





Abstract
Genetic maps provide a means to estimate the probability of the co-inheritance of linked loci as they are transmitted across generations in both experimental and natural populations. However, in the age of whole-genome sequences, physical distances measured in base pairs of DNA provide the standard coordinates for navigating the myriad features of genomes. Although genetic and physical maps are colinear, there are well-characterized and sometimes dramatic heterogeneities in the average frequency of meiotic recombination events that occur along the physical extent of chromosomes. There also are documented differences in the recombination landscape between the two sexes. We have revisited high-resolution genetic map data from a large heterogeneous mouse population and have constructed a revised genetic map of the mouse genome, incorporating 10,195 single nucleotide polymorphisms using a set of 47 families comprising 3546 meioses. The revised map provides a different picture of recombination in the mouse from that reported previously. We have further integrated the genetic and physical maps of the genome and incorporated SSLP markers from other genetic maps into this new framework. We demonstrate that utilization of the revised genetic map improves QTL mapping, partially due to the resolution of previously undetected errors in marker ordering along the chromosome.
GENETIC maps exist for hundreds of different species, and genetic map construction continues to play an important role in the characterization of genomes (Tanksley et al. 1992; Kong et al. 2002; Chowdhary and Raudsepp 2006; Stapley et al. 2008). A genetic map defines the linear order and relative distances among a set of marker loci in units that correspond to the frequency of meiotic recombination between the loci. Until recently mouse genetic maps based on simple sequence length polymorphism (SSLP) markers (Lyon 1976) have been sufficient for most experimental purposes since, unlike the hundreds of thousands of markers required in human genetic association studies, a relatively small number of markers is needed to map crosses between inbred mouse strains. However, recent developments in whole-genome high-resolution mapping in the mouse (Churchill et al. 2004; Valdar et al. 2006) and interest in examining recombination rates at an ultra-fine scale (Myers et al. 2005) have reawakened the need to develop a high-resolution genetic map in the mouse.
The current standard genetic map of the mouse has been compiled from a substantial body of historical data and maintained by the Mouse Genome Informatics (MGI) project at The Jackson Laboratory (Bult et al. 2008). We will refer to it as the MGI map. The primary sources of data used to construct the MGI map were two mapping panels, described here. However, the current map is based on a consensus developed by the 2000 Chromosome Committee using all available published data. The map has continued to be maintained by MGI with the addition of new genetic markers and data but, because the map is based on consensus, published errors may have been perpetuated.
The Jackson Laboratory developed a genetic map based on two sets of 94 progeny obtained from reciprocal backcrosses (BSB and BSS) between the inbred strains C57BL/6J and SPRET/EiJ (Rowe et al. 1994). These strains represent two different species of mouse (Mus musculus and Mus spretus). The map provides a wealth of genetic information, but problems with male fertility restrict breeding options and thus the map is female specific. The problems with male fertility may have resulted in some multi-locus distortion in the mapping panel (Montagutelli et al. 1996). Currently, 1372 and 4913 markers have been typed on the BSB and BSS backcross panels, respectively (Broman et al. 2002). Researchers at The Whitehead Institute and the Massachusetts Institute of Technology (MIT) developed a map of 4006 SSLP markers using an intercross population of 46 mice derived from strains OB (C57BL/6J-Lepob/ob) and CAST (CAST/EiJ) (Dietrich et al. 1994). Both parental strains OB and CAST are derived from M. musculus, but CAST is from a distinct subspecies, M. m. castaneus. The intercross mating strategy produces observable recombination from both male and female parents, but the two cannot be distinguished. Thus the map is sex averaged and based on 92 meioses. The Whitehead/MIT map was expanded to include 7377 SSLP markers (Dietrich et al. 1996). These are denoted as, e.g., D7Mit54, where “7” indicates the chromosome to which the marker is mapped and “54” is an arbitrary index. They are commonly referred to as “Mit” markers.
Map resolution is limited by the number of observable recombination events in each of these panels. With 94 meioses (in each backcross) and an average of 14 recombination events/haploid genome transmitted, limiting resolution is on the order of 1 cM. A much larger panel would be needed to achieve subcentimorgan resolution and to accurately position high-density sets of SNP markers.
Here we propose a new standard genetic map of the laboratory mouse based on data from a large heterogeneous stock (HS) mouse population descended from eight inbred strains (DBA/2J, C3H/HeJ, AKR/J, A/J, BALB/cJ, CBA/J, C57BL/6J, and LP/J) representing a diverse sample of the classical inbred strains (Petkov et al. 2004). Shifman et al. (2006) calculated genetic maps based on 11,247 informative SNP markers in 2293 HS individuals. The marker set is dense with 99% of the SNP intervals <500 kb, 81.2% <250 kb, and an estimated allele inheritance-based accuracy of 99.98%. Map positions were calculated separately for male and female meioses using CRIMAP software (Green et al. 1990), and the total length of the sex-averaged map is 1630 cM, as defined by the most distal SNP markers in their panel. The MGI map is 1783 cM on the basis of the most distal available marker position for each chromosome. However, on the basis of the most distal shared markers, the original Shifman map at 1612 cM is substantially longer than the MGI map at 1445 cM. It was not immediately clear if this discrepancy was due to the nature of recombination in the HS population or to their method of map estimation.
There are at least two methodological problems with the HS map reported in Shifman et al. (2006). First, the map was constructed using a sliding window of 5–15 SNPs to handle eight multi-generation families within the CRIMAP software. Ideally, one considers all markers on a chromosome simultaneously in constructing a genetic map, and we found that this could be accomplished by splitting the complex pedigrees into sibships. Although splitting the pedigree results in slightly less efficient estimates of intermarker distances, this approach should incur no bias. Maps based on the full set of markers but with the complex pedigrees split into sibships are thus arguably better than maps based on the full pedigrees but with a sliding window of 5–15 markers. Second, analysis of families with incomplete parental genotypes may have contributed to an inflated map size. Sixteen of the 72 families lack parental genotypes or have genotypes for just one parent (15 of the 72), and many of them are small (26 have six or fewer siblings). Sibships with no parental genotype data of their own can give no information about sex-specific recombination rates. In conjunction with other sibships for which parental genotypes are available, they can provide some information, but the CRIMAP software (last modified in 1990) makes some approximations that result in a large bias even in the sex-averaged genetic maps for small sibships lacking parental genotype data.
