





Abstract
Genomic integrity is threatened by multiple sources of DNA damage. DNA double-strand breaks (DSBs) are among the most dangerous types of DNA lesions and can be generated by endogenous or exogenous agents, but they can arise also during DNA replication. Sister chromatid recombination (SCR) is a key mechanism for the repair of DSBs generated during replication and it is fundamental for maintaining genomic stability. Proper repair relies on several factors, among which histone modifications play important roles in the response to DSBs. Here, we study the role of the histone H3K79 methyltransferase Dot1 in the repair by SCR of replication-dependent HO-induced DSBs, as a way to assess its function in homologous recombination. We show that Dot1, the Rad9 DNA damage checkpoint adaptor, and phosphorylation of histone H2A (γH2A) are required for efficient SCR. Moreover, we show that Dot1 and Rad9 promote DSB-induced loading of cohesin onto chromatin. We propose that recruitment of Rad9 to DSB sites mediated by γH2A and H3K79 methylation contributes to DSB repair via SCR by regulating cohesin binding to damage sites. Therefore, our results contribute to an understanding of how different chromatin modifications impinge on DNA repair mechanisms, which are fundamental for maintaining genomic stability.
IN eukaryotic cells, genomic integrity is ensured by the action of the DNA damage checkpoint. This checkpoint coordinates the cellular response to DNA damage, triggering cell cycle arrest and activating DNA repair mechanisms, thus providing time for the cell to repair the damage before resuming cell cycle progression (Harrison and Haber 2006). DNA double-strand breaks (DSBs) are among the most dangerous genomic lesions and, if they are not properly repaired, they can lead to fatal consequences. DSBs can be repaired either by homologous recombination (HR) or by nonhomologous end joining (NHEJ), but only HR with the sister chromatid ensures that the fidelity of genetic information is mantained. Thus, sister chromatid recombination (SCR) is the preferred mechanism of DSB repair in mitotic cells (Kadyk and Hartwell 1992; Johnson and Jasin 2000; González-Barrera et al. 2003). Since SCR occurs between identical DNA molecules, its analysis by genetic or physical methods is difficult but, recently, a physical assay to monitor the repair by SCR of a single DSB generated during replication has been developed in budding yeast (González-Barrera et al. 2003; Cortes-Ledesma and Aguilera 2006). This SCR assay is based on a circular minichromosome harboring an internal mini-HO site, which is cleaved mainly in one strand producing ∼10% DSBs during replication, in contrast to the direct and efficient DSB induction at the full-length HO site. In this way, upon HO induction, the DSB occurs only in one chromatid and the other one remains intact and available for repair (see Figure 1A). Although this assay has been used mainly to monitor unequal SCR events, it has been demonstrated that it is an accurate indicator of the proficiency in total SCR (González-Barrera et al. 2003; Cortes-Ledesma and Aguilera 2006). Using this physical assay, it has been established that Rad52, Rad59, Rad51, and Rad54, but not Rdh54/Tid1, are involved in SCR (Cortes-Ledesma et al. 2007b). Also, SMC (structural maintenance of chromosomes) proteins including the cohesin complex and the Smc5/6 complex are required for efficient SCR (Cortes-Ledesma and Aguilera 2006; De Piccoli et al. 2006; Cortes-Ledesma et al. 2007a).
