





Abstract
Fitness epistasis, the interaction among alleles at different loci in their effects on fitness, has potentially important consequences for adaptive evolution. We investigated fitness epistasis among amino acids of a functionally important region of the human immunodeficiency virus type 1 (HIV-1) exterior envelope glycoprotein (gp120). Seven mutations putatively involved in the adaptation of the second conserved to third variable protein region (C2–V3) to the use of an alternative host-cell chemokine coreceptor (CXCR4) for cell entry were engineered singly and in combinations on the wild-type genetic background and their effects on viral infectivity were measured. Epistasis was found to be common and complex, involving not only pairwise interactions, but also higher-order interactions. Interactions could also be surprisingly strong, changing fitness by more than 9 orders of magnitude, which is explained by some single mutations being practically lethal. A consequence of the observed epistasis is that many of the minimum-length mutational trajectories between the wild type and the mutant with highest fitness on cells expressing the alternative coreceptor are selectively inaccessible. These results may help explain the difficulty of evolving viruses that use the alternative coreceptor in culture and the delayed evolution of this phenotype in natural infection. Knowledge of common, complex, and strong fitness interactions among amino acids is necessary for a full understanding of protein evolution.
FITNESS epistasis refers to the interaction among alleles at different loci in their effects on fitness. The importance of such interactions to adaptation has been controversial. Wright (1932) argued that fitness epistasis would cause multipeaked fitness landscapes and that these would constrain adaptive evolution by attracting populations to local peaks. Fisher (1930), on the other hand, argued against the likelihood of such rugged fitness landscapes. And, although there is some indirect evidence of multipeaked fitness landscapes (e.g., Nijhuis et al. 1999; Burch and Chao 2000), it is difficult to demonstrate the existence of such landscapes conclusively. A direct demonstration would require analyzing all possible interactions in an entire genome because it is always possible that a mutation at an unstudied locus may generate a genotype that spans a fitness valley (Whitlock et al. 1995). However, even on a single-peaked fitness landscape, epistasis may produce minimum-length mutational trajectories that are unlikely to be realized during adaptive evolution because they include neutral or deleterious mutations (Weinreich et al. 2006). This will arise if the sign of the fitness effect of a mutation depends on its genetic background (sign epistasis) (Weinreich et al. 2005). Fitness epistasis may also generate linkage disequilibrium (Kimura 1956; Lewontin and Kojima 1960), with potentially important consequences for the efficiency of natural selection and the evolutionary maintenance of recombination (Felsenstein 1988; Kondrashov 1993).
Notwithstanding theoretical developments, the nature of fitness epistasis remains poorly understood. Early quantitative genetics experiments on the viability effects of epistasis in Drosophila showed these to be only weak to moderate (Spassky et al. 1965; Temin et al. 1969). More recent observations on microbes adapting to antimicrobial drugs (reviewed by Maisnier-Patin and Andersson 2004) and responding to other selection pressures (e.g., Poon and Chao 2005; Sanjuan et al. 2005) suggest strong compensatory effects of epistasis. Much of the recent work on fitness epistasis at the molecular level has involved the analysis of intergenic or intragenic interactions in microbes through the study of standing genetic variation or spontaneous mutation (Bonhoeffer et al. 2004; Maisnier-Patin et al. 2005; Bershtein et al. 2006), engineered site-specific mutations (Sanjuan et al. 2004; Lunzer et al. 2005; Weinreich et al. 2006; Pepin and Wichman 2007), and a combination of both types of data (Poon and Chao 2005; Sanjuan et al. 2005; Poon and Chao 2006). However, a systematic study of the nature and consequences of fitness epistasis within a protein has yet to be conducted. We have investigated the nature and consequences of interactions among amino acid mutations on fitness in a functionally important protein region of human immunodeficiency virus type 1 (HIV-1).
The use of site-directed mutagenesis to measure the fitness effects of mutations singly and in combination on a standard genetic background has the advantage over other approaches of allowing unambiguous attribution of fitness interactions to specific combinations of mutations. This approach was used in a recent study of protein evolution involved in the switch by HIV-1 from using its primary host-cell chemokine coreceptor to an alternative chemokine coreceptor (Pastore et al. 2006). Considerable attention has been focused on this question because HIV-1 preferentially uses the primary coreceptor in early infection, but switches to the alternative coreceptor late in infection in about 50% of patients, and this switch is associated with disease progression (Philpott 2003). The entry of an HIV-1 virus particle (virion) into a host cell requires that the exterior envelope glycoprotein, gp120, on the surface of the virion, interact with two cell-surface receptors: CD4 and one of two chemokine coreceptors, either CCR5 or CXCR4 (Wyatt and Sodroski 1998). Binding of gp120 to CD4 is thought to cause a conformational change to gp120 that exposes its third variable region (V3), allowing it to bind to one of the chemokine coreceptors (Huang et al. 2005; Huang et al. 2007). This conformational change to gp120 appears to involve its second variable region (V2), which may shield V3 (Wyatt and Sodroski 1998). V3 determines which coreceptor is used by the virus (Hwang et al. 1991) through its sequence variation (Dittmar et al. 1997; Speck et al. 1997; Cormier and Dragic 2002), although Pastore et al. (2006) have shown that the contiguous first and second variable regions (V1/V2) and the second conserved region (C2), which separates V1/V2 from V3, may also be involved.