For these reasons, we recomputed the mouse genetic map on the basis of the original data reported and discuss the differences between the original Shifman map and the revised Shifman map below. The revised Shifman map provides a markedly different picture of recombination in the mouse: the estimated sex-averaged chromosome lengths correspond more closely to those in the original MGI map; the sex difference in the overall recombination rate is greatly reduced; and numerous narrow regions of high recombination rate, apparent in the original Shifman map, have disappeared.
We propose the revised Shifman map as a new standard genetic map for the mouse. The new genetic map represents a substantial improvement over the existing MGI map due to the large number of meioses and to the genetic diversity of strains in the HS population. We have generated male, female, and sex-averaged genetic maps with physical positions and updated locus identifiers. We have established the correspondence between physical and genetic positions of 7080 Mit markers and corrected inconsistencies in the MGI map. We provide a web-based tool for the interpolation of new marker loci into the genetic map and for converting genetic map positions to NCBI mouse build 37 coordinates. Finally, we examine the effect of changing to this revised genetic map on QTL mapping in five previously published data sets (Beamer et al. 1999, 2001; Ishimori et al. 2004, 2008; Wergedal et al. 2006).
MATERIALS AND METHODS
Data cleaning:
We reviewed the quality of the raw genotype data from Shifman et al. (2006) and identified a set of 11 individuals whose genotypes were discrepant from their parents or offspring; these individuals were omitted from our analysis. We also identified 26 individuals recorded to be female but whose genotype data indicated that they were male. The sex for these individuals was switched. We omitted genotypes with a quality score <0.4. We used Pedcheck (O'Connell and Weeks 1998) to identify genotypes inconsistent between parents and offspring and omitted the genotypes deemed to be in error. The large sibships and low apparent genotyping error rate greatly simplified this task. We used the chrompic option of CRIMAP to identify unlikely tight double-recombination events indicative of genotyping errors. We omitted the offending genotype for apparent double recombinants containing a single typed marker and separated by <10 cM in sex-specific distance. We omitted 176 genotypes due to Mendelian inconsistencies and another 538 genotypes that resulted in tight double-recombination events. In all, ∼3.2/100,000 genotypes were omitted. The final data set included 22,500,431 genotypes.
The eight complex pedigrees were split into sibships (with parents and grandparents, when available). The largest sibships could not be handled in CRIMAP, and so sibships with >20 siblings were split in half. One sibship with 48 siblings was split into three sibships with 16 siblings each. Sibships with genotype data on just one parent and with eight or fewer siblings were omitted. Sibships with no parental genotype data were also omitted. While the largest of these might have been used to estimate the sex-averaged maps, we felt it best to use the same set of data for both the sex-specific and the sex-averaged maps. In all, 25 of the 72 nuclear families were omitted. The final data comprised 3546 meioses.
The revised maps were estimated with CRIMAP (Green et al. 1990), using the Kosambi map function. Recombination rates (centimorgans/megabases) were estimated using a sliding 5-Mb window.
Locating markers on the physical genome:
Physical positions of 10,202 SNPs used in the Shifman study were mapped to the mouse build 37 genome by BLAT (http://genome.ucsc.edu/cgi-bin/hgBlat) using the 100-bp flanking sequence on both sides of each SNP. Of 10,202 total SNPs, 10,196 were mapped uniquely to build 37, and 6 SNPs were mapped to more than one location; the multiply mapped SNPs were omitted from the revised map (see supporting information, Table S1).
Nomenclature for Mit markers varies across the online databases such as MGI, University of California at Santa Cruz (UCSC), and NCBI. We have identified Mit markers based on the unique MGI primer pair ID (PPID). We obtained 7080 Mit markers corresponding to 7052 PPIDs in the MGI database (ftp://ftp.informatics.jax.org/pub/reports/PRB_PrimerSeq.rpt). A single PPID may be associated with more than one Mit marker symbol, and these synonymous symbols are grouped together in our database. All marker symbols associated with the same PPID were consolidated and tabulated. There were some marker names associated with more than one primer pair, and these are flagged in our database.
Of the 7052 PPIDs, 45 did not have a primer sequence listed. The remaining 7007 unique primer pairs were mapped to the NCBI Build 37 mouse genome using the in silico PCR (isPCR) software (Hinrichs et al. 2006) with a minimum primer length of 18 bp and a minimum match size of 15 bp. The maximum product length allowed is 4000 bp, but primer pairs with amplicon lengths >1000 were not considered an alignment. In this way, we mapped 6049 primer pairs to unique positions on the NCBI mouse build 37 genome, and 144 primer pairs mapped to more than one genomic location (see Table S2). For the remaining 814 primer pairs, we again used the online version of isPCR (http://genome.ucsc.edu/cgi-bin/hgPcr?command=start), but reduced the minimum primer length to 15 bp. Using this method, we mapped 31 primer pairs to one location and 3 primer pairs to more than one location. The final remaining 780 primer pairs were deemed “unmappable” (see Table S3). They are flagged in our database, and we have retained historical genetic map positions. For each marker in the database, it is noted which of the above methods was used in mapping the primer pair. In total, we mapped 6080 primer pairs corresponding to 6388 Mit marker names.
Genetic map positions are relative. To anchor the genetic map, we assigned 0 bp in the physical map to 0 cM in the genetic map. The genetic position of the most proximal SNP was calculated on the basis of its megabase position using the average recombination rate (centimorgans/megabase) for that chromosome. We note that physical coordinates up to 3 Mb in the mouse reference sequence are placeholders for unknown or unsequenced DNA near the centromere of each chromosome.
Placing the Mit markers on the new genetic map:
We assigned male, female, and sex-averaged centimorgan positions to the Mit markers using linear interpolation based on their physical positions relative to the Shifman SNPs. Given the high density of SNPs, more sophisticated methods of interpolation should not provide any improvement. A total of 16 Mit markers at the distal ends of chromosomes 5, 8, 15, 16, 18, and X did not have flanking SNPs, and we assigned these markers to genetic map positions on the basis of their physical position by extrapolation using the chromosome-wide average recombination rate.
