





Abstract
The demography of populations and natural selection shape genetic variation across the genome and understanding the genomic consequences of these evolutionary processes is a fundamental aim of population genetics. We have developed a hierarchical Bayesian model to quantify genome-wide population structure and identify candidate genetic regions affected by selection. This model improves on existing methods by accounting for stochastic sampling of sequences inherent in next-generation sequencing (with pooled or indexed individual samples) and by incorporating genetic distances among haplotypes in measures of genetic differentiation. Using simulations we demonstrate that this model has a low false-positive rate for classifying neutral genetic regions as selected genes (i.e., ϕST outliers), but can detect recent selective sweeps, particularly when genetic regions in multiple populations are affected by selection. Nonetheless, selection affecting just a single population was difficult to detect and resulted in a high false-negative rate under certain conditions. We applied the Bayesian model to two large sets of human population genetic data. We found evidence of widespread positive and balancing selection among worldwide human populations, including many genetic regions previously thought to be under selection. Additionally, we identified novel candidate genes for selection, several of which have been linked to human diseases. This model will facilitate the population genetic analysis of a wide range of organisms on the basis of next-generation sequence data.
THE distribution of genetic variants among populations is a fundamental attribute of evolutionary lineages. Population genetic diversity shapes contemporary functional diversity and future evolutionary dynamics and provides a record of past evolutionary and demographic processes. Methods to quantify genetic diversity among populations have a long history (Wright 1951; Holsinger and Weir 2009) and provide a basis to distinguish neutral and adaptive evolutionary histories, to reconstruct migration histories, and to identify genes underlying diseases and other significant traits (Bamshad and Wooding 2003; Tishkoff and Verrelli 2003; Voight et al. 2006; Barreiro et al. 2008; Lohmueller et al. 2008; Novembre et al. 2008; Tishkoff et al. 2009; Hohenlohe et al. 2010). For example, population genetic analyses in humans have resolved a history of natural selection and independent origins of lactase persistence in adults in Europe and East Africa (Tishkoff et al. 2007). Similarly, allelic diversity at the Duffy blood group locus is consistent with the action of natural selection, and one allele that has gone to fixation in sub-Saharan populations confers resistance to malaria (Hamblin and Di Rienzo 2000; Hamblin et al. 2002). These studies of natural selection, and many others involving a diversity of organisms, utilize contrasts between genetic differentiation at putatively selected loci and the remainder of the genome. Genomic diversity within and among populations is determined primarily by mutation and neutral demographic factors, such as effective population size and rates of migration among populations (Wright 1951; Slatkin 1987). Specifically, these demographic factors determine the rates of genetic drift and population differentiation across the genome. In contrast, selection affects variation in specific regions of the genome, including the direct targets of selection and to a lesser extent genetic regions in linkage disequilibrium with these targets (Maynard-Smith and Haigh 1974; Slatkin and Wiehe 1998; Gillespie 2000; Stajich and Hahn 2005). Thus, the genomic consequences of selection are superimposed on the genomic outcomes of neutral genetic differentiation and the two must be disentangled to identify regions of the genome affected by selection.
A variety of models and methods have been proposed to identify genetic regions that have been affected by selection (Nielsen 2005), and these population genetic methods can be divided into within-population and among-population analyses. The former include widely used neutrality tests based on the site-frequency spectrum for a single locus, such as Tajima's D test (Tajima 1989), as well as recently developed tests based on the presence of extended blocks of linkage disequilibrium or reduced haplotype diversity (Andolfatto et al. 1999; Sabeti et al. 2002; Voight et al. 2006). Among-population tests for selection require multilocus data sets and identify nonneutral or outlier loci by contrasting patterns of population divergence among genetic regions (Lewontin and Krakauer 1973; Beaumont and Nichols 1996; Akey et al. 2002; Beaumont and Balding 2004; Foll and Gaggiotti 2008; Guo et al. 2009; Chen et al. 2010). The most commonly employed of these methods is the FST outlier analysis developed by Beaumont and Nichols (1996). This test contrasts FST for individual loci with an expected null distribution of FST on the basis of a neutral, infinite-island, coalescent model (Beaumont and Nichols 1996). Loci with very high levels of among-population differentiation (i.e., high FST) are considered candidates for positive or divergent selection, whereas loci with exceptionally low FST are regarded as candidates for balancing selection. However, many FST outlier analyses can be biased by violations of the assumed demographic history (Flint et al. 1999; Excoffier et al. 2009). Alternative Bayesian approaches to obtain a null distribution of population genetic differentiation assume that FST's for individual loci represent independent draws from a common, underlying distribution that characterizes the genome and that can be estimated directly from multilocus data (Beaumont and Balding 2004; Foll and Gaggiotti 2008; Guo et al. 2009). These approaches are more robust to different demographic histories, but might have reduced power to detect selection relative to methods that model the appropriate demographic history when it is known (Guo et al. 2009).
Herein we propose a hierarchical Bayesian model for estimating genomic population differentiation and detecting selection. This method improves on current methods for detecting selection in several important ways. First, unlike previous Bayesian outlier detection models (e.g., Beaumont and Balding 2004; Foll and Gaggiotti 2008; Guo et al. 2009), the likelihood component of the model captures the stochastic sampling processes inherent in next-generation sequence (NGS) data (Mardis 2008a,b). Specifically, NGS technologies result in uneven coverage among individuals, genetic regions, and homologous gene copies, including missing data for many individuals and loci, and thus, increased uncertainty in the diploid sequence of individuals relative to traditional Sanger sequencing (Lynch 2009; Gompert et al. 2010; Hohenlohe et al. 2010). This issue is most pronounced when population-level indexing is used (e.g., Gompert et al. 2010), but should persist even with individual-level indexing and high coverage data (i.e., even with high mean coverage the sequence coverage for certain combinations of individuals and genetic regions will be low). Moreover, appropriately modeling and accounting for this uncertainty is important and vastly preferable to simply ignoring it or discarding large amounts of sequence data (e.g., Hohenlohe et al. 2010). Previous methods to detect selection on the basis of genetic differentiation among populations have focused solely on allele frequency differences, as captured by FST (Nielsen 2005). Instead the model we propose measures population genetic differentiation using Excoffier et al.'s ϕ-statistics, which are DNA sequence-based measures of the proportion of molecular variation partitioned among groups or populations (Excoffier et al. 1992). Quantifying genetic differentiation using ϕ-statistics allows us to include the genetic distances among sequences in the measure of differentiation and thus take mutation rate into account, which should increase our ability to accurately identify targets of selection (Kronholm et al. 2010). Finally, the model we propose provides a novel criterion for designating outlier loci. Although we do not believe this criterion is inherently superior to alternatives (e.g., Beaumont and Nichols 1996; Foll and Gaggiotti 2008; Guo et al. 2009), we believe it accords well with the concept of statistical outliers and is well suited for genome scans for divergent selection. Specifically, the model assumes that the ϕ-statistics for each genetic region are drawn from a common, genome-level distribution and identifies outlier loci, or loci potentially affected by selection, on the basis of the probability of their locus-specific ϕ-statistic given the genome-level probability distribution of ϕ. This genome-level distribution is equivalent to the conditional probability for the locus-specific ϕ-statistics in the model.
We begin this article by fully describing the model, both verbally and mathematically. We then use coalescent simulations to generate data with a known history and investigate the performance of the model for identifying genetic regions affected by selection. To illustrate its capacity to identify exceptional genes that are likely to have experienced selection, we use the proposed model to analyze empirical data from two studies of human population genetic variation. The first of these data sets includes 316 completely sequenced genes from 24 individuals with African ancestry and 23 individuals with European ancestry (SeattleSNPs data set). The second data set was published by Jakobsson et al. (2008) and includes genotype data from 525,901 SNPs from 33 human populations distributed worldwide. We analyzed these human data with the known haplotypes model (see Bayesian models for molecular variance) and found evidence of selection affecting a total of 569 genes including genes previously identified as targets of selection in humans (e.g., CYP3A5, SLC24A5, and PKDREJ) and novel genes not previously thought to be under selection (e.g., FOXA2 and SPATA5L1). Whereas these remarkably large-scale studies do not involve NGS data, the analyses of empirical data identify a large set of genes for additional study and illustrate our analytical approach, which can be applied to a diversity of large-scale genomic data sets, including those that will result from the anticipated widespread adoption of NGS for population genomics.
METHODS
Bayesian models for molecular variance:
Our goal is to use molecular divergence among populations and groups to identify genetic regions that might have been affected by natural selection. Patterns of divergence at individual loci arise from differences in haplotype frequencies and the genetic distance among haplotypes. We assume the distances are fixed and known and we attempt to estimate the haplotype frequencies from sequence data using a hierarchical Bayesian model. The model includes a first-level likelihood for the probability of the observed haplotype counts given population haplotype frequencies, a conditional prior for the haplotype frequencies for each locus in each population given genome-level parameters and genetic distance matrix, and uninformative priors for the genome-level parameters. This conditional prior defines the distribution of ϕ-statistics across the genome, whereas locus-specific ϕ-statistics are derived from our estimate of haplotype frequencies and the genetic distance among haplotypes. From this model we are able to estimate the probability of each locus-specific ϕ-statistic given the estimated genome-level distribution, which serves as a metric for identifying outlier loci that depart from the typical level of differentiation and therefore might have been affected by selection.