Pastore et al. (2004) used selection experiments to identify the amino acid changes putatively involved in coreceptor switching. They selected for CXCR4 use in CCR5-adapted HIV-1 strains by passaging (serially transferring) virus through cell cultures containing progressively increasing proportions of CXCR4-expressing cells. They also sequenced the V1/V2 or C2 region and the V3 region for several isolates as they adapted to CXCR4. Amino acid replacements observed in these experiments were then engineered, using site-directed mutagenesis, on to CCR5-adapted wild-type genetic backgrounds and tested for their effects singly and in combination on the coreceptor usage of the virus (Pastore et al. 2006). They found that mutations in V3 are necessary for coreceptor switching, but generally reduced viral infectivity, and that mutations in V1/V2 and C2 may compensate for this loss of fitness.
Here, we investigate the nature of this epistasis and its consequences for adaptation. We have assayed the fitness of a set of HIV-1 envelope glycoprotein clones constructed by engineering amino acid mutations in the C2–V3 region singly and in combination on a CCR5-adapted wild-type background (Pastore et al. 2006). These mutations correspond to the amino acid changes of one of the CXCR4-adapted isolates evolved from a CCR5-adapted strain by Pastore et al. (2004). Thus, we describe in detail the frequency, level, direction, form, magnitude, and consequences of fitness epistasis in the context of HIV-1 adapting to CXCR4. We found that fitness epistasis is common and often very strong and compensatory and that it may be complex, involving not only pairwise interactions between residues, but also higher-order interactions. We also show that sign epistasis constrains adaptation in the C2–V3 protein region through the production of selectively inaccessible minimum-length mutational trajectories. Such constraints may help explain the difficulty of evolving CXCR4 use in culture and the delayed evolution of this phenotype in natural infection. Fitness epistasis must be considered for a full understanding of protein evolution.
MATERIALS AND METHODS
Fitness assay:
We assayed the fitness of a set of envelope glycoprotein clones that had been constructed by engineering amino acid mutations in the C2–V3 region singly and in combination on the CCR5-adapted ADA strain wild-type background (Pastore et al. 2006). These mutations correspond to the seven amino acid changes of the CXCR4-adapted ADA-1 isolate evolved from ADA (Pastore et al. 2004). Five of the these changes occurred in V3 and two in C2 (Figure 1). The available mutants included all single mutations, all possible combinations of the five V3 mutations, and a subset of combinations involving the two C2 mutations, including all seven mutations, for a total of 53 mutant envelopes (Figure 2). Pastore et al. (2006) did not construct all possible 27 − 1 = 127 mutants as they were attempting to engineer the minimum number of envelopes required to explain the evolution of ADA-1.

The HIV-1 ADA C2 (partial) and V3 gp120 protein regions. Engineered mutations are numbered and shown below the sequence. Underlined residues are putative N-linked glycosylation motifs.
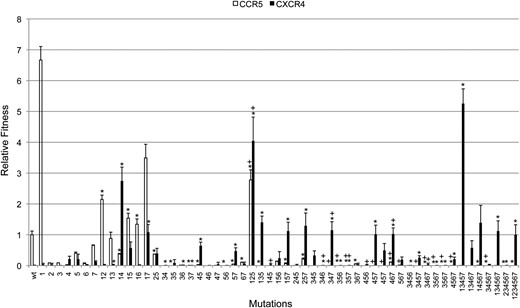
The relative fitness of each engineered envelope assayed on cells expressing CCR5 or CXCR4. Error bars indicate 1 standard deviation. An asterisk (*) indicates statistically significant overall epistasis (ε), and a plus sign (+) indicates significant net epistasis for higher-order interactions (ε′).