If available, the previous genetic map position is listed in the Mit marker database. While it is not possible to interpolate the unmapped Mit markers into the new genetic map, it may be necessary to assign updated genetic map positions for the purpose of reanalyzing historical data. We manually assigned new genetic map positions to unmapped markers on the basis of their MGI centimorgan position. This was done by determining the average base-pair positions of mapped markers with the same MGI centimorgan position and then assigning the updated centimorgan position of the mapped marker(s) to the unmapped marker at hand. In updating the QTL archive with the revised Shifman map, we assigned non-Mit markers to genetic map positions in this manner. In some cases, we found that the genotyping data from a cross are consistent with this assignment and have retained the marker; in other cases, there appear to be discrepancies and the suspect marker has been removed. We ran quality control checks on the genotypes in the archival data, but we did not revise the analysis of QTL positions.
Mit markers that aligned to multiple chromosomal positions in build 37 that were <1 Mb apart were interpolated on the basis of the average physical position of the alignments. Markers with multiple alignments to chromosomal positions >1 Mb apart or on more than one chromosome were assigned centimorgan positions in the same manner as the unmapped markers and on the basis of their MGI position. In the database, any map position not determined by the interpolation of one definitive physical position to the Shifman SNPs is flagged as unreliable.
Data sets used QTL for reanalysis:
Genotypes and phenotype data were obtained from five F2 mouse mapping crosses: B6xCAST (Beamer et al. 1999), B6xC3H (Beamer et al. 2001; Koller et al. 2003), B6x129 (Ishimori et al. 2004), NZBxRF (Wergedal et al. 2006), and NZBxSM (Ishimori et al. 2008). The Animal Care and Use Committee approval and detailed genotyping and phenotyping methods can be found in the original articles. The primary phenotype of interest here is bone mineral density (BMD). For B6xC3H and NZBxRF, average cortical bone thickness at the mid-diaphysis (bone geometry) and ultimate load at failure (strength) were also examined. These traits share some but not all QTL with BMD in the original studies.
Primary QTL analysis:
All QTL analyses were done using the R/qtl software package (Broman et al. 2003) (version 1.09-43; http://www/rqtl.org/). Phenotypic outliers were removed prior to QTL analyses. Genotype data were also examined and obvious errors were corrected in a parallel manner for both analyses. Particular attention was paid to marker-order diagnostics using the recombination fraction plot function in R/qtl (plot.rf). If a marker-order issue was identified, the issue was corrected. In all cases, the phenotype trait data were transformed to correct for any skewing of the data using the van der Waerden normal score method (Lehmann and D'Abrera 1988). For each data set and for each map version, a single-locus mainscan for QTL was performed and LOD scores were calculated at 2-cM intervals across the genome using the “imp” or imputation method in R/qtl (Sen and Churchill 2001). Thresholds for significant and suggestive QTL were determined on the basis of 1000 permutations of the data (Churchill and Doerge 1994). A QTL was considered to be suggestive if the LOD score exceeded the 37% threshold and significant if it exceeded the 95% threshold. These thresholds were chosen as widely accepted cutoffs for suggestive and significant QTL (Lander and Kruglyak 1995). The goal of this analysis was to examine the effect of the new genetic map on single QTL, so no pairwise genome scans were run and no attempt to fit the multi-locus model was done.
The NZBxSM cross was the only cross to have data available from both male and female animals. For this cross, sex was considered as both an additive and an interactive covariate. Each model was examined for the effect of the new map on the QTL.
Assessment of the effects of map change on QTL mapping:
First we determined whether a QTL exceeded the suggestive threshold for either analysis. For QTL found in both analyses, LOD score profiles were compared for correspondence in peak positioning, shape, and significance level. We scored QTL as follows:
No change between new and old map.
A shift in peak location by >5 cM, but no change in shape or marker closest to the peak.
A change in marker identified as being closest to the peak but no change in peak shape or location.
A change in QTL peak shape, which may result in a changed peak height of >1 LOD point. Marker closest to the peak may have changed.
RESULTS
The revised Shifman map:
We have constructed revised genetic maps for the mouse genome on the basis of genotype data from a large HS population (Shifman et al. 2006) as described in materials and methods. The revised maps incorporate 10,195 SNPs and are based on a total of 3546 meioses. An important feature of an integrated genetic and physical map is that it provides a characterization of recombination rate across the genome, and the revised Shifman map provides a strikingly different characterization of recombination in the mouse compared with the original Shifman map.
Differences between the MGI, revised, and original Shifman maps were assessed by comparing chromosome lengths between the most proximal and distal markers shared in common among the maps (Table 1). Mit markers with extrapolated positions were excluded from these comparisons. The revised Shifman map is notably shorter than the original Shifman map: the sex-averaged autosomal map length is 11% shorter, the female map length is 16% shorter, and the male map length is 4% shorter in the revised map compared with the original Shifman map. In fact, the sex-averaged chromosome lengths in the revised map correspond more closely to the MGI map. The average absolute difference between the chromosome lengths in the original Shifman map and the MGI map is 9.4 cM, while the corresponding number comparing the revised Shifman map and the MGI map is 4.6 cM. Particularly notable are chromosomes 4, 5, 6, and 9, whose lengths are quite similar in the revised Shifman map and the MGI map but are much longer in the original Shifman map. It should be noted, however, that chromosome 16 is quite a bit longer in the MGI map.