First-level likelihood models:
Model priors:
Designating outlier loci:
This Bayesian model gives rise to a framework for identifying outlier loci or genetic regions with an unusual proportion of molecular variation partitioned among populations or groups. Such genetic regions might have experienced selection directly, or indirectly through linkage. Genetic regions with unusual patterns of molecular variation can be identified by contrasting ϕ-statistics for each locus with the genome-level distribution for each ϕ-statistic. Specifically, we identify outlier loci by estimating the posterior probability distribution for the quantile of each locus-specific ϕ-statistic in the genome-level distribution. Formally, we define ai as the ath quantile of the posterior distribution for the locus-specific ϕ-statistic for locus i and let qn be the interval with endpoints defined as the nth/2 and 1− nth/2 quantiles of the genome-level ϕ-statistic distribution. We then consider locus i an outlier at the ath quantile with respect to a given ϕ-statistic with probability n if the interval qn does not contain ai. Values of n and a used for outlier designation will determine the stringency of the analysis, with higher values of a and lower values of n resulting in fewer loci classified as outliers. In this article we set n = 0.05 (qn = [0.025,0.975]) and use two different values of a (0.5 and 0.95 quantiles). These values for a denote the median and 95th quantile of the posterior probability distribution for the quantile of each locus-specific ϕ-statistic. When only differentiation among populations is being considered, a genetic region can be designated an outlier only with respect to ϕST, whereas a genetic region could be an outlier with respect to ϕST, ϕSC, or ϕCT when group and population structure are considered. Outlier loci can be classified as having very low ϕST, which could be indicative of balancing or purifying selection, or very high ϕST, which could be indicative of positive selection within populations or divergent selection among populations (Beaumont and Balding 2004; Beaumont 2005). Patterns of selection giving rise low or high ϕCT and ϕSC might be a bit more complex. For example, high ϕCT outliers would be expected if divergent selection occurred among groups of populations with the same alleles favored within each group and low ϕSC outliers might be expected with balancing selection within groups that favored different subsets of alleles among groups.
Several approaches have been described for identifying outlier loci using genome scans for differentiation (Beaumont and Balding 2004; Foll and Gaggiotti 2008; Guo et al. 2009). Our approach is most similar to that proposed by Guo et al. (2009), but also differs from that approach in several important ways. Guo et al. (2009) contrast an approximation of the posterior probability distribution for each locus-specific ϕST (θi's in their model) with the hyperdistribution describing among-locus variation in ϕST (equivalent to our genome-level distribution). Approximation of the posterior probability distribution for ϕST is achieved by defining a Beta distribution with first and second moments equal to those of the posterior probability distribution (Guo et al. 2009). This approximation allows Guo et al. (2009) to measure the divergence between the posterior probability distribution for each locus-specific ϕST and the genome-level distribution (also a Beta distribution, which is derived from point estimates of genome-level parameters), using Kullback–Leibler divergence (Kullback and Leibler 1951). Following calibration to determine a cutoff value for significance, the Kullback–Leibler divergence measure is used to designate outlier loci. The primary distinction between the outlier detection method proposed by Guo et al. (2009) and the outlier detection method we have implemented is that their method tests for a difference between the posterior probability distribution of each locus-specific ϕST (this distribution measures uncertainty in the parameter estimate) and a point estimate of the genome-level ϕST distribution (this distribution measures expected variation among loci in ϕST). In contrast, we test whether locus-specific ϕ-statistics are unlikely given the genome-level distribution (i.e., not that our estimates of these locus-specific ϕ-statistics simply differ significantly from the genome-level distribution). Additionally, our method differs by accounting for uncertainty in the genome-level distribution by taking the marginal distribution of the quantile of each locus-specific ϕ-statistic in the genome-level distribution, rather than using a point estimate of the genome-level distribution, which is necessary for the method of Guo et al. (2009). We do not believe that either of these methods is necessarily superior, but rather that they provide alternative criteria and definitions of outlier loci, with our method perhaps more closely reflecting common conceptions of outlier loci among evolutionary biologists and their method utilizing more information from the posterior distribution of locus-specific ϕST.
Analysis of simulated data sets:
We conducted a series of simulations to determine what proportion of genetic regions would be classified as outliers in the absence of selection. These data sets were simulated for analysis with the population structure model and for each of the three likelihood models (i.e., known haplotype model, NGS-individual model, and NGS-population model). We simulated sequence data using an infinite-sites coalescent model, using R. Hudson's software ms (Hudson 2002). Data sets were simulated with 25 or 500 genetic regions. The simulations assumed five populations split from a common ancestor τ generations in the past, where τ has units of 4Ne and was set to 0.25, 0.5, or 1.0. We conducted simulations with migration among the five populations (Nem) set to 0 or 2. The ancestral population and all five descendant populations were assigned population mutation rates θ = 4Neμ of 0.5, where μ is the per locus mutation rate. Forty gene copies were sampled from each of the five populations. For the known haplotype model analyses we treated the simulated sequences directly as the sampled data. For NGS-individual model and NGS-population model analyses we resampled the simulated sequence data sets such that coverage for each sequence was Poisson distributed (λ = 2). For the NGS-individual model analyses we retained information on which individual each sequence came from, whereas we retained only population identification for NGS-population model analyses. We generated 10 replicate simulated data sets for each combination of likelihood model, number of genetic regions (25 or 500), τ, and migration rate. Each data set was analyzed using a Markov chain Monte Carlo (MCMC) implementation of the proposed model, using the bamova software we have developed (see supporting information, File S1, MCMC algorithm; available from the authors at http://www.uwyo.edu/buerkle/software/ as stand-alone software). We calculated the distance matrix for each locus, using the number of sites by which each pair of sequences differed. Estimation of the posterior probability distribution for all parameters for each data set was based on a single MCMC algorithm run that included a 25,000-iteration burn-in followed by 50,000 samples from the posterior. Sample history plots were monitored to ensure appropriate chain mixing and convergence on the stationary distribution. We classified genetic regions as outliers on the basis of the posterior probability distribution for the quantile of each locus-specific ϕST in the genome-level ϕST distribution, as previously described (see Designating outlier loci). We then calculated the mean proportion of loci classified as outliers across the 10 replicates for each combination of parameters.
We conducted an additional series of simulations to assess the capacity of our analytical model to detect selected loci among a larger set of neutral regions. We began with a set of simulations of population structure, as above. Simulations were conducted under all combinations of conditions described previously for simulations without selection. However, we simulated selective sweeps affecting 2 (8%, for 25-locus simulations) or 25 (5%, for 500-locus simulations) of the simulated loci. We allowed selective sweeps to occur in one, three, or five populations (one set of simulations for each). Selective sweeps were simulated by selecting one haplotype from each affected population at each affected locus and increasing its frequency to 1. This was meant to simulate recent and strong selective sweeps where affected loci were in arbitrary linkage disequilibrium with the gene subject to selection. Thus, it was possible for the same or different haplotypes to be driven to fixation across populations. We calculated the mean proportion of neutral and nonneutral loci classified as outliers across the 10 replicates for each combination of parameters.
Finally, we conducted a series of simulations to determine the extent to which our group structure model could correctly identify genetic regions affected by selection. For these simulations we concentrated on the NGS-individual model and a single demographic history. We simulated two groups of five populations, with the divergence time of the populations within each group equal to 0.25 and the divergence of the two groups equal to 0.75. We allowed no migration between groups, but set the within-group migration rate to 4. We simulated data sets of 25 and 500 loci, and as with the population structure simulations, we simulated selective sweeps affecting 2 (25-locus simulations) or 25 (500-locus simulations) genetic regions. Selective sweeps occurred in all five populations within one group (sg = 1) or all five populations in both groups (sg = 2). We simulated 10 replicate data sets for each combination of simulation parameters. MCMC settings were as previously described. We identified outlier loci on the basis of ϕST (the partitioning of molecular variation among populations), ϕCT (the partitioning of molecular variation between the two groups), and ϕSC (the partitioning of molecular variation among the populations within each group). We determined the mean proportion of neutral and nonneutral loci classified as outliers on the basis of each ϕ-statistic across the 10 replicates for each combination of parameters.