Fitness was measured with a single-cycle pseudovirus infection assay, as in Pastore et al. (2006). Briefly, mutant envelope clones inserted into the pSVIII plasmid were cotransfected with env-negative, luciferase-positive (NL4-3-Luc+E-R-) reporter plasmids into 293T cells, and the resulting pseudoviruses were harvested, standardized for p24 content, and used to infect either CCR5-expressing cells or CXCR4-expressing cells. The luciferase activity from triplicate wells of a multiwell plate was measured on a luminometer after 48 hr of culture (supporting information, Figure S1). Assays were conducted in triplicate for each host-cell type: cells expressing CCR5 or CXCR4. We used NP-2-CD4-CCR5 and NP-2-CD4-CXCR4 cell lines because these cells do not express endogenous chemokine coreceptors (Soda et al. 1999) that mediate entry of some viruses via GPR1 and GPR15 (Edinger et al. 1998), thus forcing entry through CCR5 or CXCR4. Unlike other measures of fitness, such as resistance to an antiviral drug, which may trade off against other components of fitness (Mammano et al. 2000), the rate of cell infection, or infectivity, is the appropriate measure of the effect on fitness of the C2–V3 protein region. This is because the sole function of this region is in cell entry, which is unlikely to trade off against other components of fitness under culture conditions, or other steps in the replication cycle, which are controlled by other proteins (Coffin 1999). Furthermore, the interaction between V3 and the chemokine coreceptor affects the rate-limiting step in cellular infection (Platt et al. 2005). However, this protein region is targeted by antibodies (Zolla-Pazner 2004), and here we restrict our analysis to effects on the infectivity component of fitness.
Fitness epistasis:
Relative fitness, w, was calculated by dividing absolute fitness by the absolute fitness of the CCR5-adapted wild-type isolate, ADA, for assays with cells expressing CCR5, and by dividing absolute fitness by the absolute fitness of the CXCR4-adapted ADA-1 mutant isolate for assays with cells expressing CXCR4.
Equation 6 assumes that the contributing lower-order net epistatic deviations are independent.
The statistical significance of epistatic deviations (H0: ε = 0) was determined with a Z-test. To account for multiple comparisons, the experimentwise type I error rate was maintained at α′ = 0.05 using the sequential Bonferroni method (Sokal and Rohlf 1995).
This is the preferred measure of the magnitude of epistasis because it provides the order-of-magnitude change in fitness due to epistasis. Note that when applied to higher-order interactions, E is a measure of the magnitude of overall epistasis, not net epistasis. Therefore, for higher-order interactions the sign of E may differ from that of ε′.
RESULTS
Fitness:
The seven mutations in the C2–V3 region of the CXCR4-adapted isolate ADA-1 were engineered singly and in combinations on the genetic background of the CCR5-adapted wild-type strain ADA (Figure 1). The fitness of these mutants was assayed on CCR5-expressing cells and CXCR4-expressing cells. On CCR5 cells, the fitness of most single and multiple mutants was lower than that of ADA (Figure 2 and supporting information, Table S1). The exceptions are the mutant with mutation 1 and several multiple mutants containing mutation 1. Therefore, ADA does not have the highest fitness with respect to CCR5 use, possibly because of competing selection pressures in natural infection, such as antibody surveillance (Pastore et al. 2006). On CXCR4 cells, several multiple mutants had higher fitness than ADA-1 (Figure 2 and Table S1), which had been isolated after selection for CXCR4 use (Pastore et al. 2004). These mutants typically contain mutation 1. Mutant 13457 (containing mutations 1, 3, 4, 5, and 7) had the highest fitness on CXCR4 cells. The fact that a mutant with all seven mutations (ADA-1) evolved in response to selection for CXCR4 use suggests that direct mutational trajectories from ADA to mutants with higher fitness on CXCR4 cells may be selectively inaccessible because of epistasis.
Epistatic deviation:
Of the 48 interactions tested for overall epistasis on each host-cell type, 27 (56%) were statistically significant on CCR5 cells and the same number were significant on CXCR4 cells, although some of these interactions differed between the two assays (Figure 2 and Table S1). On CCR5 cells, significant overall epistatic deviations were both positive and negative, and the median epistatic deviation was 0.0002 (range −1.6857–2.5460) (Figure 3a and Table S1). In contrast, significant overall epistatic deviations on CXCR4 cells were exclusively positive, with a median of 0.4444 (0.0074–5.2552). Therefore, overall epistasis was common and ranged from −1.6857 to 5.2552.
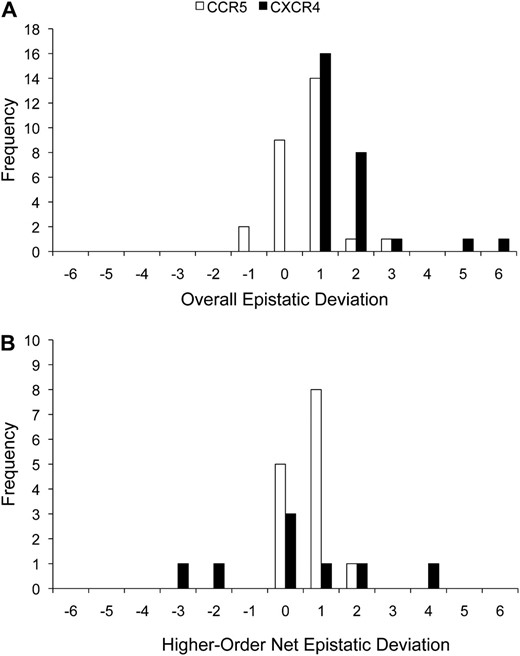
Frequency distributions of epistatic deviation for statistically significant interactions on cells expressing CCR5 or CXCR4. Values on the epistatic deviation axis are upper bounds of the intervals. (A) Overall epistatic deviation. (B) Higher-order net epistatic deviation.