Chromosome lengths and recombination rates based on positions of most proximal and most distal Mit markers, shared among the revised Shifman, original Shifman, and MGI maps
Most proximal Mit marker | Most distal Mit marker | Total map length (cM) | Chromosome average recombination rate | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Chromosome | Marker ID | Mb | Marker ID | Mb | Revised Shifmana | Original Shifmana | MGI (standard) | Revised Shifmana | Original Shifmana | MGI (standard) |
| 1 | D1Mit475 | 3.64 | D1Mit155 | 196.26 | 96.55 | 117.83 | 103.60 | 0.50 | 0.61 | 0.54 |
| 2 | D2Mit312 | 3.15 | D2Mit457 | 180.99 | 101.82 | 108.22 | 107.00 | 0.57 | 0.61 | 0.60 |
| 3 | D3Mit150 | 5.35 | D3Mit89 | 156.50 | 78.83 | 86.80 | 83.70 | 0.52 | 0.57 | 0.55 |
| 4 | D4Mit149 | 3.58 | D4Mit51 | 154.42 | 84.13 | 99.86 | 82.70 | 0.56 | 0.66 | 0.55 |
| 5 | D5Mit69 | 3.53 | D5Mit169 | 150.23 | 86.97 | 105.61 | 85.00 | 0.59 | 0.72 | 0.58 |
| 6 | D6Mit231 | 3.48 | D6Mit390 | 148.41 | 76.69 | 89.72 | 73.73 | 0.53 | 0.62 | 0.51 |
| 7 | D7Mit21 | 3.27 | D7Mit141 | 152.14 | 87.05 | 89.42 | 71.90 | 0.58 | 0.60 | 0.48 |
| 8 | D8Mit155 | 4.98 | D8Mit156 | 131.42 | 73.96 | 80.20 | 72.00 | 0.58 | 0.63 | 0.57 |
| 9 | D9Mit186 | 6.26 | D9Mit322 | 123.10 | 71.46 | 85.23 | 70.00 | 0.61 | 0.73 | 0.60 |
| 10 | D10Mit49 | 4.14 | D10Mit269 | 128.39 | 75.29 | 83.11 | 68.00 | 0.61 | 0.67 | 0.55 |
| 11 | D11Mit1 | 3.47 | D11Mit104 | 119.17 | 80.85 | 91.20 | 78.75 | 0.70 | 0.79 | 0.68 |
| 12 | D12Mit264 | 5.31 | D12Mit144 | 120.30 | 61.00 | 68.55 | 60.00 | 0.53 | 0.60 | 0.52 |
| 13 | D13Mit235 | 4.22 | D13Mit35 | 120.13 | 64.85 | 69.46 | 71.00 | 0.56 | 0.60 | 0.61 |
| 14 | D14Mit110 | 8.99 | D14Mit107 | 124.00 | 61.37 | 59.38 | 59.50 | 0.53 | 0.52 | 0.52 |
| 15 | D15Mit12 | 3.16 | D15Mit160 | 102.99 | 56.40 | 64.34 | 59.80 | 0.56 | 0.64 | 0.60 |
| 16 | D16Mit32 | 3.96 | D16Mit71 | 97.13 | 54.73 | 62.93 | 68.95 | 0.59 | 0.68 | 0.74 |
| 17 | D17Mit164 | 3.92 | D17Mit123 | 93.60 | 58.56 | 62.74 | 52.60 | 0.65 | 0.70 | 0.59 |
| 18 | D18Mit65 | 3.73 | D18Mit25 | 89.74 | 56.67 | 64.07 | 55.00 | 0.66 | 0.74 | 0.64 |
| 19 | D19Mit32 | 3.28 | D19Mit108 | 59.96 | 53.24 | 53.35 | 49.50 | 0.94 | 0.94 | 0.87 |
| X | DXMit101 | 5.68 | DXMit100 | 165.35 | 76.16 | 69.83 | 72.50 | 0.48 | 0.44 | 0.45 |
| Total | 1456.58 | 1611.84 | 1445.23 | |||||||
Most proximal Mit marker | Most distal Mit marker | Total map length (cM) | Chromosome average recombination rate | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Chromosome | Marker ID | Mb | Marker ID | Mb | Revised Shifmana | Original Shifmana | MGI (standard) | Revised Shifmana | Original Shifmana | MGI (standard) |
| 1 | D1Mit475 | 3.64 | D1Mit155 | 196.26 | 96.55 | 117.83 | 103.60 | 0.50 | 0.61 | 0.54 |
| 2 | D2Mit312 | 3.15 | D2Mit457 | 180.99 | 101.82 | 108.22 | 107.00 | 0.57 | 0.61 | 0.60 |
| 3 | D3Mit150 | 5.35 | D3Mit89 | 156.50 | 78.83 | 86.80 | 83.70 | 0.52 | 0.57 | 0.55 |
| 4 | D4Mit149 | 3.58 | D4Mit51 | 154.42 | 84.13 | 99.86 | 82.70 | 0.56 | 0.66 | 0.55 |
| 5 | D5Mit69 | 3.53 | D5Mit169 | 150.23 | 86.97 | 105.61 | 85.00 | 0.59 | 0.72 | 0.58 |
| 6 | D6Mit231 | 3.48 | D6Mit390 | 148.41 | 76.69 | 89.72 | 73.73 | 0.53 | 0.62 | 0.51 |
| 7 | D7Mit21 | 3.27 | D7Mit141 | 152.14 | 87.05 | 89.42 | 71.90 | 0.58 | 0.60 | 0.48 |
| 8 | D8Mit155 | 4.98 | D8Mit156 | 131.42 | 73.96 | 80.20 | 72.00 | 0.58 | 0.63 | 0.57 |
| 9 | D9Mit186 | 6.26 | D9Mit322 | 123.10 | 71.46 | 85.23 | 70.00 | 0.61 | 0.73 | 0.60 |
| 10 | D10Mit49 | 4.14 | D10Mit269 | 128.39 | 75.29 | 83.11 | 68.00 | 0.61 | 0.67 | 0.55 |
| 11 | D11Mit1 | 3.47 | D11Mit104 | 119.17 | 80.85 | 91.20 | 78.75 | 0.70 | 0.79 | 0.68 |
| 12 | D12Mit264 | 5.31 | D12Mit144 | 120.30 | 61.00 | 68.55 | 60.00 | 0.53 | 0.60 | 0.52 |
| 13 | D13Mit235 | 4.22 | D13Mit35 | 120.13 | 64.85 | 69.46 | 71.00 | 0.56 | 0.60 | 0.61 |
| 14 | D14Mit110 | 8.99 | D14Mit107 | 124.00 | 61.37 | 59.38 | 59.50 | 0.53 | 0.52 | 0.52 |
| 15 | D15Mit12 | 3.16 | D15Mit160 | 102.99 | 56.40 | 64.34 | 59.80 | 0.56 | 0.64 | 0.60 |
| 16 | D16Mit32 | 3.96 | D16Mit71 | 97.13 | 54.73 | 62.93 | 68.95 | 0.59 | 0.68 | 0.74 |
| 17 | D17Mit164 | 3.92 | D17Mit123 | 93.60 | 58.56 | 62.74 | 52.60 | 0.65 | 0.70 | 0.59 |
| 18 | D18Mit65 | 3.73 | D18Mit25 | 89.74 | 56.67 | 64.07 | 55.00 | 0.66 | 0.74 | 0.64 |
| 19 | D19Mit32 | 3.28 | D19Mit108 | 59.96 | 53.24 | 53.35 | 49.50 | 0.94 | 0.94 | 0.87 |
| X | DXMit101 | 5.68 | DXMit100 | 165.35 | 76.16 | 69.83 | 72.50 | 0.48 | 0.44 | 0.45 |
| Total | 1456.58 | 1611.84 | 1445.23 | |||||||
Position on sex-averaged map except for chromosome X; these centimorgan positions are based on the female map.