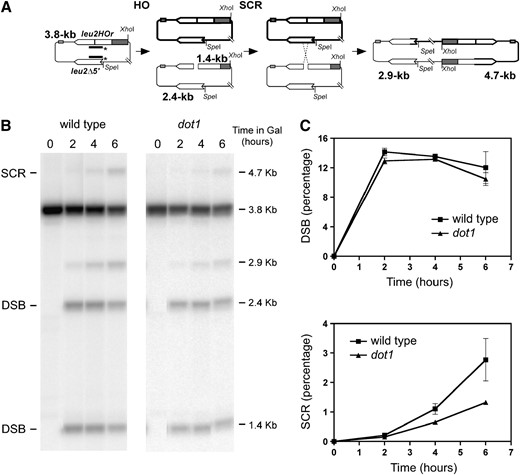
Dot1 is required for efficient SCR. (A) Schematic of the physical assay used to monitor repair by SCR of an HO-induced DSB in the centromeric plasmid pRS316-TINV. Fragments generated after XhoI–SpeI digestion, detected by the LEU2 probe (line with asterisks) are indicated with their corresponding sizes. Since other recombination events can also lead to the 2.9-kb fragment, only the 4.7-kb band is used to measure SCR. (B) Kinetics of HO-induced DSB formation and its repair in wild-type (MKOS-3C) and dot1 (YP764) cells incubated in galactose for the indicated times. A representative Southern blot is presented showing the different fragments detected. The 3.8-kb band corresponds to the intact plasmid and equal SCR events, the 1.4-kb and 2.4-kb fragments arise after HO cut, the 2.9-kb band results from unequal SCR and IC-BIR and the 4.7-kb band is specific for unequal SCR. (C) Quantification of DSBs (1.4-kb plus 2.4-kb bands) and SCR (4.7-kb band) relative to the total DNA. Averages and standard deviations are shown. In some cases, such as the dot1 SCR values, the error bars are hidden by the graph symbols.
Detection, signaling and repair of genomic lesions occur in the context of chromatin; therefore, factors regulating chromatin structure, such as histone modifications and chromatin remodelers, play important roles in the DNA damage response (Peterson and Cote 2004; Lydall and Whitehall 2005; van Attikum and Gasser 2005; Downs et al. 2007). Mec1- and Tel1-dependent phosphorylation of histone H2A at serine 129 (hereafter referred to as γH2A) is required for DSB repair by NHEJ and likely HR (Downs et al. 2000) and also mediates recruitment of cohesin to DSB sites (Unal et al. 2004). Another chromatin modification involved in the DNA damage response is the methylation of lysine 79 of histone H3 (H3K79) mediated by Dot1 (van Leeuwen et al. 2002). During meiosis, Dot1 is required for the meiotic recombination checkpoint (San-Segundo and Roeder 2000) and, in mitotic cells, Dot1-dependent H3K79 methylation is involved in the Rad9-mediated activation of the Rad53 checkpoint kinase (Giannattasio et al. 2005; Wysocki et al. 2005). Moreover, genetic analyses of the response to different DNA damaging agents, such as ionizing radiation (IR), methyl methanesulfonate (MMS), and UV, have suggested that Dot1 modulates multiple DNA repair pathways (Game et al. 2006; Toh et al. 2006; Bostelman et al. 2007; Conde and San-Segundo 2008) and also controls DSB resection (Lazzaro et al. 2008). To gain further insight in the molecular mechanisms of DNA repair impacted by Dot1 function we have used a physical assay to monitor DSB repair by SCR as a manifestation of HR repair. We provide molecular and genetic evidence indicating that Dot1, together with γH2A, promotes SCR by Rad9-mediated recruitment of cohesin to DSB sites.
MATERIALS AND METHODS
Strains and plasmids:
Yeast strains used in this work are listed in Table 1. All strains are in the W303 or the JKM179 genetic background, as indicated. SCC1-9Myc and DDC2-GFP epitope tagging, as well as rad9∷kanMX6, rad9∷hphMX4, and dot1∷kanMX6 gene deletions, were performed using standard PCR-based approaches (Longtine et al. 1998; Goldstein and McCusker 1999; Knop et al. 1999). Plasmids pSS30 and pSS44 were used to generate dot1∷URA3 and dot1∷TRP1, respectively (San-Segundo and Roeder 2000). The YP933 strain carrying the H2A-S129* mutations in MKOS-3C for SCR analysis was derived from strain JDY22 (Downs et al. 2000) by genetic crosses always in a W303 isogenic background. Strain W4638-2C (a gift of Rodney Rothstein) has been described (Lisby et al. 2004). The JKM179 strain, lacking HMRa and HMLα, was provided by Jim Haber (Lee et al. 1998). The centromeric monocopy plasmid pRS316-TINV has been previously described (González-Barrera et al. 2003). The mini-HO site inserted at the EcoRI site of LEU2 in pRS316-TINV has been generally referred to as a 21-bp HO site (Cortes-Ledesma and Aguilera 2006), but the flanking nucleotides make it to be a 24-bp HO site.