To give specific examples, we focus on two statistically significant pairwise interactions, one on CCR5 cells and the other on CXCR4 cells. On CCR5 cells, mutation 1 had fitness (w) 6.6671 (variance = 0.1926) and mutation 5, fitness 0.3998 (0.0015), relative to ADA (w = 1) (Figure 2 and Table S1). The envelope with both mutations had fitness 1.5381 (0.0258), whereas the expected fitness of the double mutant with independent effects of the mutations is 6.6671 × 0.3998 = 2.6655, giving an epistatic deviation of ε = 1.5381 − 2.6655 = −1.1271 (0.1252). On CXCR4 cells, mutation 4 had fitness 0.2129 (0.0082) and mutation 5 had fitness 0.2071 (0.0267), relative to ADA-1. Therefore, the expected fitness of the double mutant in the absence of an interaction is 0.0441. However, the double mutant had fitness 0.6439 (0.0138), giving ε = 0.5998 (0.0156).
Of the 31 envelopes engineered with more than two mutations, higher-order net interactions could be tested for only 23 with each cell-type assay because some lower-order mutants were not constructed. Of these 23 higher-order net interactions that could be tested, 14 (61%) were statistically significant on CCR5 cells and 8 (35%) were significant on CXCR4 cells (Figure 2 and Table S1). Significant higher-order net interactions on both CCR5 and CXCR4 cells were both positive and negative (Figure 3b and Table S1). For CCR5 cells, the median net epistatic deviation was 0.0604 (range −0.2856–1.7724), and for CXCR4 cells, the median net epistatic deviation was −0.2750 (−3.8550–3.0768). The highest-order (involving the most residues) significant net interaction occurred on CCR5 cells and involved five mutations (3, 4, 5, 6, and 7) (Figure 2 and Table S1). Therefore, higher-order net interactions were common and could be complex. In addition, higher-order net epistatic deviation on CXCR4 cells had a substantially broader range than on CCR5 cells.
The form of epistasis:
The form of fitness epistasis may be classified in various ways according to the signs of the fitness effects of the interacting mutations and the epistatic deviation (Phillips et al. 2000). Essentially, if the sign of the fitness effects is opposite to that of the epistatic deviation, epistasis is antagonistic, and if the signs are the same, epistasis is synergistic. Antagonistic epistasis may be further classified as either compensatory or decompensatory. If the fitness effects of individual mutations are negative and epistasis is positive, then the fitness effect of the combined mutations is less negative than expected from the independent effects of the mutations, and the interaction is compensatory. If the reverse, that is, the fitness effects of the individual mutations are positive and epistasis is negative, then the fitness effect of the combined mutations is less positive than expected from the independent effects of the mutations, and the interaction is decompensatory. A simple classification that accommodates interacting mutations having fitness effects with different signs and that accommodates higher-order interactions was used: if the fitness effects of interacting mutations differ in sign, then, if epistasis is positive, the interaction is compensatory, and if epistasis is negative, the interaction is decompensatory.
On CCR5 cells, fitness was calculated relative to the CCR5-adapted ADA wild type, and the 27 significant cases of overall epistasis were mostly compensatory (59%), although there were also some cases of decompensatory (11%) and negative synergistic (30%) epistasis. On CXCR4 cells, fitness was calculated relative to the CXCR4-adapted mutant ADA-1, and the 27 significant cases of overall epistasis were exclusively compensatory. This reflects the consistently negative fitness effects of interacting mutations and the consistently positive overall epistasis of interactions on CXCR4 cells.
The magnitude of epistasis:
The magnitude of epistasis was measured as the number of orders-of-magnitude change in fitness due to overall epistasis. On CCR5 cells, the magnitude for significant overall epistasis ranged from −2.65 to 2.04 (median = 0.46), and on CXCR4 cells, the magnitude ranged from 0.86 to 9.04 (median = 3.15) (Figure 4 and Table S1). These values match the signs and ranges of significant overall epistatic deviations (Figure 3a and Table S1). The large values, especially for effects measured on CXCR4 cells, reflect that although some single mutations are practically lethal, in combinations they produce large increases in fitness (Figure 2 and Table S1). For example, the CXCR4-adapted mutant, ADA-1, which carries all seven mutations, exhibited the highest magnitude on CXCR4 cells (9.04), but four of the mutations had only negligible fitness when measured singly. Therefore, overall epistasis on CXCR4 cells was consistently positive, increasing fitness by a median value of over 3 orders of magnitude, even though the net effects of higher-order interactions were sometimes negative (Figure 3b and Table S1).
