QEHprfABT8wQR5xTHZ5hnnu-RUoJxfRoANsCgygl7eY5JcWICac4YlcuV8RCRJu1ETnc0ibwBHK18jj5JQ8SjH8KyyNoKCzvLIuUrllQX5e-wD-~GO3cnVwrAEeo4DcY8eYfv2M6CMXqO0PYdXc4V9rI8ouheQhP589fU6FiPSgteSBbsgwJYioK8-P0KTINPBYat7QYaN7yDQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
A sample cross-section of tumor heterogeneity. (A) The composition of a tumor cell population at time units after tumor initiation. Each “wave” of cells, defined as the set of cells harboring the same number of (epi)genetic alterations, is represented by a different color. Individual clones of cells with identical genotype are shown as circles, positioned on the horizontal axis according to fitness (i.e., birth rate) and on the vertical axis according to clone size. Note that later waves tend to have larger fitness values. (B) The time at which individual clones of cells were created during tumor progression. The color scale depicts the time of emergence of each clone. In A and B, parameters are , , , and
The determination of the distribution v in the context of bacteria and viruses has been the subject of several experimental studies (Imhof and Schlotterer 2001; Sanjuan et al. 2004), which have generally produced results leading to the conclusion that v has an exponential distribution. However, more recently Rokyta and colleagues (Rokyta et al. 2008) presented studies of bacteriophages, in which the distribution of beneficial mutational effects appears to have a truncated right tail. One possible explanation for this result is that the experiments were done at an elevated temperature that might have led to a limited number of available beneficial mutations. A similar scenario might arise during tumorigenesis when only a limited number of (epi)genetic alterations allow a cell to progress to a more aggressive phenotype.
RESULTS
There are two sources of heterogeneity present in the population: variability in the number of mutations per cell (heterogeneity between generations) and genotypic variation between members of the same generation (heterogeneity within a generation). We investigate these two sources of heterogeneity and derive analytic results that quantify the relationship between model parameters—e.g., mutation rate and mutational fitness distribution—and the amount of genotypic variation present in the population over time.
Between-generation heterogeneity:
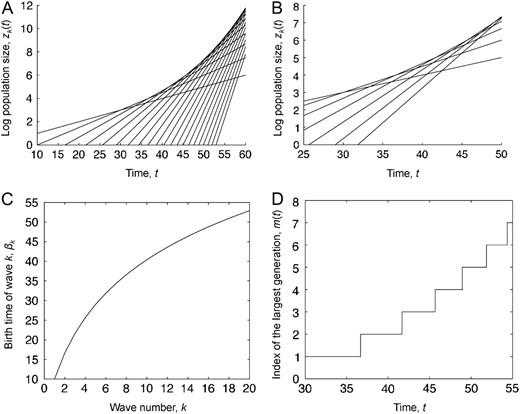
The process for the small mutation limit. The limiting process is shown for the case in which the mutation rate goes to zero. (A) The time, t, on the horizontal axis vs. the log number of cells of wave k, , as given by Equation 3. Both time and space are given in units of L = log(1/u). This plot shows the first 20 waves started at i.e., the time that type-1 cells begin to be born. (B) A closer look at the first 7 waves from a, showing the changes in the dominant type. (C) The birth times of the first 20 generations as a function of the generation number. (D) The dominant type in the population as a function of time. The index of the largest generation at time t is defined as . In A–D, parameters are and .
Bozic et al. (2010) observed a similar acceleration of waves in their model of mutation accumulation. On the basis of approximations by Beerenwinkel et al. (2007), they concluded that this acceleration was an artifact of the presence of both passenger and driver mutations and that it does not occur when only driver mutations conferring a fixed selective advantage are considered—i.e., when the fitness increments are deterministic. In contrast to these conclusions, we find that the acceleration of waves occurs regardless of the choice of mutational fitness distribution and is due to the difference in growth rates between successive generations: type-k cells arise when generation k − 1 reaches size and since the asymptotic growth rate of generation k is larger than the asymptotic growth rate of generation k − 1, generation k + 1 reaches size faster than generation k. We observed another example of this phenomenon earlier during our study of a related Moran model for tumor growth in which the total population of cells grows at a fixed exponential rate (Durrett and Mayberry 2010). In this model, the cause of acceleration was similarly related to growth rates—later generations take longer to achieve dominance in the expanding population of cells, and hence new types are born with a higher fitness advantage compared to the population bulk, allowing them to grow more rapidly.