Yeast strains
Strain | Genotype |
|---|---|
| W303-1A | MATa leu2-3, 112 trp1-1 ura3-1 ade2-1 his3-11,15 can1-100 rad5-G535R |
| W303-1B | MATα leu2-3, 112 trp1-1 ura3-1 ade2-1 his3-11,15 can1-100 rad5-G535R |
| MKOS-3C | W303-1B leu2Δ∷SFA1 ade3∷GAL-HO |
| YP764 | MKOS-3C dot1∷TRP1 |
| YP817 | MKOS-3C rad9∷kanMX6 |
| YP818 | MKOS-3C rad9∷kanMX6 dot1∷TRP1 |
| YP933 | MKOS-3C hta1-S129* hta2-S129* |
| YP934 | MKOS-3C hta1-S129* hta2-S129* dot1∷TRP1 |
| W4638-2C | W303-1A ADE2 bar1∷LEU2 RAD5 RAD9-YFP |
| YP759 | W303-1A ADE2 bar1∷LEU2 RAD5 RAD9-YFP dot1∷kanMX6 |
| JKM179 | MATα hml∷ADE1 hmr∷ADE1 ade1-110 leu2, 3-112 lys5 trp1∷hisG ura3-52 ade3∷GAL10-HO |
| CCG2876 | JKM179 SCC1-9Myc∷TRP1 DDC2-GFP∷kanMX4 |
| YP960 | JKM179 SCC1-9Myc∷TRP1 DDC2-GFP∷kanMX4 dot1∷URA3 |
| YP1150 | JKM179 SCC1-9Myc∷TRP1 DDC2-GFP∷kanMX4 rad9∷hphMX4 |
Strain | Genotype |
|---|---|
| W303-1A | MATa leu2-3, 112 trp1-1 ura3-1 ade2-1 his3-11,15 can1-100 rad5-G535R |
| W303-1B | MATα leu2-3, 112 trp1-1 ura3-1 ade2-1 his3-11,15 can1-100 rad5-G535R |
| MKOS-3C | W303-1B leu2Δ∷SFA1 ade3∷GAL-HO |
| YP764 | MKOS-3C dot1∷TRP1 |
| YP817 | MKOS-3C rad9∷kanMX6 |
| YP818 | MKOS-3C rad9∷kanMX6 dot1∷TRP1 |
| YP933 | MKOS-3C hta1-S129* hta2-S129* |
| YP934 | MKOS-3C hta1-S129* hta2-S129* dot1∷TRP1 |
| W4638-2C | W303-1A ADE2 bar1∷LEU2 RAD5 RAD9-YFP |
| YP759 | W303-1A ADE2 bar1∷LEU2 RAD5 RAD9-YFP dot1∷kanMX6 |
| JKM179 | MATα hml∷ADE1 hmr∷ADE1 ade1-110 leu2, 3-112 lys5 trp1∷hisG ura3-52 ade3∷GAL10-HO |
| CCG2876 | JKM179 SCC1-9Myc∷TRP1 DDC2-GFP∷kanMX4 |
| YP960 | JKM179 SCC1-9Myc∷TRP1 DDC2-GFP∷kanMX4 dot1∷URA3 |
| YP1150 | JKM179 SCC1-9Myc∷TRP1 DDC2-GFP∷kanMX4 rad9∷hphMX4 |
Yeast strains
Strain | Genotype |
|---|---|
| W303-1A | MATa leu2-3, 112 trp1-1 ura3-1 ade2-1 his3-11,15 can1-100 rad5-G535R |
| W303-1B | MATα leu2-3, 112 trp1-1 ura3-1 ade2-1 his3-11,15 can1-100 rad5-G535R |
| MKOS-3C | W303-1B leu2Δ∷SFA1 ade3∷GAL-HO |
| YP764 | MKOS-3C dot1∷TRP1 |
| YP817 | MKOS-3C rad9∷kanMX6 |
| YP818 | MKOS-3C rad9∷kanMX6 dot1∷TRP1 |
| YP933 | MKOS-3C hta1-S129* hta2-S129* |
| YP934 | MKOS-3C hta1-S129* hta2-S129* dot1∷TRP1 |
| W4638-2C | W303-1A ADE2 bar1∷LEU2 RAD5 RAD9-YFP |
| YP759 | W303-1A ADE2 bar1∷LEU2 RAD5 RAD9-YFP dot1∷kanMX6 |
| JKM179 | MATα hml∷ADE1 hmr∷ADE1 ade1-110 leu2, 3-112 lys5 trp1∷hisG ura3-52 ade3∷GAL10-HO |
| CCG2876 | JKM179 SCC1-9Myc∷TRP1 DDC2-GFP∷kanMX4 |