Analysis of human SeattleSNP data:
To illustrate the application of the proposed model to real genetic data we analyzed 316 fully sequenced genes (exons and introns) from the SeattleSNPs data set (http://pga.gs.washington.edu; downloaded May 2010). Each gene was sequenced in 24 individuals with African ancestry [either the African-American (AA) panel or the HapMap Yoruba (YRI) population] and 23 individuals with European ancestry [either the Centre d'Étude du Polymorphisme Humain (CEPH) population or the HapMap Uthah residents with European ancestry (CEU) population]. The phase of polymorphisms in these sequences has been estimated statistically using PHASE v2.0 (Stephens et al. 2001) to produce haplotypes. Similar to NGS data sets, these data are DNA sequences, but these data lack the sampling uncertainty associated with NGS data and might represent fewer (but much longer) genetic regions than would be typical for current NGS studies. We used these data and the bamova software to identify loci with exceptional ϕST estimates that were consistent with divergent selection between or balancing selection within these European and African ancestry populations. The mean number of SNPs per gene was 62.98 (SD = 50.73), resulting in a large number of haplotypes per gene. This large number of SNPs per gene, which resulted from these data being complete gene sequences, was considerably greater than typical for the short reads generated by NGS (e.g., Gompert et al. 2010; Hohenlohe et al. 2010) and resulted in poor MCMC mixing. Therefore we based our analysis on the first five SNPs in each gene (see File S1, Human SeattleSNP data: alternative data subsets, for analyses using other SNPs). All insertion–deletion polymorphisms were ignored. We used the known haplotypes model with population structure. We ran a 25,000-iteration burn-in followed by 50,000 iterations to estimate posterior probabilities. We identified outlier loci using the criteria described for the simulated data sets. We then examined variation in genetic differentiation among SNPs within each outlier locus, as well as several loci that had typical, “neutral” ϕST estimates, by calculating point estimates of FST at each SNP.
Analysis of worldwide SNP data:
We obtained data for genomic diversity across 33 widely distributed human populations (including Africa, Eurasia, East Asia, Oceania, and America) from Jakobsson et al. (2008) (version 1.3; http://neurogenetics.nia.nih.gov/paperdata/public/). These data include statistically phased haplotypes for 597 individuals, based on 525,901 SNPs. These data differ from NGS data as they are not DNA sequences, but the large number of genetic regions in this data set might be similar to what will be acquired in NGS studies. For each SNP in the HumanHap550 genotyping panel we obtained annotations and information for location relative to genes from the manufacturer of the genotyping technology (Illumina, San Diego). To focus our analysis on haplotypic variation within transcribed, genic regions, we retained only those SNPs that were annotated as being within coding, intron, 5′-UTR, 3′-UTR, or UTR regions. If for a particular gene this included >4 SNPs (minimally, for inclusion we required 2 SNPs per locus), we retained SNPs that were annotated as within coding or intron sequence plus any neighboring SNPs to bring the total to 4 SNPs per locus. Finally, if there were >4 SNPs within coding or intron sequence at a locus, we retained the first and last SNP and randomly chose 2 intervening SNPs in coding or intron sequence. We utilized a maximum of 4 SNPs per locus because this was similar to the number of variable sites expected in short NGS data (Gompert et al. 2010; Hohenlohe et al. 2010) and led to a sufficiently small number of haplotypes across populations, relative to the numbers of individuals per population, to yield informative haplotype frequencies for populations. Some isolated SNPs had annotations to genes elsewhere in the genome and conflicted with neighboring SNPs and were excluded. However, we did allow these SNPs to break genes into separate genic regions for analysis, so that we utilized haplotypic data from 12,649 regions and 11,866 distinct genes. We excluded the limited amount of data from the mitochondrion, the Y chromosome, and the pseudoautosomal region of the X and Y chromosomes and focused on the autosomes and the X chromosome.
We conducted a separate analysis for each chromosome to test for evidence of selection on the basis of levels of molecular differentiation among these human populations, using the bamova software. This allowed us to contrast patterns of genetic variation among chromosomes and identify outlier loci relative to levels of genetic differentiation for the chromosome on which they are found. We used the known haplotypes likelihood model with population structure and estimated posterior probabilities on the basis of 50,000 MCMC iterations following a 25,000-iteration burn-in. We classified outlier loci for each data set using the criteria described for the simulated data sets.
RESULTS
Results from simulated data sets:
When data were simulated in the absence of selection, few genetic regions were classified as outliers and thus as being associated with targets of selection (Table 1). This result suggests that the method has a low false-positive rate (mean proportion = 0.012, SD = 0.030). The proportion of genetic regions classified as outliers was similar across all likelihood models, but tended to be slightly lower when migration among populations was simulated (particularly for high ϕST outliers). With a = 0.5 the mean proportion of genetic regions classified as outliers across 10 replicate simulations varied from 0.003 (known haplotype model, no migration, 500 sequence loci, high ϕST outliers) to 0.034 (NGS-population model, no migration, 500 sequence loci, low ϕST outliers). Using a = 0.95, the mean proportion of outliers detected across 10 replicate simulations varied from 0 (many simulation conditions) to 0.004 (NGS-individual model, no migration, 500 sequence loci, high ϕST outliers).
Proportion of outlier loci in simulated neutral data
m = 0 | m = 2 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| a = 0.5 | a = 0.95 | a = 0.5 | a = 0.95 | ||||||
| No. loci | τ | Low | High | Low | High | Low | High | Low | High |
| Known haplotypes model | |||||||||
| 25 | 0.25 | 0.012 | 0.028 | 0.000 | 0.000 | 0.016 | 0.020 | 0.000 | 0.000 |
| 0.50 | 0.028 | 0.028 | 0.000 | 0.000 | 0.016 | 0.012 | 0.000 | 0.000 | |
| 1.00 | 0.024 | 0.004 | 0.004 | 0.000 | 0.012 | 0.016 | 0.000 | 0.000 | |
| 500 | 0.25 | 0.0132 | 0.0212 | 0.0004 | 0.0036 | 0.0108 | 0.0170 | 0.0000 | 0.0012 |
| 0.50 | 0.0240 | 0.0164 | 0.0028 | 0.0004 | 0.0130 | 0.0158 | 0.0000 | 0.0012 | |
| 1.00 | 0.0320 | 0.0032 | 0.0080 | 0.0000 | 0.0136 | 0.0188 | 0.0002 | 0.0024 | |
| NGS-individual model | |||||||||
| 25 | 0.25 | 0.012 | 0.012 | 0.000 | 0.000 | 0.020 | 0.016 | 0.000 | 0.000 |
| 0.50 | 0.004 | 0.024 | 0.000 | 0.000 | 0.020 | 0.012 | 0.000 | 0.000 | |
| 1.00 | 0.020 | 0.024 | 0.000 | 0.000 | 0.040 | 0.008 | 0.000 | 0.000 | |
| 500 | 0.25 | 0.0136 | 0.0242 | 0.0005 | 0.0042 | 0.0098 | 0.0156 | 0.0004 | 0.0020 |
| 0.50 | 0.0220 | 0.0202 | 0.0022 | 0.0042 | 0.0112 | 0.0182 | 0.0004 | 0.0020 | |
| 1.00 | 0.0280 | 0.0074 | 0.0068 | 0.0006 | 0.0140 | 0.0176 | 0.0008 | 0.0036 | |
| NGS-population model | |||||||||
| 25 | 0.25 | 0.016 | 0.020 | 0.000 | 0.000 | 0.008 | 0.012 | 0.000 | 0.000 |
| 0.50 | 0.024 | 0.040 | 0.000 | 0.000 | 0.008 | 0.012 | 0.000 | 0.000 | |
| 1.00 | 0.032 | 0.004 | 0.000 | 0.000 | 0.004 | 0.008 | 0.000 | 0.004 | |
| 500 | 0.25 | 0.0112 | 0.0202 | 0.0002 | 0.0024 | 0.0054 | 0.0166 | 0.0000 | 0.0018 |
| 0.50 | 0.0190 | 0.0164 | 0.0006 | 0.0012 | 0.0100 | 0.0174 | 0.0000 | 0.0012 | |
| 1.00 | 0.0340 | 0.0064 | 0.0050 | 0.0000 | 0.0088 | 0.0180 | 0.0000 | 0.0022 | |
m = 0 | m = 2 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| a = 0.5 | a = 0.95 | a = 0.5 | a = 0.95 | ||||||
| No. loci | τ | Low | High | Low | High | Low | High | Low | High |
| Known haplotypes model | |||||||||
| 25 | 0.25 | 0.012 | 0.028 | 0.000 | 0.000 | 0.016 | 0.020 | 0.000 | 0.000 |
| 0.50 | 0.028 | 0.028 | 0.000 | 0.000 | 0.016 | 0.012 | 0.000 | 0.000 | |
| 1.00 | 0.024 | 0.004 | 0.004 | 0.000 | 0.012 | 0.016 | 0.000 | 0.000 | |
| 500 | 0.25 | 0.0132 | 0.0212 | 0.0004 | 0.0036 | 0.0108 | 0.0170 | 0.0000 | 0.0012 |
| 0.50 | 0.0240 | 0.0164 | 0.0028 | 0.0004 | 0.0130 | 0.0158 | 0.0000 | 0.0012 | |
| 1.00 | 0.0320 | 0.0032 | 0.0080 | 0.0000 | 0.0136 | 0.0188 | 0.0002 | 0.0024 | |
| NGS-individual model | |||||||||
| 25 | 0.25 | 0.012 | 0.012 | 0.000 | 0.000 | 0.020 | 0.016 | 0.000 | 0.000 |
| 0.50 | 0.004 | 0.024 | 0.000 | 0.000 | 0.020 | 0.012 | 0.000 | 0.000 | |
| 1.00 | 0.020 | 0.024 | 0.000 | 0.000 | 0.040 | 0.008 | 0.000 | 0.000 | |
| 500 | 0.25 | 0.0136 | 0.0242 | 0.0005 | 0.0042 | 0.0098 | 0.0156 | 0.0004 | 0.0020 |
| 0.50 | 0.0220 | 0.0202 | 0.0022 | 0.0042 | 0.0112 | 0.0182 | 0.0004 | 0.0020 | |
| 1.00 | 0.0280 | 0.0074 | 0.0068 | 0.0006 | 0.0140 | 0.0176 | 0.0008 | 0.0036 | |
| NGS-population model | |||||||||
| 25 | 0.25 | 0.016 | 0.020 | 0.000 | 0.000 | 0.008 | 0.012 | 0.000 | 0.000 |
| 0.50 | 0.024 | 0.040 | 0.000 | 0.000 | 0.008 | 0.012 | 0.000 | 0.000 | |
| 1.00 | 0.032 | 0.004 | 0.000 | 0.000 | 0.004 | 0.008 | 0.000 | 0.004 | |
| 500 | 0.25 | 0.0112 | 0.0202 | 0.0002 | 0.0024 | 0.0054 | 0.0166 | 0.0000 | 0.0018 |
| 0.50 | 0.0190 | 0.0164 | 0.0006 | 0.0012 | 0.0100 | 0.0174 | 0.0000 | 0.0012 | |
| 1.00 | 0.0340 | 0.0064 | 0.0050 | 0.0000 | 0.0088 | 0.0180 | 0.0000 | 0.0022 | |
Mean proportion of loci is shown over 10 replicates identified as outliers in the absence of selection for each of the three different likelihoods (known haplotypes model, NGS-individual model, and NGS-population model). Three times since divergence (τ) and two levels of migration (m) were simulated. Outlier loci were identified as low or high outliers relative to the genome-wide distribution and based on two different quantiles (a = 0.5 or 0.95) of their posterior distribution.