Chromosome lengths and recombination rates based on positions of most proximal and most distal Mit markers, shared among the revised Shifman, original Shifman, and MGI maps
Most proximal Mit marker | Most distal Mit marker | Total map length (cM) | Chromosome average recombination rate | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Chromosome | Marker ID | Mb | Marker ID | Mb | Revised Shifmana | Original Shifmana | MGI (standard) | Revised Shifmana | Original Shifmana | MGI (standard) |
| 1 | D1Mit475 | 3.64 | D1Mit155 | 196.26 | 96.55 | 117.83 | 103.60 | 0.50 | 0.61 | 0.54 |
| 2 | D2Mit312 | 3.15 | D2Mit457 | 180.99 | 101.82 | 108.22 | 107.00 | 0.57 | 0.61 | 0.60 |
| 3 | D3Mit150 | 5.35 | D3Mit89 | 156.50 | 78.83 | 86.80 | 83.70 | 0.52 | 0.57 | 0.55 |
| 4 | D4Mit149 | 3.58 | D4Mit51 | 154.42 | 84.13 | 99.86 | 82.70 | 0.56 | 0.66 | 0.55 |
| 5 | D5Mit69 | 3.53 | D5Mit169 | 150.23 | 86.97 | 105.61 | 85.00 | 0.59 | 0.72 | 0.58 |
| 6 | D6Mit231 | 3.48 | D6Mit390 | 148.41 | 76.69 | 89.72 | 73.73 | 0.53 | 0.62 | 0.51 |
| 7 | D7Mit21 | 3.27 | D7Mit141 | 152.14 | 87.05 | 89.42 | 71.90 | 0.58 | 0.60 | 0.48 |
| 8 | D8Mit155 | 4.98 | D8Mit156 | 131.42 | 73.96 | 80.20 | 72.00 | 0.58 | 0.63 | 0.57 |
| 9 | D9Mit186 | 6.26 | D9Mit322 | 123.10 | 71.46 | 85.23 | 70.00 | 0.61 | 0.73 | 0.60 |
| 10 | D10Mit49 | 4.14 | D10Mit269 | 128.39 | 75.29 | 83.11 | 68.00 | 0.61 | 0.67 | 0.55 |
| 11 | D11Mit1 | 3.47 | D11Mit104 | 119.17 | 80.85 | 91.20 | 78.75 | 0.70 | 0.79 | 0.68 |
| 12 | D12Mit264 | 5.31 | D12Mit144 | 120.30 | 61.00 | 68.55 | 60.00 | 0.53 | 0.60 | 0.52 |
| 13 | D13Mit235 | 4.22 | D13Mit35 | 120.13 | 64.85 | 69.46 | 71.00 | 0.56 | 0.60 | 0.61 |
| 14 | D14Mit110 | 8.99 | D14Mit107 | 124.00 | 61.37 | 59.38 | 59.50 | 0.53 | 0.52 | 0.52 |
| 15 | D15Mit12 | 3.16 | D15Mit160 | 102.99 | 56.40 | 64.34 | 59.80 | 0.56 | 0.64 | 0.60 |
| 16 | D16Mit32 | 3.96 | D16Mit71 | 97.13 | 54.73 | 62.93 | 68.95 | 0.59 | 0.68 | 0.74 |
| 17 | D17Mit164 | 3.92 | D17Mit123 | 93.60 | 58.56 | 62.74 | 52.60 | 0.65 | 0.70 | 0.59 |
| 18 | D18Mit65 | 3.73 | D18Mit25 | 89.74 | 56.67 | 64.07 | 55.00 | 0.66 | 0.74 | 0.64 |
| 19 | D19Mit32 | 3.28 | D19Mit108 | 59.96 | 53.24 | 53.35 | 49.50 | 0.94 | 0.94 | 0.87 |
| X | DXMit101 | 5.68 | DXMit100 | 165.35 | 76.16 | 69.83 | 72.50 | 0.48 | 0.44 | 0.45 |
| Total | 1456.58 | 1611.84 | 1445.23 | |||||||
Most proximal Mit marker | Most distal Mit marker | Total map length (cM) | Chromosome average recombination rate | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Chromosome | Marker ID | Mb | Marker ID | Mb | Revised Shifmana | Original Shifmana | MGI (standard) | Revised Shifmana | Original Shifmana | MGI (standard) |
| 1 | D1Mit475 | 3.64 | D1Mit155 | 196.26 | 96.55 | 117.83 | 103.60 | 0.50 | 0.61 | 0.54 |
| 2 | D2Mit312 | 3.15 | D2Mit457 | 180.99 | 101.82 | 108.22 | 107.00 | 0.57 | 0.61 | 0.60 |
| 3 | D3Mit150 | 5.35 | D3Mit89 | 156.50 | 78.83 | 86.80 | 83.70 | 0.52 | 0.57 | 0.55 |
| 4 | D4Mit149 | 3.58 | D4Mit51 | 154.42 | 84.13 | 99.86 | 82.70 | 0.56 | 0.66 | 0.55 |
| 5 | D5Mit69 | 3.53 | D5Mit169 | 150.23 | 86.97 | 105.61 | 85.00 | 0.59 | 0.72 | 0.58 |
| 6 | D6Mit231 | 3.48 | D6Mit390 | 148.41 | 76.69 | 89.72 | 73.73 | 0.53 | 0.62 | 0.51 |
| 7 | D7Mit21 | 3.27 | D7Mit141 | 152.14 | 87.05 | 89.42 | 71.90 | 0.58 | 0.60 | 0.48 |
| 8 | D8Mit155 | 4.98 | D8Mit156 | 131.42 | 73.96 | 80.20 | 72.00 | 0.58 | 0.63 | 0.57 |
| 9 | D9Mit186 | 6.26 | D9Mit322 | 123.10 | 71.46 | 85.23 | 70.00 | 0.61 | 0.73 | 0.60 |
| 10 | D10Mit49 | 4.14 | D10Mit269 | 128.39 | 75.29 | 83.11 | 68.00 | 0.61 | 0.67 | 0.55 |
| 11 | D11Mit1 | 3.47 | D11Mit104 | 119.17 | 80.85 | 91.20 | 78.75 | 0.70 | 0.79 | 0.68 |
| 12 | D12Mit264 | 5.31 | D12Mit144 | 120.30 | 61.00 | 68.55 | 60.00 | 0.53 | 0.60 | 0.52 |
| 13 | D13Mit235 | 4.22 | D13Mit35 | 120.13 | 64.85 | 69.46 | 71.00 | 0.56 | 0.60 | 0.61 |
| 14 | D14Mit110 | 8.99 | D14Mit107 | 124.00 | 61.37 | 59.38 | 59.50 | 0.53 | 0.52 | 0.52 |
| 15 | D15Mit12 | 3.16 | D15Mit160 | 102.99 | 56.40 | 64.34 | 59.80 | 0.56 | 0.64 | 0.60 |
| 16 | D16Mit32 | 3.96 | D16Mit71 | 97.13 | 54.73 | 62.93 | 68.95 | 0.59 | 0.68 | 0.74 |
| 17 | D17Mit164 | 3.92 | D17Mit123 | 93.60 | 58.56 | 62.74 | 52.60 | 0.65 | 0.70 | 0.59 |
| 18 | D18Mit65 | 3.73 | D18Mit25 | 89.74 | 56.67 | 64.07 | 55.00 | 0.66 | 0.74 | 0.64 |
| 19 | D19Mit32 | 3.28 | D19Mit108 | 59.96 | 53.24 | 53.35 | 49.50 | 0.94 | 0.94 | 0.87 |
| X | DXMit101 | 5.68 | DXMit100 | 165.35 | 76.16 | 69.83 | 72.50 | 0.48 | 0.44 | 0.45 |
| Total | 1456.58 | 1611.84 | 1445.23 | |||||||
Position on sex-averaged map except for chromosome X; these centimorgan positions are based on the female map.