| YP960 | JKM179 SCC1-9Myc∷TRP1 DDC2-GFP∷kanMX4 dot1∷URA3 |
| YP1150 | JKM179 SCC1-9Myc∷TRP1 DDC2-GFP∷kanMX4 rad9∷hphMX4 |
Strain | Genotype |
|---|---|
| W303-1A | MATa leu2-3, 112 trp1-1 ura3-1 ade2-1 his3-11,15 can1-100 rad5-G535R |
| W303-1B | MATα leu2-3, 112 trp1-1 ura3-1 ade2-1 his3-11,15 can1-100 rad5-G535R |
| MKOS-3C | W303-1B leu2Δ∷SFA1 ade3∷GAL-HO |
| YP764 | MKOS-3C dot1∷TRP1 |
| YP817 | MKOS-3C rad9∷kanMX6 |
| YP818 | MKOS-3C rad9∷kanMX6 dot1∷TRP1 |
| YP933 | MKOS-3C hta1-S129* hta2-S129* |
| YP934 | MKOS-3C hta1-S129* hta2-S129* dot1∷TRP1 |
| W4638-2C | W303-1A ADE2 bar1∷LEU2 RAD5 RAD9-YFP |
| YP759 | W303-1A ADE2 bar1∷LEU2 RAD5 RAD9-YFP dot1∷kanMX6 |
| JKM179 | MATα hml∷ADE1 hmr∷ADE1 ade1-110 leu2, 3-112 lys5 trp1∷hisG ura3-52 ade3∷GAL10-HO |
| CCG2876 | JKM179 SCC1-9Myc∷TRP1 DDC2-GFP∷kanMX4 |
| YP960 | JKM179 SCC1-9Myc∷TRP1 DDC2-GFP∷kanMX4 dot1∷URA3 |
| YP1150 | JKM179 SCC1-9Myc∷TRP1 DDC2-GFP∷kanMX4 rad9∷hphMX4 |
Physical analysis of recombination:
Sister chromatid recombination assays were carried out essentially as described (González-Barrera et al. 2003). Briefly, cells carrying pRS316-TINV were grown to mid-log phase in SC −Ura 3% glycerol 2% lactate; then, galactose (2%) was added to induce HO expression. Samples were collected at different time points and DNA was purified, digested with XhoI–SpeI, and analyzed by Southern blotting using Hybond N+ (GE Healthcare) membranes. A radioactively labeled 0.6-kb ClaI–EcoRV LEU2 fragment from pRS315 was used as a probe. The radioactive signal on the membrane was detected with a Personal Molecular Imager (Bio-Rad) and quantified using the Quantity One software (Bio-Rad). The signal of each band relative to the total signal on each lane was measured. For the SCR analysis described in Figures 3 and 5, cells were first grown in SC −Ura 3% glycerol 2% lactate; then, galactose (2%) was added and after 2 hr of HO induction, glucose (2%) was added to repress HO expression. Samples were collected at different time points after addition of glucose and processed as described above. SCR experiments were repeated at least two or three times for each strain. To calculate statistical significance of differences in SCR levels, a two-tailed unpaired Student t-test was used. The GraphPad Prism version 4.0 software was used to perform the calculation of P-values.