Proportion of outlier loci in simulated neutral data
m = 0 | m = 2 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| a = 0.5 | a = 0.95 | a = 0.5 | a = 0.95 | ||||||
| No. loci | τ | Low | High | Low | High | Low | High | Low | High |
| Known haplotypes model | |||||||||
| 25 | 0.25 | 0.012 | 0.028 | 0.000 | 0.000 | 0.016 | 0.020 | 0.000 | 0.000 |
| 0.50 | 0.028 | 0.028 | 0.000 | 0.000 | 0.016 | 0.012 | 0.000 | 0.000 | |
| 1.00 | 0.024 | 0.004 | 0.004 | 0.000 | 0.012 | 0.016 | 0.000 | 0.000 | |
| 500 | 0.25 | 0.0132 | 0.0212 | 0.0004 | 0.0036 | 0.0108 | 0.0170 | 0.0000 | 0.0012 |
| 0.50 | 0.0240 | 0.0164 | 0.0028 | 0.0004 | 0.0130 | 0.0158 | 0.0000 | 0.0012 | |
| 1.00 | 0.0320 | 0.0032 | 0.0080 | 0.0000 | 0.0136 | 0.0188 | 0.0002 | 0.0024 | |
| NGS-individual model | |||||||||
| 25 | 0.25 | 0.012 | 0.012 | 0.000 | 0.000 | 0.020 | 0.016 | 0.000 | 0.000 |
| 0.50 | 0.004 | 0.024 | 0.000 | 0.000 | 0.020 | 0.012 | 0.000 | 0.000 | |
| 1.00 | 0.020 | 0.024 | 0.000 | 0.000 | 0.040 | 0.008 | 0.000 | 0.000 | |
| 500 | 0.25 | 0.0136 | 0.0242 | 0.0005 | 0.0042 | 0.0098 | 0.0156 | 0.0004 | 0.0020 |
| 0.50 | 0.0220 | 0.0202 | 0.0022 | 0.0042 | 0.0112 | 0.0182 | 0.0004 | 0.0020 | |
| 1.00 | 0.0280 | 0.0074 | 0.0068 | 0.0006 | 0.0140 | 0.0176 | 0.0008 | 0.0036 | |
| NGS-population model | |||||||||
| 25 | 0.25 | 0.016 | 0.020 | 0.000 | 0.000 | 0.008 | 0.012 | 0.000 | 0.000 |
| 0.50 | 0.024 | 0.040 | 0.000 | 0.000 | 0.008 | 0.012 | 0.000 | 0.000 | |
| 1.00 | 0.032 | 0.004 | 0.000 | 0.000 | 0.004 | 0.008 | 0.000 | 0.004 | |
| 500 | 0.25 | 0.0112 | 0.0202 | 0.0002 | 0.0024 | 0.0054 | 0.0166 | 0.0000 | 0.0018 |
| 0.50 | 0.0190 | 0.0164 | 0.0006 | 0.0012 | 0.0100 | 0.0174 | 0.0000 | 0.0012 | |
| 1.00 | 0.0340 | 0.0064 | 0.0050 | 0.0000 | 0.0088 | 0.0180 | 0.0000 | 0.0022 | |
m = 0 | m = 2 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| a = 0.5 | a = 0.95 | a = 0.5 | a = 0.95 | ||||||
| No. loci | τ | Low | High | Low | High | Low | High | Low | High |
| Known haplotypes model | |||||||||
| 25 | 0.25 | 0.012 | 0.028 | 0.000 | 0.000 | 0.016 | 0.020 | 0.000 | 0.000 |
| 0.50 | 0.028 | 0.028 | 0.000 | 0.000 | 0.016 | 0.012 | 0.000 | 0.000 | |
| 1.00 | 0.024 | 0.004 | 0.004 | 0.000 | 0.012 | 0.016 | 0.000 | 0.000 | |
| 500 | 0.25 | 0.0132 | 0.0212 | 0.0004 | 0.0036 | 0.0108 | 0.0170 | 0.0000 | 0.0012 |
| 0.50 | 0.0240 | 0.0164 | 0.0028 | 0.0004 | 0.0130 | 0.0158 | 0.0000 | 0.0012 | |
| 1.00 | 0.0320 | 0.0032 | 0.0080 | 0.0000 | 0.0136 | 0.0188 | 0.0002 | 0.0024 | |
| NGS-individual model | |||||||||
| 25 | 0.25 | 0.012 | 0.012 | 0.000 | 0.000 | 0.020 | 0.016 | 0.000 | 0.000 |
| 0.50 | 0.004 | 0.024 | 0.000 | 0.000 | 0.020 | 0.012 | 0.000 | 0.000 | |
| 1.00 | 0.020 | 0.024 | 0.000 | 0.000 | 0.040 | 0.008 | 0.000 | 0.000 | |
| 500 | 0.25 | 0.0136 | 0.0242 | 0.0005 | 0.0042 | 0.0098 | 0.0156 | 0.0004 | 0.0020 |
| 0.50 | 0.0220 | 0.0202 | 0.0022 | 0.0042 | 0.0112 | 0.0182 | 0.0004 | 0.0020 | |
| 1.00 | 0.0280 | 0.0074 | 0.0068 | 0.0006 | 0.0140 | 0.0176 | 0.0008 | 0.0036 | |
| NGS-population model | |||||||||
| 25 | 0.25 | 0.016 | 0.020 | 0.000 | 0.000 | 0.008 | 0.012 | 0.000 | 0.000 |
| 0.50 | 0.024 | 0.040 | 0.000 | 0.000 | 0.008 | 0.012 | 0.000 | 0.000 | |
| 1.00 | 0.032 | 0.004 | 0.000 | 0.000 | 0.004 | 0.008 | 0.000 | 0.004 | |
| 500 | 0.25 | 0.0112 | 0.0202 | 0.0002 | 0.0024 | 0.0054 | 0.0166 | 0.0000 | 0.0018 |
| 0.50 | 0.0190 | 0.0164 | 0.0006 | 0.0012 | 0.0100 | 0.0174 | 0.0000 | 0.0012 | |
| 1.00 | 0.0340 | 0.0064 | 0.0050 | 0.0000 | 0.0088 | 0.0180 | 0.0000 | 0.0022 | |
Mean proportion of loci is shown over 10 replicates identified as outliers in the absence of selection for each of the three different likelihoods (known haplotypes model, NGS-individual model, and NGS-population model). Three times since divergence (τ) and two levels of migration (m) were simulated. Outlier loci were identified as low or high outliers relative to the genome-wide distribution and based on two different quantiles (a = 0.5 or 0.95) of their posterior distribution.