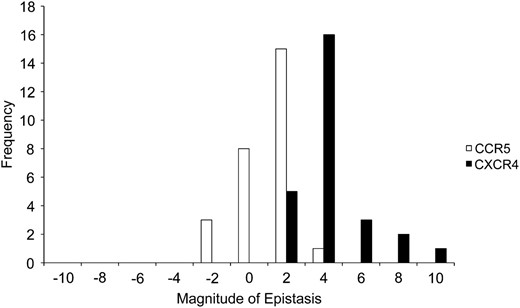
The frequency distribution of the magnitude of significant overall epistasis, E, on cells expressing CCR5 or CXCR4. Values on the magnitude axis are upper bounds of the intervals and are in units of orders of magnitude.
The evolutionary consequences of epistasis:
The observed common, complex, and strong fitness epistasis is expected to have a significant impact on the dynamics of adaptation. A useful way to identify this impact is to construct the minimum-length mutational trajectories between two alleles. A minimum-length mutational trajectory is one that involves only single-mutation steps and no reversals and is therefore the most direct evolutionary path between alleles. Sign epistasis, in which the sign of a mutation's fitness effect depends on its genetic background, will make a minimum-length mutational trajectory selectively inaccessible, thereby constraining adaptation (Weinreich et al. 2005; Weinreich et al. 2006; DePristo et al. 2007). Figure 5 shows the observable minimum-length mutational trajectories on CXCR4 cells, given the constructed mutant envelopes, from the CCR5-adapted wild-type ADA to the envelope with the highest fitness on CXCR4 cells, mutant 13457 (Figure 2 and Table S1). Because the five mutational differences between ADA and mutant 13457 may in principle occur in any order, these mutations produce 5! = 120 minimum-length trajectories (permutations) between these alleles. However, because not all mutant envelopes were constructed, only 24 complete trajectories are observable. Single-mutation steps in these trajectories were considered to increase fitness relative to the preceding mutant regardless of whether the increase was statistically significant. This makes the identification of mutations that do not increase fitness conservative. Of the 51 single-mutation steps in the observable trajectories from ADA to mutant 13457, 19 do not increase fitness. Indeed, none of the 24 observable minimum-length mutational trajectories from ADA to mutant 13457 are selectively accessible because each contains at least one single-mutation step that does not increase fitness. Other trajectories from ADA to mutant 13457, involving mutants that were not constructed, may be selectively accessible. These trajectories would necessarily involve a four-mutation envelope containing mutation 1 (Figure 5). The mutants are possibly 1345, 1347, 1357, and 1457, some of which may be selectively accessible from the accessible mutants 135, 157, and 457, or other possibly accessible triple mutants that were not constructed. An example would be the trajectory 135 → 1345 → 13457. However, any selectively accessible trajectories between ADA and mutant 13457 that may exist could not be traced because not all of the necessary mutants were constructed.
Minimum-length mutational trajectories on CXCR4 cells. The shortest observable mutational trajectories linking the CCR5-adapted wild-type ADA allele (wt), and the CXCR4-adapted ADA-1 allele (1234567), to the allele with the highest fitness when infecting cells expressing CXCR4 (13457). Mutations are numbered as in Figure 1. Only those mutation combinations that were engineered are shown. Solid arrows indicate single mutations that increase fitness. Shaded arrows indicate single mutations that do not increase fitness.
Sign epistasis constrains adaptation for the simple reason that in the absence of sign epistasis the mutations that characterize the allele with highest fitness must be advantageous singly and in any combination. In this case, every minimum-length mutational trajectory to the fittest allele is selectively accessible. Even with magnitude epistasis, in which only the magnitude of the fitness effect (not the sign) depends on the genetic background (the other mutations), every minimum-length trajectory will be selectively accessible (Weinreich et al. 2005). A change in the sign of a mutation's fitness effect dependent on the other mutations with which it is found is equivalent to one or more nonbeneficial mutational steps in a trajectory because the mutation must be beneficial in the final allele. Sign epistasis is evident in Figure 5. For example, the fitness effect of adding mutation 4 to the wild-type ADA background (wt → 4) is positive, increasing relative fitness from 0.0109 to 0.2129 (Figure 2 and Table S1). However, the fitness effect of adding mutation 4 to a background containing mutation 7 (7 → 47) is negative, decreasing fitness from 0.1283 to 0.0459. Therefore, although not all 120 possible minimum-length trajectories between ADA and mutant 13457 could be observed, because not all of the necessary envelopes were constructed, the fact that none of the observable trajectories were selectively accessible indicates severe constraints on adaptation.