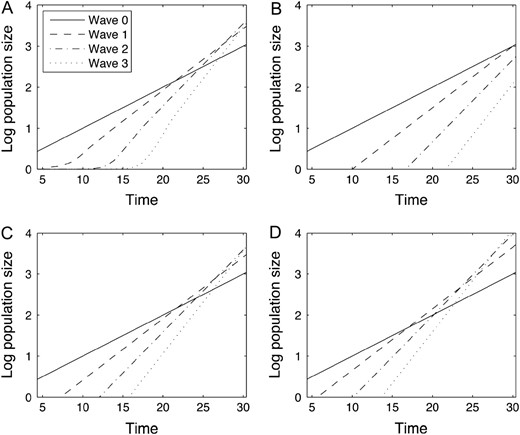
The size of the first four generations of cells. The log size of generations 1–4 is shown as a function of time t. Both time and space are plotted in units of . (A) The average values of the log generation sizes over sample simulations. (B) The limiting approximation from Equation 3 for the log size of the generations. (C) The approximation from Equation 6 using the mean of . (D) The approximation from Equation 6 using a value two standard deviations above the mean of to demonstrate an extreme scenario. Parameters are , , , , and
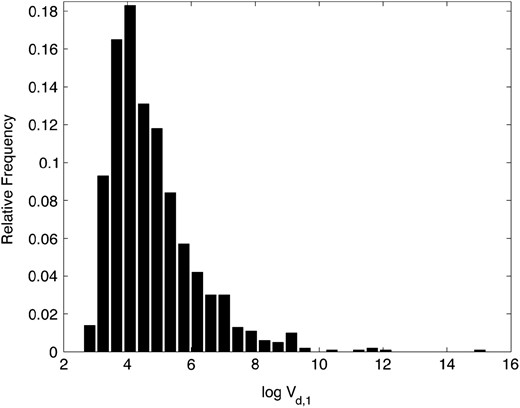
The distribution of log Vd,1. The relative frequency histogram of 1000 random samples from the distribution of is shown, as specified by Equation 1.
Means and standard deviations for log Vd,k for k = 1, 2, 3, as specified by Equation 1
| Generation | Mean | Standard deviation |
| 1 | 4.7638 | 1.3738 |
| 2 | 10.6010 | 2.2434 |
| 3 | 17.0519 | 3.0282 |
| Generation | Mean | Standard deviation |
| 1 | 4.7638 | 1.3738 |
| 2 | 10.6010 | 2.2434 |
| 3 | 17.0519 | 3.0282 |
Means and standard deviations for log Vd,k for k = 1, 2, 3, as specified by Equation 1
| Generation | Mean | Standard deviation |
| 1 | 4.7638 | 1.3738 |
| 2 | 10.6010 | 2.2434 |
| 3 | 17.0519 | 3.0282 |
| Generation | Mean | Standard deviation |
| 1 | 4.7638 | 1.3738 |
| 2 | 10.6010 | 2.2434 |
| 3 | 17.0519 | 3.0282 |
While the limit in (3) depends on the fitness distribution only through the maximum attainable fitness increase b, the distribution of also depends on the fitness distribution through the probability of attaining a fitness advance of b if v is discrete and the value of the probability density function at b if v is continuous (see Equation 4.9 in Durrett et al. 2010). As a consequence, our finite time approximation (6) takes into consideration the shape of the fitness distribution near b and the corrector term log accounts for variations in the likelihood of attaining the maximum possible fitness advance.