Simulations that included selection typically resulted in an increased number of genetic regions being classified as outliers. As expected, we found that the ability to correctly classify nonneutral genetic regions as outlier loci was dependent upon the extent of selection. Few outliers were detected when only one population was affected (e.g., high ϕST, a = 0.5, NGS-individual model mean = 0.057, SD = 0.040), but most selected loci were correctly classified as outliers when all five populations were affected (e.g., high ϕST, a = 0.5, NGS-individual model mean = 0.612, SD = 0.310; Table S1, Table S2, and Table S3). Selection was easiest to detect when data were simulated and analyzed in accordance with the known haplotype model (in this case, when all five populations were affected by selection, all selected loci were identified as outliers even with a = 0.95; Table S1). Selection was generally easier to detect when migration was simulated. For example, under the known haplotype model the proportion of selected loci correctly classified as high outliers increased from 0.429 to 0.492 when simulated populations experienced the homogenizing effects of migration (means across all simulation conditions). For the complete set of simulations, the proportion of selected genetic regions that were correctly classified as outlier loci was greater than the proportion of neutral genetic regions incorrectly classified as outlier loci (Table S1, Table S2, and Table S3).
The ability of the method to correctly identify genetic regions affected by selective sweeps in the presence of group structure (i.e., when populations are organized into groups of populations) was highly dependent on the extent of the selective sweeps. When selective sweeps were confined to a single group, outliers were most often detected as genetic regions with high differentiation among populations within a group (i.e., high ϕSC; Table S4). The proportion of swept genetic regions correctly classified as ϕSC outliers was generally small and did not exceed 0.150, but was much greater than the proportion of neutral genetic regions classified as ϕSC outliers. In contrast, when both groups of populations were affected by selective sweeps, nearly all swept loci were classified as high ϕST and ϕSC outliers (92.5–100% using a = 0.5) and a smaller proportion (0.100–0.225) were classified as low or high ϕCT outliers. Thus, the selective sweeps were most readily detected on the basis of high differentiation among all populations (ϕST) or among populations within each group (ϕSC), but could also be detected on the basis of low or high differentiation between the two groups (ϕCT). This variety of outcomes arises from the stochastic nature used to determine the specific haplotypes that were swept to fixation. The false-positive rate for these group analyses was very low (between 0 and 0.032), similar to the population structure analyses (Table S2 and Table S4).
Divergent selection among African and European populations:
Mean genome-level ϕST between African ancestry and European ancestry populations based on the SeattleSNPs data set was 0.080 [95% equal tail probability interval (ETPI) = 0.065–0.097]. This means that ∼8% of DNA sequence variation was partitioned between the African and European ancestry populations.
We classified three genes as high ϕST outliers (using a = 0.5) on the basis of the first five SNPs data subset (Figure 1 and Figure 2). One of these genes was HSD11B2. Approximately 32% of molecular variation at this gene was partitioned between African and European ancestry populations (ϕST = 0.317, 95% ETPI = 0.159–0.482, Figure 1). Allelic variants of this gene produce an inherited form of hypertension and an end-stage renal disease (Quinkler and Stewart 2003). A weak association has also been detected between an intronic microsatellite in this gene and type 1 diabetes mellitus and diabetic nephropathy (Lavery et al. 2002). FOXA2 was also identified as an outlier, with ϕST = 0.321 (95% ETPI= 0.124–0.513, Figure 1). FOXA2 regulates insulin sensitivity and controls hepatic lipid metabolism in fasting and type 2 diabetes mice (Wolfrum et al. 2004; Puigserver and Rodgers 2006). A genome-wide association study detected a SNP near FOXA2 (rs1209523) that was associated with fasting glucose levels in European- and African-Americans (Xing et al. 2010). This outlier is particularly interesting as type 2 diabetes is 1.2–2.3 times more common in African-Americans than in European-Americans (Harris 2001). The third outlier locus was POLG2, with an estimated ϕST of 0.327 (95% ETPI = 0.175–0.477, Figure 1). This gene was also classified as a target of selection in humans in a recent study (Barreiro et al. 2008).
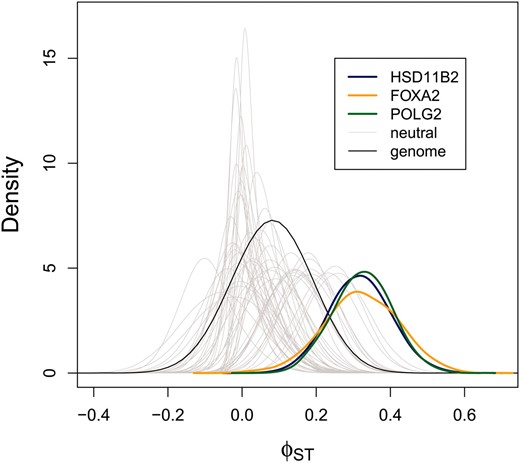
Locus-specific ϕST stimates for Africans and Europeans (SeattleSNPs data set). A point estimate of the genome-level ϕST distribution (based on the median from the posterior probability distributions of αST and βST) is denoted with a solid black line. The posterior probability distributions for the three outlier loci (colored lines) and 50 additional, randomly chosen genetic regions (gray lines) are also shown. These results are based on the first five SNPs in each gene; additional results are shown in Figure S3.
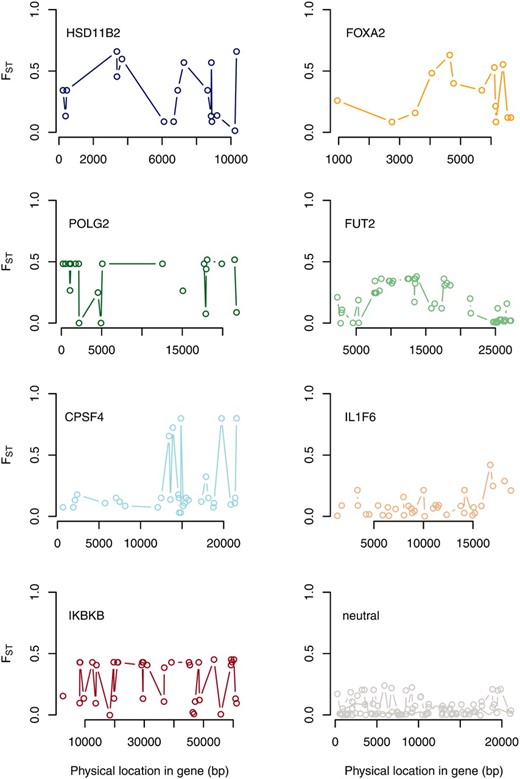
Point estimates of FST along specific genes (SeattleSNPs data set). Point estimates of FST are shown at each SNP for seven genes identified as outliers in the comparison of human populations with African and European ancestry as well as three randomly chosen neutral genes.
Selection among worldwide human populations:
For the worldwide human SNP data, estimates of mean chromosome-level ϕST were similar for the autosomal chromosomes and ranged from 0.083 (95% ETPI = 0.0752–0.091; chromosome 22) to 0.113 (95% ETPI = 0.104–0.120; chromosome 16; Table 2 and Figure 3). The estimate of ϕST for the X chromosome was considerably higher (0.139, 95% ETPI = 0.128–0.149).