Since the variant that evolved in response to selection by CXCR4, ADA-1, contains all seven mutations, in contrast to the observed envelope with the highest fitness on CXCR4 cells, mutant 13457, which contains only five mutations, it is possible that ADA-1 sits on a local fitness peak, implying a fitness landscape with multiple peaks. However, this is not the case since there is a selectively accessible minimum-length mutational trajectory from ADA-1 to mutant 13457 (Figure 5). The evolution of ADA-1 from ADA, together with the numerous selectively inaccessible minimum-length trajectories from ADA to mutant 13457, suggests that ADA-1 lies on an indirect, but selectively accessible, mutational trajectory from ADA to mutant 13457. Such a trajectory would involve reversals of mutations 2 and 6. This trajectory could not be traced with the constructed envelopes. Other envelopes that were not constructed may have even higher fitnesses than mutant 13457. However, the evolution of ADA-1 suggests strong constraints on the evolution of any mutant with higher fitness.
DISCUSSION
Although the statistical test for fitness epistasis was conservative, 56% of interactions tested for overall epistasis on each cell type were statistically significant. Interactions involved not only pairs of amino acids, but also higher-order epistasis above pairwise effects. Previous studies have reported direct evidence of fitness epistasis for pairs of residues, either intergenically (e.g., Sanjuan et al. 2004; Poon and Chao 2005; Sanjuan et al. 2005; Poon and Chao 2006) or intragenically (e.g., Bonhoeffer et al. 2004; Lunzer et al. 2005; Pepin and Wichman 2007). The present study demonstrates directly and unequivocally net higher-order fitness epistasis among amino acids within a protein region, that is, epistasis among three or more residues in addition to any lower-order interactions occurring among the same residues. The highest-level significant net interaction occurred among five amino acids. We tested whether data from a recent study showing sign epistasis in the evolution of antibiotic resistance in Escherichia coli β-lactamase (Weinreich et al. 2006) also provide evidence of higher-order epistasis. Using our approach, we found that out of 16 mutants with more than two mutations, out of a possible five, 12 exhibit statistically significant higher-order epistasis. The highest-level interactions occurred with quadruple mutants. Therefore, higher-order epistasis may be common among proteins.
Interactions occurred across the 134-amino acid C2–V3 protein region, although five of the seven mutations were within the 35-amino acid V3 region. A high density of interactions has also been reported in a survey of published data on suppressor mutations in viruses, prokaryotes and eukaryotes (Poon et al. 2005; Davis et al. 2009). In some cases suppressor mutations recover fitness in individuals with a deleterious mutation by suppressing the phenotypic effect of the deleterious mutation and are therefore compensatory. For viruses, Poon et al. (2005) estimate approximately nine compensatory mutations for every deleterious mutation and that about 64% of interactions occur intragenically rather than intergenically. In an experimental analysis of the DNA bacteriophage ϕX174, Poon and Chao (2005) report approximately nine compensatory mutations for each deleterious mutation, that about half of compensatory mutations are intragenic, and that the average intragenic compensatory mutation clusters significantly within 20% of the protein's length from the deleterious mutation. Shapiro et al. (2006) inferred the coevolutionary history of amino acid replacements in 177 RNA virus genes to detect positive fitness epistasis. They found that interactions most often occur within a distance of 15 amino acids. The concentration of epistasis within short stretches of amino acids suggests that interactions occur directly between residues or indirectly through local conformational effects on protein structure. This conclusion is supported by a recent study of the structural mechanism of epistasis within a protein (Ortlund et al. 2007).
Overall epistasis was both positive and negative on CCR5 cells, but was exclusively positive on CXCR4 cells. The positive epistasis on CXCR4 cells may be explained by the fact that the single mutations analyzed tend to increase fitness relative to ADA, but are deleterious relative to ADA-1 (Figure 2 and Table S1). Fitness on CXCR4 cells was measured relative to that of ADA-1 and deleterious mutations tend to generate positive epistasis in microorganisms (Burch and Chao 2004; de Visser and Elena 2007; Jasnos and Korona 2007; Kouyos et al. 2007; Maisnier-Patin et al. 2005; Sanjuan and Elena 2006; Sanjuan et al. 2004). An explanation for this may be that mutations within the same gene or protein interact antagonistically on fitness because they are affecting the same functional unit or fitness component. Antagonistic interactions, in which the combined effect of mutations is less than expected from their independent, multiplicative effects, generate positive epistasis with deleterious mutations and negative epistasis with beneficial mutations (Phillips et al. 2000). A similar argument is made by Sanjuan and Elena (2006) for genome-wide interactions. They argue that for simple, compact genomes, such as those of RNA viruses, antagonistic epistasis is expected to predominate because of the high probability that different mutations will affect the same functional module. The association of positive epistasis with deleterious mutations is also predicted by the biophysics of protein structure (DePristo et al. 2005).