Within-generation heterogeneity:
We begin our investigation of within-generation heterogeneity by examining the extent of diversity present in the first generation of cells. We use two statistical measures to assess heterogeneity: (i) Simpson's index, which is given by the probability that two randomly chosen cells from the first generation stem from the same clone, and (ii) the fraction of individuals in the first generation that stem from the largest family of cells. To obtain these results, we derive an alternate formulation of the limit in Equation 1 that shows the limit is the sum of points in a nonhomogeneous Poisson process (see the appendix for more details). Each point in the limiting process represents the contribution of a different mutant lineage to so that it suffices to calculate i and ii for the points in the limiting process.
Simpson's index:
![The expected value of Simpson's index for the first wave of cells. The sample mean of Simpson's index (dots) over time t and the expected value of Simpson's index for the limiting point process (line) are shown, for two different values of α. Parameters are λ0=0.1, a0=0.2, and v∼U([0,b]), where b=0.01 in a and b=0.05 in b.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/genetics/188/2/10.1534_genetics.110.125724/6/m_461fig5.jpeg?Expires=1716554805&Signature=Ij3zwjUygFomF2sOWvNZ570fX-PF2z4lo37sKCDiF2BX-KF8V0A2uXMiMmj0XJBlfHAuZ5FGCMwQLEdKlSId8ZuMcx4M16KnqkK~W3Z1vBf2mpDDqyyYKcn8PGl2SK8vz2IqKzGbU7xnGVUdkwHTdjQ~8sJvvEELiVQUbHuiIfPIZJAoR7hxPfaYTPBjbknUm8xJWCKBeXtZgOrO19roXk3keAXNySkvIhvuvlYNdClgWqWJUu1RUf1IHDu2LNArjIWOcFml7m9CTU~0O6mbwkHPH4PgcY~OkNQTSYS9DN-E5xuh5Vvbtoo8B2efV8j80k3w5DtdBBGSeoUK9cRh9Q__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
The expected value of Simpson's index for the first wave of cells. The sample mean of Simpson's index (dots) over time t and the expected value of Simpson's index for the limiting point process (line) are shown, for two different values of . Parameters are , , and , where in a and in b.
![The empirical distribution of Simpson's index for the first wave of cells. Individual plots of Simpson's index are shown for the branching process at times t=70, 90, 110, and 130 along with Simpson's index for the limiting point process (t=∞). The histograms show the average over 1000 sample simulations. For the limiting point process, we approximate Simpson's index by examining the largest 104 points in the process. Parameters are λ0=0.1, a0=0.2, and v∼U([0,0.01]).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/genetics/188/2/10.1534_genetics.110.125724/6/m_461fig6.jpeg?Expires=1716554805&Signature=kzZf5IUi2nMrZ~4xi5qHE1ItTWLxxy3WAGXV8h231w6357YEtt9Mn7Tl6FwImFn3ukLgLpkx~14PeIqjUH8tOri~uQbmG5fHNHRzNV7h4FBSYlQ2X7SBduNXz1cd1EwRVH9fMQ3~gCPEfZawnFXTcL1Ir5YBmAFRDgZMyqgb04uMgIEUCu5d60N3pfwxDa5W-zfA2y4kDXlrJgb1y2eaTnaSyVOPCsbp2FbaCJE-3-Vol1oRn5K4Qcierxe7WxoEY5exxy2t5eFg6eL15zcrfaP0ZCtDeqYJv9Q1VIxLgGsecrx7VpoS3sXsIWFocsAbIWY4jlGFsz9O9aY5Rsd7bQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
The empirical distribution of Simpson's index for the first wave of cells. Individual plots of Simpson's index are shown for the branching process at times , 90, 110, and 130 along with Simpson's index for the limiting point process . The histograms show the average over 1000 sample simulations. For the limiting point process, we approximate Simpson's index by examining the largest points in the process. Parameters are , , and .