Summary of worldwide human HapMap data and results
High ϕST | Low ϕST | ||||||
|---|---|---|---|---|---|---|---|
| Chromosome | Chromosome ϕST | No. loci | a = 0.5 | a = 0.95 | a = 0.5 | a = 0.95 | |
| 1 | 0.098 | (0.095–0.103) | 1402 | 59 | 28 | 8 | 1 |
| 2 | 0.101 | (0.097–0.106) | 956 | 32 | 11 | 5 | 1 |
| 3 | 0.099 | (0.095–0.104) | 723 | 29 | 13 | 2 | 0 |
| 4 | 0.100 | (0.094–0.106) | 563 | 22 | 12 | 1 | 0 |
| 5 | 0.095 | (0.089–0.100) | 565 | 20 | 10 | 3 | 2 |
| 6 | 0.091 | (0.087–0.095) | 710 | 23 | 8 | 6 | 0 |
| 7 | 0.098 | (0.092–0.103) | 679 | 25 | 14 | 2 | 0 |
| 8 | 0.092 | (0.087–0.098) | 438 | 24 | 6 | 2 | 0 |
| 9 | 0.095 | (0.089–0.099) | 569 | 22 | 9 | 2 | 0 |
| 10 | 0.092 | (0.087–0.098) | 526 | 20 | 6 | 3 | 2 |
| 11 | 0.096 | (0.092–0.101) | 686 | 24 | 7 | 1 | 0 |
| 12 | 0.097 | (0.092–0.102) | 683 | 36 | 14 | 2 | 0 |
| 13 | 0.090 | (0.082–0.099) | 243 | 8 | 7 | 1 | 0 |
| 14 | 0.100 | (0.093–0.107) | 388 | 16 | 8 | 2 | 0 |
| 15 | 0.109 | (0.101–0.117) | 380 | 20 | 10 | 0 | 0 |
| 16 | 0.113 | (0.104–0.120) | 426 | 21 | 9 | 1 | 0 |
| 17 | 0.107 | (0.101–0.114) | 603 | 27 | 12 | 1 | 0 |
| 18 | 0.092 | (0.084–0.100) | 225 | 8 | 4 | 3 | 0 |
| 19 | 0.099 | (0.094–0.104) | 729 | 34 | 10 | 4 | 0 |
| 20 | 0.101 | (0.093–0.109) | 367 | 15 | 8 | 0 | 0 |
| 21 | 0.083 | (0.075–0.091) | 158 | 6 | 3 | 1 | 0 |
| 22 | 0.102 | (0.093–0.111) | 283 | 11 | 6 | 1 | 0 |
| X | 0.139 | (0.128–0.149) | 347 | 16 | 7 | 0 | 0 |
High ϕST | Low ϕST | ||||||
|---|---|---|---|---|---|---|---|
| Chromosome | Chromosome ϕST | No. loci | a = 0.5 | a = 0.95 | a = 0.5 | a = 0.95 | |
| 1 | 0.098 | (0.095–0.103) | 1402 | 59 | 28 | 8 | 1 |
| 2 | 0.101 | (0.097–0.106) | 956 | 32 | 11 | 5 | 1 |
| 3 | 0.099 | (0.095–0.104) | 723 | 29 | 13 | 2 | 0 |
| 4 | 0.100 | (0.094–0.106) | 563 | 22 | 12 | 1 | 0 |
| 5 | 0.095 | (0.089–0.100) | 565 | 20 | 10 | 3 | 2 |
| 6 | 0.091 | (0.087–0.095) | 710 | 23 | 8 | 6 | 0 |
| 7 | 0.098 | (0.092–0.103) | 679 | 25 | 14 | 2 | 0 |
| 8 | 0.092 | (0.087–0.098) | 438 | 24 | 6 | 2 | 0 |
| 9 | 0.095 | (0.089–0.099) | 569 | 22 | 9 | 2 | 0 |
| 10 | 0.092 | (0.087–0.098) | 526 | 20 | 6 | 3 | 2 |
| 11 | 0.096 | (0.092–0.101) | 686 | 24 | 7 | 1 | 0 |
| 12 | 0.097 | (0.092–0.102) | 683 | 36 | 14 | 2 | 0 |
| 13 | 0.090 | (0.082–0.099) | 243 | 8 | 7 | 1 | 0 |
| 14 | 0.100 | (0.093–0.107) | 388 | 16 | 8 | 2 | 0 |
| 15 | 0.109 | (0.101–0.117) | 380 | 20 | 10 | 0 | 0 |
| 16 | 0.113 | (0.104–0.120) | 426 | 21 | 9 | 1 | 0 |
| 17 | 0.107 | (0.101–0.114) | 603 | 27 | 12 | 1 | 0 |
| 18 | 0.092 | (0.084–0.100) | 225 | 8 | 4 | 3 | 0 |
| 19 | 0.099 | (0.094–0.104) | 729 | 34 | 10 | 4 | 0 |
| 20 | 0.101 | (0.093–0.109) | 367 | 15 | 8 | 0 | 0 |
| 21 | 0.083 | (0.075–0.091) | 158 | 6 | 3 | 1 | 0 |
| 22 | 0.102 | (0.093–0.111) | 283 | 11 | 6 | 1 | 0 |
| X | 0.139 | (0.128–0.149) | 347 | 16 | 7 | 0 | 0 |
Summary of data and results for each chromosome are shown, including chromosome-level ϕST (median and 95% ETPI), the number of loci, and the number of loci classified as outliers (data from Jakobsson et al. 2008).
Summary of worldwide human HapMap data and results
High ϕST | Low ϕST | ||||||
|---|---|---|---|---|---|---|---|
| Chromosome | Chromosome ϕST | No. loci | a = 0.5 | a = 0.95 | a = 0.5 | a = 0.95 | |
| 1 | 0.098 | (0.095–0.103) | 1402 | 59 | 28 | 8 | 1 |
| 2 | 0.101 | (0.097–0.106) | 956 | 32 | 11 | 5 | 1 |
| 3 | 0.099 | (0.095–0.104) | 723 | 29 | 13 | 2 | 0 |
| 4 | 0.100 | (0.094–0.106) | 563 | 22 | 12 | 1 | 0 |
| 5 | 0.095 | (0.089–0.100) | 565 | 20 | 10 | 3 | 2 |
| 6 | 0.091 | (0.087–0.095) | 710 | 23 | 8 | 6 | 0 |
| 7 | 0.098 | (0.092–0.103) | 679 | 25 | 14 | 2 | 0 |
| 8 | 0.092 | (0.087–0.098) | 438 | 24 | 6 | 2 | 0 |
| 9 | 0.095 | (0.089–0.099) | 569 | 22 | 9 | 2 | 0 |
| 10 | 0.092 | (0.087–0.098) | 526 | 20 | 6 | 3 | 2 |
| 11 | 0.096 | (0.092–0.101) | 686 | 24 | 7 | 1 | 0 |
| 12 | 0.097 | (0.092–0.102) | 683 | 36 | 14 | 2 | 0 |
| 13 | 0.090 | (0.082–0.099) | 243 | 8 | 7 | 1 | 0 |
| 14 | 0.100 | (0.093–0.107) | 388 | 16 | 8 | 2 | 0 |
| 15 | 0.109 | (0.101–0.117) | 380 | 20 | 10 | 0 | 0 |
| 16 | 0.113 | (0.104–0.120) | 426 | 21 | 9 | 1 | 0 |
| 17 | 0.107 | (0.101–0.114) | 603 | 27 | 12 | 1 | 0 |
| 18 | 0.092 | (0.084–0.100) | 225 | 8 | 4 | 3 | 0 |
| 19 | 0.099 | (0.094–0.104) | 729 | 34 | 10 | 4 | 0 |
| 20 | 0.101 | (0.093–0.109) | 367 | 15 | 8 | 0 | 0 |
| 21 | 0.083 | (0.075–0.091) | 158 | 6 | 3 | 1 | 0 |
| 22 | 0.102 | (0.093–0.111) | 283 | 11 | 6 | 1 | 0 |
| X | 0.139 | (0.128–0.149) | 347 | 16 | 7 | 0 | 0 |
High ϕST | Low ϕST | ||||||
|---|---|---|---|---|---|---|---|
| Chromosome | Chromosome ϕST | No. loci | a = 0.5 | a = 0.95 | a = 0.5 | a = 0.95 | |
| 1 | 0.098 | (0.095–0.103) | 1402 | 59 | 28 | 8 | 1 |
| 2 | 0.101 | (0.097–0.106) | 956 | 32 | 11 | 5 | 1 |
| 3 | 0.099 | (0.095–0.104) | 723 | 29 | 13 | 2 | 0 |
| 4 | 0.100 | (0.094–0.106) | 563 | 22 | 12 | 1 | 0 |
| 5 | 0.095 | (0.089–0.100) | 565 | 20 | 10 | 3 | 2 |
| 6 | 0.091 | (0.087–0.095) | 710 | 23 | 8 | 6 | 0 |
| 7 | 0.098 | (0.092–0.103) | 679 | 25 | 14 | 2 | 0 |
| 8 | 0.092 | (0.087–0.098) | 438 | 24 | 6 | 2 | 0 |
| 9 | 0.095 | (0.089–0.099) | 569 | 22 | 9 | 2 | 0 |
| 10 | 0.092 | (0.087–0.098) | 526 | 20 | 6 | 3 | 2 |
| 11 | 0.096 | (0.092–0.101) | 686 | 24 | 7 | 1 | 0 |
| 12 | 0.097 | (0.092–0.102) | 683 | 36 | 14 | 2 | 0 |
| 13 | 0.090 | (0.082–0.099) | 243 | 8 | 7 | 1 | 0 |
| 14 | 0.100 | (0.093–0.107) | 388 | 16 | 8 | 2 | 0 |
| 15 | 0.109 | (0.101–0.117) | 380 | 20 | 10 | 0 | 0 |
| 16 | 0.113 | (0.104–0.120) | 426 | 21 | 9 | 1 | 0 |
| 17 | 0.107 | (0.101–0.114) | 603 | 27 | 12 | 1 | 0 |
| 18 | 0.092 | (0.084–0.100) | 225 | 8 | 4 | 3 | 0 |
| 19 | 0.099 | (0.094–0.104) | 729 | 34 | 10 | 4 | 0 |
| 20 | 0.101 | (0.093–0.109) | 367 | 15 | 8 | 0 | 0 |
| 21 | 0.083 | (0.075–0.091) | 158 | 6 | 3 | 1 | 0 |
| 22 | 0.102 | (0.093–0.111) | 283 | 11 | 6 | 1 | 0 |
| X | 0.139 | (0.128–0.149) | 347 | 16 | 7 | 0 | 0 |
Summary of data and results for each chromosome are shown, including chromosome-level ϕST (median and 95% ETPI), the number of loci, and the number of loci classified as outliers (data from Jakobsson et al. 2008).