Chromatin immunoprecipitation:
Cells grown in YP −3% glycerol 2% lactate were arrested in G2/M with 15 μg/ml nocodazole. After 1.5 hr, more nocodazole (7.5 μg/ml) was added to the cultures and after 30 min or 1 hr, galactose (2%) was added to induce the HO cut at MAT for 2 or 4 hr. The presence of Ddc2-GFP foci in most cells was used to assess the efficiency of HO induction. Cells were fixed with 1% formaldehyde for 30 min. Fixed cells were washed twice, resuspended in ice-cold buffer I (140 mm NaCl, 1mm EDTA, 1% Triton-X 100, 0.1% sodium deoxycholate and 50 mm HEPES/KOH pH 7.5) for experiments presented in Figure 6A or in SDS-lysis buffer (EZ-ChIP kit; Millipore) for the experiments presented in Figures 6B and 7B, and broken in a FastPrep (Qbiogene). Chromatin fragmentation to an average size of ∼500 bp was performed with a Bioruptor (Diagenode) by two cycles of sonication during 10 min with pulses of 30 sec “on” and 60 sec “off” in iced water. For the experiment presented in Figure 6A, ChIP was carried out essentially as described (De Piccoli et al. 2006). For the experiments presented in Figures 6B and 7B, ChIP of Scc1-9Myc was carried out with 5 μg of anti-myc monoclonal antibody (clone 4A6, Millipore) using the EZ-ChIP kit (Millipore, cat. no. 17-371) following the manufacturer's protocol. Immunoprecipitated DNA was analyzed by quantitative PCR in a DNA Engine Opticon 2 (Bio-Rad/MJ Research) or in an ABI7000 machine (Applied Biosystems) using primer pairs previously described (Shroff et al. 2004). Each sample of input and immunoprecipitated DNA was analyzed in duplicate or triplicate in every experiment.
Other techniques:
Fluorescence microscopy to detect Rad9-YFP foci was performed using an Olympus IX71 inverted microscope, equipped with a 100× 1.40 NA UPPlanSApo objective and a YFP filter set, as part of a DeltaVision system (Applied Precision). For each field, images from 11 z-sections at 0.4-μm intervals were collected and deconvoluted using the SoftWorX 3.6.2 DeltaVision software. Projections of the deconvolved images are presented. Western blot analysis was performed as described (Conde and San-Segundo 2008). Antibodies that specifically recognize the mono-, di- and trimethylated forms of histone H3K79 (Abcam ab2886, ab3594, and ab2621, respectively) were used at 1:1000 dilution. Flow cytometry (FACS) analysis was performed as described previously (Perez-Hidalgo et al. 2003).