The magnitude of overall fitness epistasis could be very high, causing changes in fitness of over 2 orders of magnitude on CCR5 cells and over 9 orders of magnitude on CXCR4 cells. These results contrast with those from early quantitative genetics experiments on the viability effects of epistasis in Drosophila, which showed only weak to moderate effects (Spassky et al. 1965; Temin et al. 1969). The difference is very likely due to the early experiments dealing with many unknown mutations of small effect and ignoring lethal mutations, whereas in this study we analyzed few mutations of large effect, some of which were effectively lethal. Our results argue for epistasis being a dominant force in adaptive dynamics.
Fitness epistasis may restrict the minimum-length mutational trajectories taken during adaptive evolution. Such constraints will arise when the sign of a mutation's fitness effect depends on its genetic background (Weinreich et al. 2005, 2006). In the present study, the main consequence of such sign epistasis is that all 24 observable minimum-length mutational trajectories from the CCR5-adapted wild-type ADA strain to the engineered envelope with the highest fitness on CXCR4 cells are selectively inaccessible. Although these 24 observable trajectories are only a small portion of the 120 minimum-length trajectories possible involving five mutations, their selective inaccessibility indicates severe constraints on adaptation. Any selectively accessible minimum-length trajectory must have included mutation 1 from C2 in a four-mutation intermediate before reaching the final five-mutation envelope with the highest fitness, mutant 13457. However, the fact that not all envelopes were constructed prevents us from tracing any of the potentially selectively accessible minimum-length trajectories between ADA and mutant 13457. This effect of sign epistasis was first shown for the evolution of antibiotic resistance in E. coli β-lactamase, in which only a minority of all minimum-length mutational trajectories are selectively accessible (Weinreich et al. 2006).
Alternatively, sign epistasis may have constrained the evolutionary trajectory from ADA to mutant 13457 to be indirect, involving more than the minimum possible number of mutations. The evolution of ADA-1, the isolate with all seven mutations, from ADA in response to selection by CXCR4 (Pastore et al. 2004) may have occurred as an intermediate in an indirect chain of selectable mutational steps to mutant 13457. Such a selectively accessible, but indirect, trajectory would require a minimum of four additional mutational steps over a direct trajectory: two additional mutations to reach ADA-1 and two mutational reversions. To reach mutants with even higher fitness, which may exist but were not constructed, would also require an indirect trajectory through ADA-1. Mutational trajectories involving reversions were shown to potentially comprise a large proportion of selectively accessible trajectories in the evolution of E. coli β-lactamase antibiotic resistance (DePristo et al. 2007).
Nonbeneficial single-mutation steps are unlikely to be circumvented by the spread to fixation of a double mutant, the probability of which is proportional to the product of the square of the mutation rate and the effective population size (Gillespie 1984; Weinreich and Chao 2005), because of the small within-patient effective population size of HIV-1. Although the within-patient census population size of HIV-1 is large, on the order of 107 infected cells (Chun et al. 1997), estimates of the effective population size range from 102 to 105 (e.g., Leigh Brown 1997; Rouzine and Coffin 1999; Seo et al. 2002; Achaz et al. 2004; Shriner et al. 2004; Kouyos et al. 2006). A high rate of recombination, as observed for HIV-1 (Jung et al. 2002; Levy et al. 2004), is also unlikely to help because high recombination is expected to reduce the rate of adaptation on rugged fitness landscapes (Weinreich and Chao 2005; de Visser et al. 2009). However, because V3 is the primary target of neutralizing antibodies (Zolla-Pazner 2004), fluctuating selection by antibodies (Richman et al. 2003; Wei et al. 2003) could change the fitness landscape so that some trajectories become selectively accessible.
Nevertheless, severe constraints on adaptation to CXCR4 are consistent with the apparent difficulty of evolving a CXCR4-utilizing variant in culture (Pastore et al. 2004) and may explain why switching from CCR5 use to CXCR4 use tends to occur only late in infection (Philpott 2003). In selecting for CXCR4 use in CCR5-adapted virus strains, Pastore et al. (2004) report that only one out of four strains evolved exclusive CXCR4 use, the remaining three strains evolving dual coreceptor use only. In addition, evolutionary intermediates in these experiments are more sensitive to cell entry inhibitors (CCR5 or CXCR4 ligands) than either the CCR5-adapted parental viruses or the endpoint viruses (Pastore et al. 2007), suggesting a decrease in fitness along the evolutionary path between phenotypes. This could mean that in an environment with both chemokine coreceptors available, each coreceptor-usage phenotype represents a fitness peak. Furthermore, adaptation to CXCR4 involved on average two to four mutations, mainly within V3, which occurred in parallel for isolates from the same viral strain (Pastore et al. 2004), implying strong constraints on evolutionary pathways. Finally, although mutations in V3 are necessary for coreceptor switching, they generally reduce viral infectivity and mutations in V1/V2 or C2 may be necessary to compensate for this loss of fitness (Pastore et al. 2006).