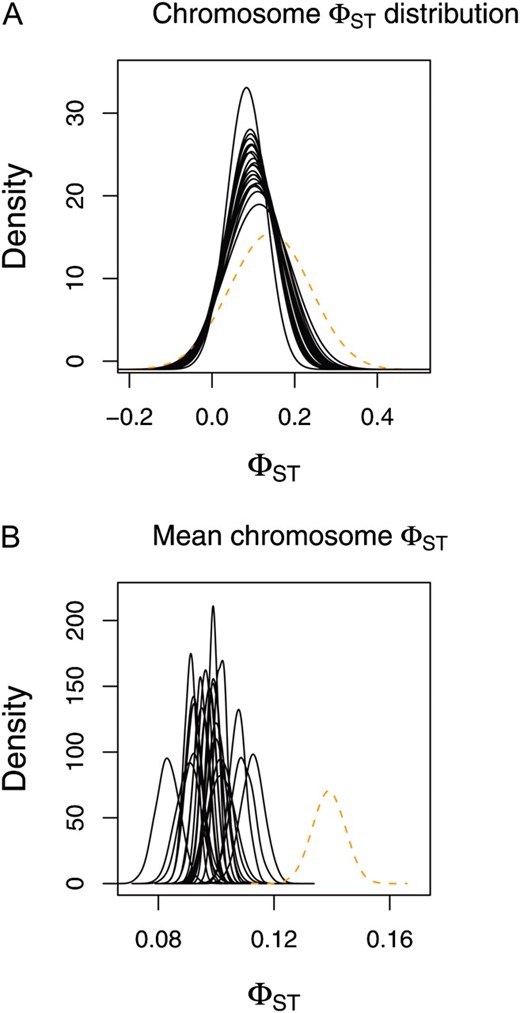
Chromosome-level estimates of ϕST for the large sample of human genetic diversity in 33 populations (data from Jakobsson et al. 2008). (A and B) Point estimates of each chromosome-level ϕST distribution (based on the median from the posterior probability distributions of αST and βST) are denoted with solid black lines (autosomes) or a dashed orange line (A; X chromosome). (B) Posterior probability distributions for the mean chromosome-level ϕST for each chromosome.
We detected remarkable variation in ϕST along each of the chromosomes (Figure 4). We detected 569 unique ϕST outlier loci, which we designate as candidate genes for selection among worldwide human populations (Table S5). Of these 569 genes, 518 were high ϕST outlier loci (including 222 with a = 0.95) and 51 were low ϕST outlier loci (including 6 with a = 0.95). Outlier loci were detected on all chromosomes, with the greatest number identified on chromosome 1 (67 outlier loci) and the fewest on chromosome 13 (9 outlier loci; Table 2 and Figure 4). Some of the genes that we classified as outliers have previously been implicated as genes experiencing selection in human populations. These include CYP3A4 and CYP3A5, which are cytochrome P450 genes found near one another on chromosome 7 (a = 0.95; CYP3A4 ϕST = 0.293, 95% ETPI = 0.245–0.319 and CYP3A5 ϕST = 0.359, 95% ETPI = 0.315–0.401). These genes are important for detoxification of plant secondary compounds and are involved in metabolism of some prescribed drugs. Additional evidence that these genes have experienced positive selection in human populations exists from previous studies based on distortions of the site frequency spectrum in African, European, and Chinese populations; population differentiation between the CEU and YRI populations; and extended haplotype homozygosity in HapMap populations (Carlson et al. 2005; Voight et al. 2006; Nielsen et al. 2007; Chen et al. 2010). Another example is the PKDREJ gene on chromosome 22, which is a candidate sperm receptor gene of mammalian egg-coat proteins and had one of the highest estimates of ϕST (0.455, 95% ETPI = 0.396–0.504). Variation in this gene is consistent with positive selection among primate lineages, although evidence suggests that balancing selection might act to maintain diversity at this gene in human populations (Hamm et al. 2007). We also found evidence of divergent selection acting on SLC24A5, which has been associated with differences in skin pigmentation among humans and was classified as a candidate for positive selection in several studies (Barreiro et al. 2008). Finally, three of the high ϕST outliers at a = 0.95 (RTTN, MSX2, and CDAN1) were among seven human skeletal genes identified by Wu and Zhang as genes with elevated FST at nonsynonymous SNPs between African and non-African (European and East Asian) populations and candidates for recent positive selection in Europeans and East Asians (Wu and Zhang 2010).
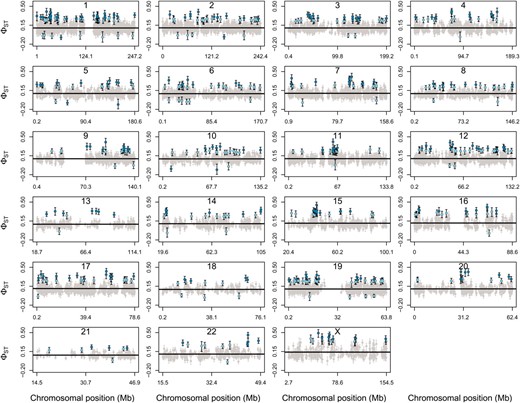
Estimates of ϕST across the genome of worldwide human populations (data from Jakobsson et al. 2008). Each panel depicts a different human chromosome and is labeled accordingly. The solid black line denotes the point estimate (median of the posterior distribution) of the mean chromosome-level ϕST for a chromosome. Estimates of ϕST for individual genes are shown in gray for nonoutlier genes and light (at a = 0.5) or dark blue (at a = 0.95) for outlier genes. For each gene the solid circle gives the median from the posterior distribution of ϕST for that locus and the bars denote the 95% ETPI.
We detected additional genes with very high ϕST that have not, to the best of our knowledge, been previously implicated as experiencing selection in human populations but have been found to be associated with disease traits. For example, the estimate of ϕST for spermatogenesis-associated 5-like 1 (SPATA5L1) on chromosome 15 was 0.347 (95% ETPI = 0.246–0.430). This gene was classified as a high ϕST outlier in the analysis and has been linked to renal function and kidney disease, but has not been identified in previous tests for selection (Köttgen et al. 2009).
We also identified novel candidate targets of balancing selection in worldwide human populations. Interestingly, none of the genes that we classified as low ϕST outlier loci were implicated as targets of balancing selection in European- and African-American populations in a recent study by Andrés et al. (2009). Several of these genes have been linked to diseases. For example, RIF1 was classified as a low ϕST outlier at the a = 0.95 level (ϕST = −0.065, 95% ETPI = −0.105– −0.023). RIF1 is an anti-apoptotic factor involved in DNA repair that is necessary for S-phase progression and is heavily expressed in breast cancer tumors (Wang et al. 2009). Another example is the AKT3 gene found on chromosome 1 (ϕST = −0.046, 95% ETPI = −0.102–0.020; outlier at a = 0.5). This gene is involved in cell-cycle regulation and is highly expressed in malignant melanoma, but is also important for attainment of normal organ size, including brain size in mice (Stahl et al. 2004; Easton et al. 2005).
DISCUSSION
We have presented a novel model to quantify genome-wide population genetic structure and identify genetic regions that are likely to have experienced natural selection. Unlike previous methods for quantifying structure and detecting genetic signatures of selection, the proposed method accurately models the stochastic sampling of sequences that is inherent in current NGS instruments and incorporates genetic distances among sequences in estimates of genetic differentiation. Although few population genomics studies based on NGS of individuals have been published to date (hence the analysis of Sanger sequence and SNP human data sets instead of NGS data sets), various large-scale projects are currently underway to obtain these data in large samples of humans (e.g., 1000 genomes project, http://www.1000genomes.org) and a few recent studies suggest that these data will soon be available for many nonhuman (including nonmodel) species (Gompert et al. 2010; Hohenlohe et al. 2010). However, beyond data acquisition, substantial biological insights will be possible only if accompanied by models and methods designed to take full advantage of these data, while accurately modeling sources of error. Along with a few other recent reports of models for NGS (Guo et al. 2009; Lynch 2009; Futschik and Schlötterer 2010; Gompert et al. 2010), we believe our analytical methods help to address this need.
Model properties:
The analyses of simulated and human genetic data sets suggest that the model provides statistically sound estimates of population differentiation for large sets of loci (see File S1, Figure S1, and Figure S2, Simulations: estimation of ϕ-statistics). For example, the estimates of genome-level ϕST for the SeattleSNPs human sequence data and chromosome-level ϕST for the worldwide human SNP data (0.080–0.139) were similar to mean levels of genetic differentiation among human populations based on FST [e.g., FST = 0.09–0.14 for Yoruba, European, Han Chinese, and Japanese populations (Weir et al. 2005; Barreiro et al. 2008)]).