RESULTS
Dot1 contributes to efficient SCR:
To gain insights into how chromatin modifications affect the DNA damage response, we have investigated the role of the histone H3 methylase Dot1 in sister chromatid recombination (SCR), which is a major DSB repair mechanism in mitotic yeast cells. To address whether Dot1 is required for DSB repair by SCR, we have used a physical assay that allows one to monitor the SCR-mediated repair of a single DSB created by the HO endonuclease (Figure 1A). After Southern blot analysis, the HO cleavage products are detected as 2.4-kb and 1.4-kb bands. A 4.7-kb fragment specifically results from repair of the DSB by unequal SCR (Figure 1, A and B). A 2.9-kb fragment that can arise either from SCR or from intrachromatid break-induced replication (IC-BIR) is also detected. Quantification of the relative intensity of the 1.4-kb plus 2.4-kb products and the 4.7-kb product gives an estimate of HO cleavage and SCR efficiencies, respectively (Figure 1C). It is worth noticing that the 4.7-kb product only measures the unequal fraction of all SCR products. However, we have previously shown that there is a strict correlation between the proportion of unequal vs. equal SCR events in this recombination system (González-Barrera et al. 2003). For this reason, the conclusions drawn for these unequal SCR products in this system can be extended to total SCR events.
When we analyzed the dot1 mutant with the SCR physical assay, we observed similar levels of DSB formation in both wild-type and dot1 cells; however, the repair by SCR, monitored as the relative percentage of the 4.7-kb fragment, was reduced ∼2.5-fold in dot1 compared to the wild type after 6 hr of HO induction (Figure 1C). Thus, Dot1 function is important for recombinational DSB repair and, in particular, it is necessary for efficient repair by SCR of a DSB generated during DNA replication.
The checkpoint adaptor Rad9 functions in SCR:
Using genetic assays based on his3 truncated genes it has been proposed that Rad9 promotes DSB repair by SCR (Fasullo et al. 1998) and genetic studies of DNA damage sensitivity have placed DOT1 in the RAD9 epistasis group (Wysocki et al. 2005; Conde and San-Segundo 2008). In addition, it has been reported that Dot1-mediated histone H3K79 methylation is required for the recruitment of the Rad9 checkpoint adaptor protein to an irreparable HO-induced DSB at the chromosomal MAT locus (Wysocki et al. 2005). Moreover, the formation of IR-induced Rad9 foci in G2 cells is impaired in the absence of Dot1 (Toh et al. 2006; Grenon et al. 2007), and the formation of MMS-induced Rad9 foci in G1 cells is also compromised in the dot1 mutant (supporting information, Figure S1). Therefore, it was possible that the SCR defect of dot1 results from failures to effectively recruit Rad9. To explore this possibility we analyzed the wild-type strain and the rad9, dot1, and rad9 dot1 mutants using the SCR physical assay. Like dot1, the rad9 and rad9 dot1 mutants displayed reduced repair by SCR upon continuous HO induction; however, DSB levels were also slightly reduced in the absence of Rad9 (Figure 2).
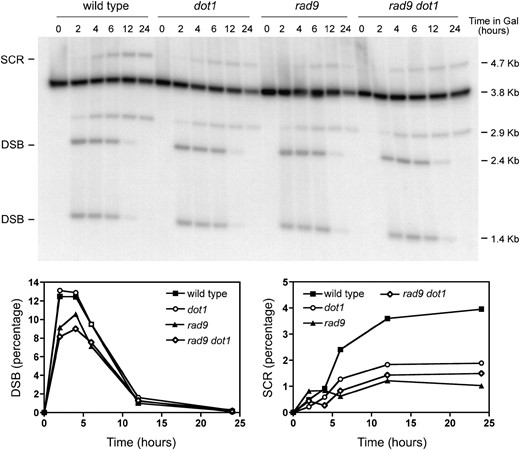
Rad9 functions in SCR. Kinetics and quantification of HO-induced DSB formation and its repair by SCR in wild-type (MKOS-3C), dot1 (YP764), rad9 (YP817), and rad9 dot1 (YP818) cells incubated in galactose for the indicated times. Other details as in Figure 1.