Largest clones:
To further investigate heterogeneity properties of the point process, we examine the fraction of cells descended from the largest family of first-generation mutants defined as . This quantity reveals the degree of dominance of the largest clone in the first wave of mutants. For large n, values of Vn near one indicate that the population is largely dominated by a single clone, while values near zero indicate a highly heterogeneous population where no single clone contributes significantly to the total. Using a similar approach to that in the previous section, we again consider this calculation in the context of a random walk. Consider a sequence of independent, identically distributed random variables Yi with partial sums Wn. Define the maximum value of this sequence from 1 to n to be Y(1). Then classical results regarding one-dimensional random walks characterize the limiting characteristic function of Wn/Y(1) (Darling 1952). In the appendix, we demonstrate that these results can be applied to the study the largest clone contributions in the limiting point process.
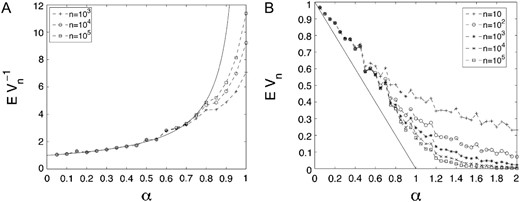
The largest clones in the population of cells. (A) A comparison between Monte Carlo estimates for and the limit (B) A comparison of the Monte Carlo estimates for and the curve . The Monte Carlo estimates are averaged over 100 sample simulations.
Equation 9 implies that Vn converges to a nontrivial limit V = W−1 and Jensen's inequality applied to the strictly convex function 1/x implies that This result provides a lower bound on the expected value of the limit of Vn and indicates that for values of α close to zero, the population is eventually dominated by a single clone. Even though this result indicates only a lower bound, simulations suggest that deviations of the mean from 1−α are small, as illustrated in Figure 7b.
Extensions to generation k:
Total population heterogeneity:
DISCUSSION
In this article, we investigated the evolution of intratumor heterogeneity in a stochastic model of tumor cell expansion. Our model incorporates random mutational advances conferred by (epi)genetic alterations and our analysis focused on the extent of heterogeneity present in the tumor. We first considered heterogeneity between tumor subpopulations with varying numbers of alterations and obtained limiting results, as the mutation rate approaches zero, for the contribution of each wave of mutants to the total tumor cell population. We showed that in the limit, this intergeneration heterogeneity depends on the maximum attainable fitness advance conferred by (epi)genetic alterations, but not on the specific form of the mutational fitness distribution. Our analysis also led to analytical expressions for the arrival time of the first cell with k mutations and showed that the rate of accumulation of new genetic alterations accelerates over time due to the increasing growth rates of successive generations. We demonstrated with stochastic simulations that for small but positive mutation rates, our limiting approximations provide good predictions of the model behavior (see Figure 3). These simulations also suggest that as time increases, multiple waves of mutants coexist without a single, largely dominant wave. For large t, the mean growth rate of the kth wave is given by , showing that variation in fitness within a particular wave is a transient property. The extent to which this variation affects tumor dynamics at small times is the subject of ongoing work.
We also investigated the genotypic diversity present within the kth generation of mutants by considering two measures of diversity: Simpson's index, which is given by the probability that two randomly selected cells stem from the same family, and the fraction of individuals in generation k that stem from the largest family of individuals. We obtained limiting expressions for the mean of Simpson's index as well as the form of its density near the origin. Interestingly, the limiting mean of Simpson's index is given by the quantity 1−α, where α is the ratio between the maximum attainable fitness values of type-(k − 1) and type-k individuals. We then observed that, as time increases, the mass of the distribution of Simpson's index moves closer to 0, indicating higher levels of diversity in the tumor at later times (see Figure 6). This behavior was also observed via direct numerical simulation of the branching process—the distribution and mean of Simpson's index converged to the predicted limiting values.
Finally, we investigated the ratio between the total population size of the kth wave of mutants and the size of the largest family. We showed that this ratio can be approximated by a random variable with mean (1−α)−1. An explicit formula for the characteristic function of this random variable was also obtained (see Equation 9). Note that as α approaches 1, the mean of the ratio grows to infinity—i.e., the largest family of cells constitutes a vanishing proportion of the total population of wave-k cells as the maximum possible fitness advance goes to zero.