The simulation results indicate that the model only rarely identifies neutral loci incorrectly as outliers (i.e., it has a low false-positive rate). Using a = 0.5, the false-positive rate for high or low ϕST outliers was never >0.04, and using a = 0.95 this rate was never >0.01. Low false-positive rates are particularly important for genome-wide scans in which a large number of genes could otherwise show spurious evidence of selection. In contrast, false-positive rates as high as 0.343 have been reported for FST outlier analyses of selection that use coalescent simulations under an incorrect demographic history to derive a neutral distribution (Excoffier et al. 2009). Moreover, the low false-positive rate for the method held across all simulated demographic scenarios (i.e., different population divergence times, variation in migration rates, and the presence or absence of group structure). This is expected as the null, genome-level distribution we estimate is based on the observed data rather than an assumed demographic history and most demographic histories should be appropriately captured by this genome-level distribution (Beaumont and Balding 2004; Guo et al. 2009). Similarly low false-positive rates were reported by Guo et al. (2009), using a hierarchical Bayesian model based on FST. Nonetheless, it is important to note that these hierarchical models generally identify outlier loci as those that are inconsistent with the genome as a whole and thus will not work well if most of the genome has recently experienced selection of a similar magnitude.
Simulation results further indicate that the method has the ability to detect genetic regions under selection and to a greater extent when selective sweeps affect multiple populations or migration occurs. When a simulated genetic region was affected by selection in at least three populations, the selected locus was generally at least 10 times more likely to be classified as an outlier than a randomly chosen neutral locus. This suggests a high true-positive rate (at least under favorable conditions) and that candidate selected genes identified by the method will be enriched substantially for genes actually experiencing selection. This is particularly true when genes are classified as outliers using the more stringent criterion of a = 0.95 (although this will also decrease the total number of selected genes identified relative to a = 0.5). One benefit of quantifying genetic divergence on the basis of haplotypes (using ϕST) instead of SNP, microsatellite, or AFLP alleles (using FST) is that multiple selective sweeps should result in increased genetic distances among haplotypes in different populations, making them easier to detect than genes that experienced a single selective sweep, as was used for the simulations.
Despite these generally promising results from the simulations, many selected genes were not classified as outliers using the method and constitute false negatives. There are several synergistic reasons that genes affected by selection might not be classified as outliers, which include inherent limitations in outlier-based tests for selection and difficulties detecting selection (which does not always leave a clear signal), as well as specific details of the simulations (Nielsen 2005; Kelley et al. 2006). First, as pointed out previously, outlier analyses will detect only genes that stand out from the genome-wide distribution and thus might not detect genes experiencing weak selection or be applicable when much of the genome is under selection (Michel et al. 2010). However, this particular issue is more likely to affect empirical data than the simulated data sets. The data we simulated experienced selective sweeps with arbitrary linkage between the affected genetic region and the gene under selection. This means that selection could have favored the same or different haplotypes in each population. Thus, a clear molecular signature was not always left by the simulated selective sweeps. We simulated selection in this manner for computational efficiency and because it accurately reflects the effect of a recent and complete selective sweep. Specifically, we believe that this is a realistic model for selection as researchers will often detect selection on the basis of genetic regions linked to the gene under selection rather than the actual gene under selection and patterns of linkage might vary among populations. Nonetheless, when evidence of selection comes directly from the target of selection or there is tight linkage with consistent patterns of linkage disequilibrium between the selected gene and a sequenced genetic region, a stronger signal of selection should be evident and selection should be easier to detect.
Evidence of selection in human populations:
A diverse array of studies (Akey et al. 2002; Nielsen et al. 2007; Barreiro et al. 2008; Nielsen et al. 2009) has found that natural selection has played an important role in shaping functional genetic variation in humans. Analyses based on our model for quantifying the distribution of genetic variation among populations similarly find substantial evidence for the action of natural selection. We classified ∼10 times as many genes as candidates for positive or divergent selection (high ϕST outlier loci) than as candidates for balancing selection (low ϕST outlier loci). However, this does not mean that divergent and positive selection more commonly affects human genetic variation than balancing selection. Evidence from other studies suggests that weak negative selection is prevalent in the human genome and balancing selection is also fairly common (Bustamante et al. 2005; Andrés et al. 2009). Instead this difference in the prevalence of different forms of selection might indicate that there is more variation in the strength of positive selection, resulting in a greater number of extreme genetic regions, than there is in the strength of negative or balancing selection with many genetic regions weakly affected by these factors. However, this cannot be explicitly addressed by our study. Finally, previous simulation studies suggest that it is difficult to detect balancing selection using outlier analyses (Beaumont and Balding 2004; Guo et al. 2009), which might reflect the different molecular signals left by divergent and balancing selection or the bounded nature of FST and ϕST.
Specific genes identified as outliers by our analysis include several genes that have been repeatedly implicated as targets of positive selection in humans, such as POLG2, CYP3A5, and SLC24A5. Nonetheless, we also detected novel candidate genes for selection in humans (e.g., FOXA2) and failed to detect selection on genes expected to have experienced strong selection on the basis of previous studies, such as the lactase (LCT) gene (Bersaglieri et al. 2004; Nielsen et al. 2007; Tishkoff et al. 2007). A lack of complete concordance with earlier studies is not surprising as a general lack of concordance among studies of selection in humans has been noted (Nielsen et al. 2007). This lack of concordance likely reflects the sensitivity of different methods to different signatures of selection, which affects whether methods are more likely to detect recent, ongoing, or more ancient selective sweeps, as well as the specific nucleotides and populations analyzed. For example, the lack of evidence from the worldwide human data set for selection on LCT appears to reflect the SNPs included in this study. Previous studies of human genetic variation have detected high values of FST at specific SNPs in the LCT gene (e.g., 0.53 for SNPs rs4988235 and rs182549), but have also shown that FST varies across this gene (Bersaglieri et al. 2004). Previous estimates of FST for the two genic SNPs in LCT included in our analysis that were also investigated by Bersaglieri et al. (2004) were 0 (rs2874874) and 0.17 (rs2322659), which do not stand out markedly from background levels of differentiation.
Conclusions:
Technological advances in DNA sequencing are ushering in a new era of population genomics. Whereas researchers previously were constrained to various, relatively low-throughput molecular markers for genotyping, it is now possible to rapidly generate very large volumes of DNA sequence data for any group of organisms (Mardis 2008a; Gilad et al. 2009). This new capability will allow researchers to address long-standing and fundamental questions in evolutionary biology, which were once considered nearly intractable because of limited genetic data (Mardis 2008b). However, most published NGS studies have been primarily descriptive (e.g., transcriptome characterization; Vera et al. 2008; Parchman et al. 2010) or have been forced to discard valuable data because of analytical limitations (Hohenlohe et al. 2010) and have not taken full advantage of the potential of the sequence data. To do so, researchers need robust and accessible methods and models that can be applied to NGS data to test evolutionary hypotheses. The model presented here helps fill this gap, as it allows us to quantify heterogeneous genomic divergence among populations and identify genetic regions affected by selection. The Bayesian analysis of molecular variance illustrates the potential of combining appropriate models and NGS to address important questions in evolutionary biology and genetics and is a critical step toward utilizing the growing abundance of sequence data for population genomics.
Footnotes
Supporting information is available online at http://www.genetics.org/cgi/content/full/genetics.110.124693/DC1.
Footnotes
Communicating editor: L. Excoffier
Acknowledgements
We thank J. Fordyce, M. Forister, C. Nice, P. Nosil, T. Parchman, and R. Safran for discussion and comments on previous drafts of this article. Comments from L. Excoffier and two anonymous reviewers helped to improve the article. We are indebted to R. Williamson for his contributions to the development of the bamova software. This work was supported by National Science Foundation Division of Biological Infrastructure (DBI) award 0701757 (to C.A.B.).
References
Beaumont, M. A., and R. A. Nichols,
Flint, J., J. Bond, D. C. Rees, A. J. Boyce, J. M. Roberts-Thomson et al.,
Gompert, Z., M. L. Forister, J. A. Fordyce, C. C. Nice, R. Williamson et al.,
Kronholm, I., O. Loudet and J. de Meaux,
Kullback, S., and R. A. Leibler,
Lavery, G. G., C. L. McTernan, S. C. Bain, T. A. Chowdhury, M. Hewison et al.,
Meyer, M., and M. Kircher,
Michel, A. P., S. Sim, T. H. Q. Powell, M. S. Taylor, P. Nosil et al.,
Nielsen, R., I. Hellmann, M. Hubisz, C. Bustamante and A. G. Clark,
Parchman, T. L., K. S. Geist, J. A. Grahnen, C. W. Benkman and C. A. Buerkle,
Stajich, J. E., and M. H. Hahn,
Voight, B. F., S. Kudaravalli, X. Q. Wen and J. K. Pritchard,

{kind=link}
{kind=link}
{kind=link}
{kind=link}