In this study, mutation 1 in C2 increased fitness by over sixfold compared to the CCR5-adapted wild-type ADA on CCR5 cells. A similar effect of this mutation is reported by Pastore et al. (2006). This may be explained by mutation 1 altering a putative N-linked glycosylation motif. These motifs are sometimes advantageous under selection by neutralizing antibodies (Wei et al. 2003) and are known to affect coreceptor usage (e.g., Ogert et al. 2001; Pollakis et al. 2001). Mutation 1 alone does not appreciably increase fitness on CXCR4 cells in comparison to the wild type, but does increase fitness substantially in combination with other mutations that are moderately beneficial relative to the wild type (e.g., mutants 14 and 125; Figure 2). Measuring fitness relative to the CXCR4-adapted isolate ADA-1, these combinations exhibit significant positive epistasis and are compensatory. This supports our observation that the evolutionary trajectory from the wild type to the variant with the highest fitness on CXCR4 cells must involve mutation 1 and supports the observation by Pastore et al. (2006) that mutation 1 compensates for the loss of infectivity caused by V3 mutations.
Mutation 3 in V3 also eliminates a putative N-linked glycosylation motif, and there is a significant compensatory interaction between mutations 1 and 3 on CXCR4 cells. V3 mutations 4 and 7 have been implicated in affecting coreceptor usage in functional studies (de Jong et al. 1992; Fouchier et al. 1992; Hung et al. 1999; Pastore et al. 2006) and structural modeling studies (Cardozo et al. 2007; Gorry et al. 2007; Rosen et al. 2006). These mutations had significant pairwise and higher-order interactions with mutations 1 and 3, as well as other mutations. The V3 mutations studied here have been reported for subtype B viruses infecting patients (Kuiken et al. 2009), with mutations 4 and 7 more common in CXCR4-using viruses than in CCR5-using viruses (da Silva 2006). Mutation 4 has also been reported to be positively selected during the switch from CCR5 use to CXCR4 use in a patient (Coetzer et al. 2008). In addition, linkage disequilibrium between amino acids at the sites of mutations 4 and 7 and other sites has been reported frequently for HIV-1 subtype B sequences sampled from patients (e.g., Bickel et al. 1996; Korber et al. 1993; Poon et al. 2007). This disequilibrium may be caused by the fitness epistasis reported here.
We have shown that fitness epistasis is common among the amino acids of a short protein region and that it may be complex, involving not only pairwise interactions but also higher-order interactions. The interactions are mostly compensatory, often very strong and appear to severely constrain the adaptation of the HIV-1 C2–V3 region to the chemokine coreceptor CXCR4. Sign epistasis may help explain the difficulty in evolving CXCR4 use in culture and the delayed evolution of this phenotype in natural infection. These results support the view that an understanding of protein evolution requires knowledge of the common, complex, and strong fitness interactions among amino acids.
Footnotes
Supporting information is available online at http://www.genetics.org/cgi/content/full/genetics.109.112458/DC1.
Footnotes
Communicating editor: J. Lawrence
Acknowledgements
This manuscript was improved by the suggestions of three reviewers. We acknowledge the support of the School of Molecular and Biomedical Science, and its Discipline of Genetics, of the University of Adelaide. This work was supported by grants R01 AI052778 from the National Institute of Allergy and Infectious Diseases (NIAID). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIAID or the National Institutes of Health.
References
Coffin, J. M.,
Davis, B. H., A. F. Y. Poon and M. C. Whitlock,
de Visser, J. A. G. M., and S. F. Elena,
Felsenstein, J.,
Goodman, L. A.,
Kimura, M.,
Korber, B. T., R. M. Farber, D. H. Wolpert and A. S. Lapedes,
Kuiken, C., T. Leitner, B. Foley, B. Hahn, P. Marx et al. (Editors),
Leigh Brown, A. J.,
Levy, D. N., G. M. Aldrovandi, O. Kutsch and G. M. Shaw,
Lewontin, R. C., and K.-i. Kojima,
Ogert, R. A., M. K. Lee, W. Ross, A. Buckler-White, M. A. Martin et al.,
Phillips, P. C., S. P. Otto and M. C. Whitlock,
Poon, A. F., and L. Chao,
Richman, D. D., T. Wrin, S. J. Little and C. J. Petropoulos,
Rosen, O., M. Sharon, S. R. Quadt-Akabayov and J. Anglister,
Rouzine, I. M., and J. M. Coffin,
Sanjuan, R., and S. F. Elena,
Sanjuan, R., A. Moya and S. F. Elena,
Sokal, R. R., and F. J. Rohlf,
Weinreich, D. M., R. A. Watson and L. Chao,
Whitlock, M. C., P. C. Phillips, F. B. Moore and S. J. Tonsor,
Wright, S.,

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}