In the context of tumorigenesis, where a tumor originally starting from a single cell reaches cell numbers of ≥1012, the limit as t goes to infinity is indeed the appropriate regime of study. We have compared our results regarding Simpson's index with numerical simulations at finite times (see, e.g., Figures 5 and 6) and have found good qualitative agreement with the asymptotic limits. Estimates of overall mutation rates in evolving cancer cell populations range from 10−9 to 10−2. In Figure 3 we demonstrate good agreement between our results regarding interwave heterogeneity in the small mutation rate limit and numerical simulations when the mutation rate is ; thus, analysis in this limiting regime captures the dynamics for mutation rates < per cell division.
In conclusion, our analysis indicates that tumor diversity is strongly dependent upon the age of the tumor and the maximum attainable fitness advance of mutant cells. If only small fitness advances are possible, then the tumor population is expected to have a larger extent of diversity compared to situations in which fitness advances are large. The acceleration of waves observed in our studies of intergeneration heterogeneity provides evidence that an older tumor has a higher level of diversity than a young tumor. In addition, we have shown that the mean of Simpson's index for generation k is a decreasing function of the generation number (see Equation 11), indicating a larger extent of diversity in later generations and suggesting a further increase in the total extent of heterogeneity present in the tumor at later times.
Possible extensions of our model include spatial considerations and the effects of tissue organization on the generation of intratumor heterogeneity as well as the inclusion of other cell types, such as immune system cells and the microenvironment. Furthermore, alternative growth dynamics should be considered to test the extent of heterogeneity arising in populations that follow logistic, Gompertzian, or other growth models. We have neglected these aspects in the current version of the model to focus on the dynamics of tumor diversity in an exponentially growing population of cells. Our model provides a rational understanding of the extent and dynamics of intratumor heterogeneity and is useful for obtaining an accurate picture of its generation during tumorigenesis.
Acknowledgements
The authors thank Theresa Edmonds for exceptional research assistance. This work is supported by National Cancer Institute grant U54CA143798 (to J.F., K.L., and F.M.), National Science Foundation (NSF) grant DMS 0704996 (to R.D.), and NSF RTG grant DMS 0739164 (to J.M.).
Footnotes
Communicating editor: M. W. Feldman
LITERATURE CITED
APPENDIX
Convergence in distribution of Zk(t):
Point process limit:
In this section, we discuss the point process representation for the limit in Equations A1 and A2 in the case where k = 1. The limit is the sum of points in a nonhomogeneous Poisson process. Before stating this result, we introduce some terminology. Here and in what follows, we use to denote the number of points in the set A. We say that Λ is a Poisson process on (0, ∞) with mean measure μ if Λ is a random set of points in (0, ∞) with the following properties:
For any A⊂(0, ∞), N(A) = is a Poisson random variable with mean μ(A).
For any k ≥ 1, if A1, … , Ak are disjoint subsets of (0, ∞), then N(Ai), 1 ≤ i ≤ k are independent.
We also let α = λ0/λ1 ∈ (0, 1) denote the ratio of the growth rate of type-0 cells to the maximal growth rate of type-1 cells and note that 1 + p1 = 1/α.
For more details, see Durrett et al. (2010), Theorem 3, and Durrett and Moseley (2010), Corollary to Theorem 3. Note that the mean measure for Λ has tail m(x, ∞) = x−α.
Lemma 1.
Proof. Since Tn has a Gamma(n, 1) distribution, we have Stirling's approximation implies that Γ(n − 1/α)/ Γ(n) ∼ n−1/α and the second conclusion follows. ■
Simpson's index:
As a consequence of this lemma, the fact that Rn(ε) ≤ 1, and the bounded convergence theorem, we have the following:
It thus remains to establish the following:
We can now complete the proof of Equation 7 by letting n → ∞ and then ε → 0 in (A4) and applying Lemmas 2 and 4 and Corollary 1.
Largest clones:
The form of the characteristic function is the same as the characteristic function for limn→∞Tn/Y(1), where the Yi are iid random variables with power law tails, Y(1) = maxi≤nYi, and (see, for example, Darling 1952). Again, this agreement is a consequence of the previously discussed connection between Δn and the limiting Poisson point process.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}