The overall sex difference in recombination rate is greatly attenuated in the revised map. In the original Shifman map, the autosomal map is 26% longer in females than in males; in the revised map, the female autosomal map is only 9% longer than the male map.
Perhaps the most striking differences between the original and the revised Shifman maps are seen in an examination of recombination rates across individual chromosomes (Figure 1 and Figure S1). While many regions of apparently high recombination remain in the revised maps, their intensity is often attenuated compared to the original Shifman map. In addition, the numerous narrow regions of unusually high recombination rate, apparent in the original Shifman map, are largely eliminated in the revised map. For example, on chromosome 1 (Figure 1), some peaks in the recombination rates remain, but several prominent ones have disappeared. Particularly notable is the region at ∼125 Mb, which disappears entirely in males and is greatly attenuated in females; similar artifacts are seen on almost all chromosomes (see Figure S1). Figure 2 depicts the cumulative sex-specific genetic maps along chromosome 1. All the chromosomes are depicted in Figure S2.
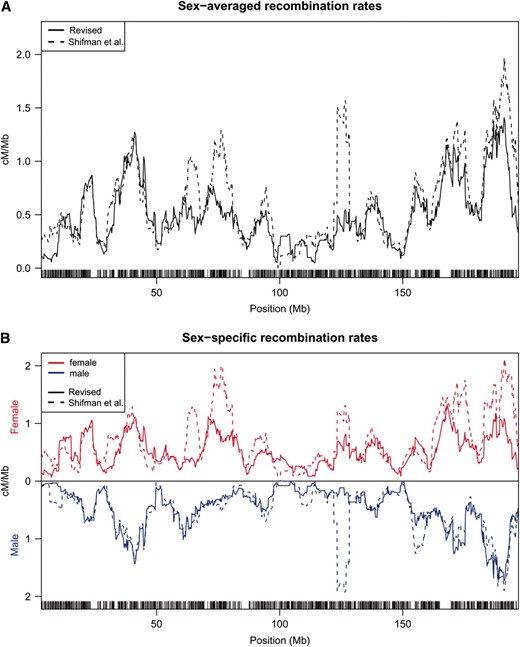
Comparison of the original and revised genetics maps. Sex-averaged recombination rates (A) and sex-specific recombination rates (B) for the original and revised genetic maps of chromosome 1. Maps are based on data from Shifman et al. (2006) as described in materials and methods. Figures showing all chromosomes can be found in Figure S1.
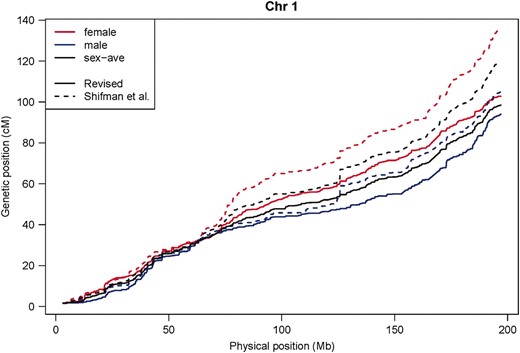
Cumulative genetic maps of chromosome 1. Dotted lines show the original Shifman map; solid lines are from the revised Shifman map. Female, male, and sex-averaged maps are shown in red, blue, and black, respectively. See Figure S2 for the cumulative maps for each chromosome.
Mit marker map:
There are 7380 distinct Mit marker symbols associated with 7052 primer pairs. Conflicts and ambiguities in marker symbols between MGI, UCSC, and NCBI were resolved as described in materials and methods. Of the 7052 primer pairs, 6080 mapped to one unique genomic location, and these were assigned centimorgan positions on the basis of the interpolation in the revised Shifman map; 147 primer pairs mapped to more than one genomic location, and 780 primer pairs could not be mapped to the build 37 mouse genome using any of the methods described above. Forty-five PPIDs do not have known primer sequences according to the online databases. Markers that map to more than one location are not recommended for use in future studies. However, the status of these markers may be revised in light of future releases of the mouse genome assembly.
The relationships between physical and genetic map positions of Mit markers as reported in the MGI map and the revised Shifman map are shown in Figure 3. The most important difference is that markers in the revised Shifman map are consistent with the physical order of markers along each chromosome.
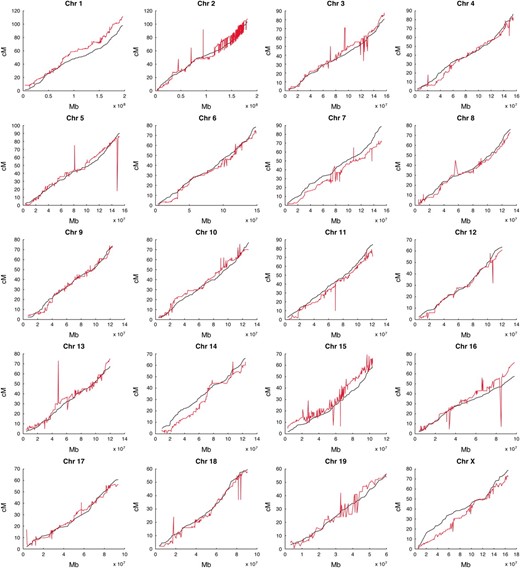
Physical and genetic positions of markers. Genetic marker positions (in centimorgans) are plotted against their physical positions (in megabases) for the MGI genetic map (red) and the revised Shifman map (black). Marker-ordering errors in the MGI map are indicated by non-monotone fluctuations. In contrast, the curves for the revised map are smooth and monotone.
Impact of the revised Shifman map on QTL localization:
Incorrect order of markers in the MGI map represents a substantial problem for mapping and identification of QTL. To investigate this further, we examined the effect on QTL mapping of changing to the revised Shifman map in five previously published data sets. First, a genome scan for primary QTL was done using the marker position information provided with the original data set (i.e., used in the published analysis). Second, an analysis was done using the new genetic map. Care was taken to ensure that the only difference in the analyses was the genetic map. For example, the removal of phenotypic outliers and phenotypic data normalization was identical in both analyses. As such, the effect of changing the genetic map on QTL mapping could be discerned independently of the different QTL mapping algorithms used in the original analyses.
In total, 78 QTL were examined in this study. Half of these QTL were not affected by the change in the genetic map. For the remainder, a variety of changes were noted (summarized in Table 2). For 28% of the QTL, no change in the shape of the QTL peak was identified, but the location of the peak shifted by ≥5 cM. We found 15 QTL with substantial changes (Table S4). For 6 QTL, a different marker closest to the peak was identified. For 9 QTL, a new shape for the QTL peak was noted (Figure 4). An example of such a peak shape change is presented in Figure 4, A and B. In this example, the third and fourth markers are reversed in order when comparing the two maps. This switch in marker order was not detected using standard marker quality control checks during either analysis. The LOD score associated with the peak was different in 2 of the QTL for which a change in peak shape was noted. Specifically, for one QTL, the change in LOD resulted in a failure to identify the QTL in the analysis utilizing the traditional map. In summary, for 15 of 78 QTL (19%), a mapping issue was identified that could impact the identification of the underlying gene.
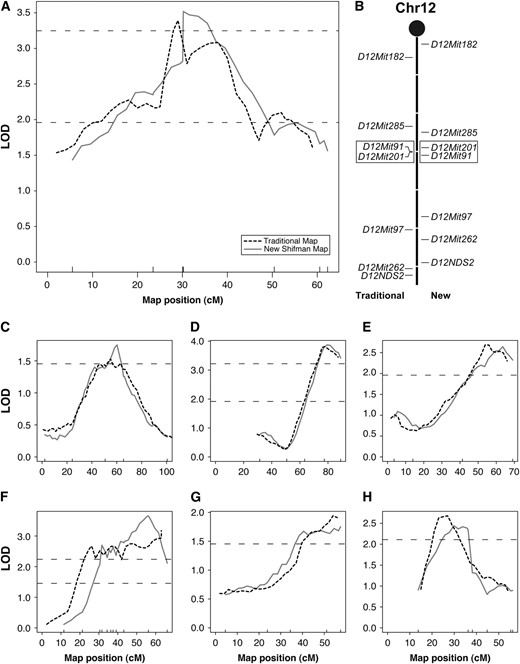
QTL With changes in peak shape. Seven QTL were found where the peak shape was altered due to subtle differences between the original and the revised Shifman maps. (A) The QTL for femoral BMD on chromosome 12 in the NZBxRF cross appears as a double peak when analyzed using the Mouse Genome Database (MGD)/traditional genetic map (dashed line), suggesting the presence of two closely linked QTL. When reanalyzed using the new genetic map, this double peak is collapsed into a single peak (solid line). (B) This double peak is the result of two flipped neighboring markers. Seven markers were typed in this cross for chromosome 12. The markers are placed on the centimorgan scale (center line) in relation to the MGD/traditional map (left) and the new genetic map (right). Note the difference in spacing of the markers when comparing the two maps. The other QTL with a change in peak shape were found for: (C) B6xCAST on chromosome 2, (D) B6xC3H on chromosome 2 for vBMD, (E) NZBxRF on chromosome 4 for vBMD, (F) B6xCAST on chromosome 14, (G) B6xCAST on chromosome 18, and (H) NZBxSM on chromosome 19.
Types of changes in QTL resulting from the new map
Change in QTL | No. of QTL |
|---|---|
| No change | 41 |
| Shift in location of >5 cM | 22 |
| Different marker closest to the peak | 6 |
| Significant change in QTL shape | 9 |
| Total | 78 |
Change in QTL | No. of QTL |
|---|---|
| No change | 41 |
| Shift in location of >5 cM | 22 |
| Different marker closest to the peak | 6 |
| Significant change in QTL shape | 9 |
| Total | 78 |
Types of changes in QTL resulting from the new map
Change in QTL | No. of QTL |
|---|---|
| No change | 41 |
| Shift in location of >5 cM | 22 |
| Different marker closest to the peak | 6 |
| Significant change in QTL shape | 9 |
| Total | 78 |
Change in QTL | No. of QTL |
|---|---|
| No change | 41 |
| Shift in location of >5 cM | 22 |
| Different marker closest to the peak | 6 |
| Significant change in QTL shape | 9 |
| Total | 78 |
Resources:
The revised genetic map and the updated Mit marker database are available in a tab-delimited text format online at http://cgd.jax.org/mousemapconverter/. A web interface allows the user to convert between genetic and physical map coordinates and to query the positions of mouse markers. Male, female, and sex-averaged positions are available. The original data from Shifman et al. (2006) and the cleaned data are also posted here. We will periodically update these resources as new versions of the mouse genome assembly are released.
Historical QTL data have been generated with a changing standard genetic map or, quite often, with a genetic map constructed de novo on the basis of the cross data. This inconsistency presents a roadblock to integrated analysis of common QTL. We are actively curating available historical QTL data sets by updating marker positions to the revised Shifman map. This resource also provides the original phenotype and genotype data and references to the original publications. These data are available at http://qtlarchive.org or through the mouse phenome database (http://www.jax.org/phenome/QTL).
DISCUSSION
We have constructed a genetic map of the mouse genome based on data from a single large experiment and integrated it with the physical positions of markers along each chromosome. Mouse SSLP markers (Mit markers), which were not used in the genetic map construction, have been assigned new genetic map positions by interpolation on the basis of their physical positions. This allowed us to correct inconsistent assignment of positions in the MGI genetic map. The new map provides a common framework for candidate gene identification.
The MGI map has served as the standard reference map of the mouse genome for many years. However, it is a consensus map compiled largely before the availability of whole-genome sequence data. Moreover, the MGI map combined recombination data from male and female meioses to produce a sex-averaged composite despite well-established sex differences in the recombination process.
In the revised Shifman map, errors in marker order and spacing have been corrected, suggesting that this new map will affect QTL mapping. Indeed, for 15 of the 78 QTL examined in this study, updating the genetic map changed the QTL peak localization. For 4 QTL, a major change in peak shape resulted from a change in marker order between the two maps. In each of these cases, it was not obvious in the diagnostic plots that there was an error in marker order. This suggests that marker errors are not always apparent in cross data and underscores the importance of having an accurate and independent genetic map. For 4 additional QTL, a major change in peak shape was found to be caused by a large difference in relative distance between markers near the peak in the revised Shifman map. In interval mapping, the inference of genotype between two known markers is partially a function of the defined distance between these two markers. These results demonstrate that changing the distance over which genetic data is inferred will affect mapping. Usually, it is not possible in a publication to show the LOD plot for all QTL. For this reason, QTL are often published as lists in tables. Sometimes the peak location in centimorgans is provided, but often only the marker found to be closest to the peak is published. For 6 of the 78 QTL examined here, the marker closest to the peak was different between the two analyses. On the basis of these results, caution is advised when comparing published QTL for which you have only the name of the marker closest to the peak. On the basis of these findings, we recommend the recalculation of historical QTL mapping data sets that are the basis of ongoing gene discovery programs.
In accord with the original Shifman map (Shifman et al. 2006) and earlier sex-specific genetic maps from mice (Reeves et al. 1990; Roderick and Hilyard 1990), the revised Shifman map displays clear distributional differences between male and female recombination. Male recombination events tend to cluster in subtelomeric intervals and are virtually absent in peri-centromeric regions, whereas female recombination events are more uniformly distributed across chromosomes. These distributional trends have been noted in several other mammalian species, including humans (Broman et al. 1998; Stapley et al. 2008), but despite their phylogenetic pervasiveness, an explanation for these fundamental sex differences is still wanting. Recently, Petkov et al. (2007) suggested that differences in crossover interference distances underlie the overall differences in recombination rate. Additionally, the total genetic map is 9% longer in females than in males, although this overall pattern masks finer-scale variation in the ratio of the male-to-female recombination rate (Figure 1, Figure S1, and Figure S2). Together, these sexually dimorphic features of the mouse recombination landscape strongly motivate the use of sex-specific genetic maps in backcross QTL analyses.
The HS pedigrees are composed of animals with admixed genomes bearing contributions from eight classical laboratory strains. It is well established that individuals and different inbred mouse strains vary with respect to both the distribution and the overall intensity of recombination (Koehler et al. 2002; Coop and Przeworski 2007). This fact carries important implications for the interpretation of the genetic maps presented here. Most notably, the recombination fractions that we report represent an average over the eight founding strains and are likely to differ from recombination frequencies calculated in independent mouse crosses. These interstrain differences in recombination are due to the cumulative effects of trans-acting recombination modifying loci and their interaction with cis-acting DNA sequences (Grey et al. 2009; Parvanov et al. 2009); the precise identification of these genetic variants poses an exciting and important challenge to the field of genetics (Paigen et al. 2008).
Despite the presence of individual-level variation in recombination rate, at the scale of resolution required for most genetic studies (∼1 Mb), recombination rates are conserved across different strain combinations (2000 Chromosome Committee; Duret and Arndt 2008). Paigen et al. (2008) found that, at an ∼2-Mb interval, their B6xCAST high-resolution (minimum 225 kb) chromosome 1 map correlates highly with the original Shifman map; this interval is likely decreased to closer to 1 Mb in comparison with the revised map presented here. Heterogeneity in individual recombination rates for this HS population is described in an accompanying article in this issue (Dumont et al. 2009). In addition, in comparing the two maps, the regions of low and high recombination occur in parallel. Thus, the genetic map based on an HS population presented here will serve as a useful and practical approximation to the distribution and intensity of recombination events in any mouse strain. The large number of meioses surveyed across the HS pedigree and the extraordinary number of genetic loci placed on this map combine to present the mouse community with a genetic resource unparalleled in other experimental model systems. This map is equipped with sufficient precision for most types of genetic crosses, including backcross, intercross, recombinant inbred, and advanced intercross designs. It provides consistent genetic map coordinates that are directly tied to the physical genome.
Footnotes
Supporting information available online at: http://www.genetics.org/cgi/content/full/genetics.109.105486/DC1.
Footnotes
Communicating editor: L. Siracusa
Acknowledgements
The authors thank Ben King, Randy von Smith, Stephen Grubb, Nazira Bektassova, and Keith Sheppard for advice and assistance with informatics and database tools. We thank Ken Paigen, Janan Eppig, Petko Petkov, Thomas Gridley, and Lucy Rowe for critical discussion and review. This work was supported by the National Institutes of Health with grants GM070683 (G.A.C.), GM074244 (K.W.B.), HL077796 (B.P.), 5T32HD07065 (C.L.A.-B.), and AR053992 (C.L.A.-B.); by the National Institute of General Medical Sciences Centers of Excellence in systems biology grant GM076468 (G.A.C.); and by the Federal Ministry for Education and Research, Germany (National Genome Research Network project 01GS0829, G.A.B.). The Shared Scientific Services were supported in part by a Basic Cancer Center Core Grant (CA34196) from the National Cancer Institute to the Jackson Laboratory.
References
Bult, C. J., J. T. Eppig, J. A. Kadin, J. E. Richardson and J. A. Blake,
Coop, G., and M. Przeworski,
Green, P., K. Falls and S. Crooks,
Ishimori, N., I. M. Stylianou, R. Korstanje, M. A. Marion, R. Li et al.,
Lehmann, E., and H. D'Abrera,
Parvanov, E. D., S. H. Ng, P. M. Petkov and K. Paigen,
Roderick, T. H., and A. L. Hilyard,

{kind=link}
{kind=link}
{kind=link}
{kind=link}