To rule out the possibility that the decreased proportion of SCR products simply reflects the reduced efficiency of HO cleavage, we carried out a modification of the SCR physical assay. Instead of continuous induction of HO with galactose, we induced the endonuclease for 2 hr, after which, the expression was turned off by adding glucose. This point was taken as the zero time point for the experiment. Samples were collected throughout time to monitor the repair by SCR. The percentage of the 4.7-kb SCR-specific product at each time point was normalized to the initial DSB levels to correct for the different cleavage efficiencies in the different strains (Figure 3). Even after this normalization, the rad9 mutant was found to be defective in SCR. Furthermore, the rad9 dot1 double mutant displayed the same reduced efficiency in DSB repair by SCR than the rad9 single mutant. This implies that Dot1 and Rad9 act in the same pathway, which is consistent with the Dot1 requirement for Rad9 recruitment to DSB sites. Although the SCR defect in the rad9 dot1 double mutant appears to be slightly increased compared to the dot1 single mutant (Figure 3D), the difference is not statistically significant (P = 0.380).
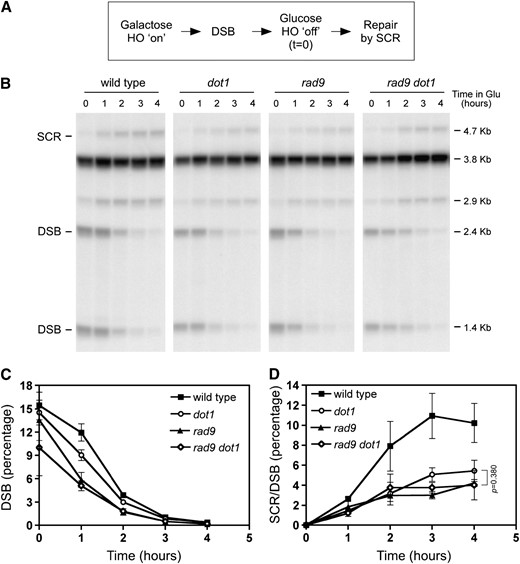
The dot1, rad9, and rad9 dot1 mutants display similar defects in SCR. (A) Scheme of the procedure used to follow repair of HO-induced DSBs. HO was induced for 2 hr in galactose and then glucose was added to repress HO expression. Repair by SCR was monitored by Southern blot analysis as explained in Figure 1. (B) Representative Southern blot of kinetics of DSB repair by SCR in wild-type (MKOS-3C), dot1 (YP764), rad9 (YP817), and rad9 dot1 (YP818). Note that the DSB-specific bands present at time zero (immediately after turning off HO expression) disappear as SCR (4.7-kb band) is taking place. (C) Quantification of DSB disappearance. (D) Quantification of SCR levels relative to the initial DSB percentage for each strain at time zero. Before normalization to the initial DSB values, the values of the SCR-specific 4.7-kb band present at time zero were subtracted from the values obtained at the other time points to exclude the small fraction of SCR products that have been generated during HO induction for 2 hr before adding glucose. Averages, standard deviations, and P-value of the statistical comparison are shown. In some cases, the error bars are hidden by the graph symbols.
γH2A contributes to SCR:
γH2A is necessary for accumulation of Rad9 at IR-induced foci in G2 (Toh et al. 2006) and, together with Dot1, promotes chromatin association of Rad9 at DSB sites in G1 cells (Javaheri et al. 2006; Hammet et al. 2007). γH2A also mediates recruitment of cohesin to DSB sites (Unal et al. 2004), which is involved in repair by SCR (Cortes-Ledesma and Aguilera 2006). Therefore, to further understand the regulation of SCR by histone modifications, we examined DSB repair by SCR in an hta1-S129* hta2-S129* mutant lacking the S129 phosphorylation site in both copies of histone H2A (Downs et al. 2000) (hereafter referred to as H2A-S129* mutant), as well as in a dot1 H2A-S129* double mutant. Like dot1, the H2A-S129* mutant displayed a reduction in SCR levels compared to the wild type (Figure 4).
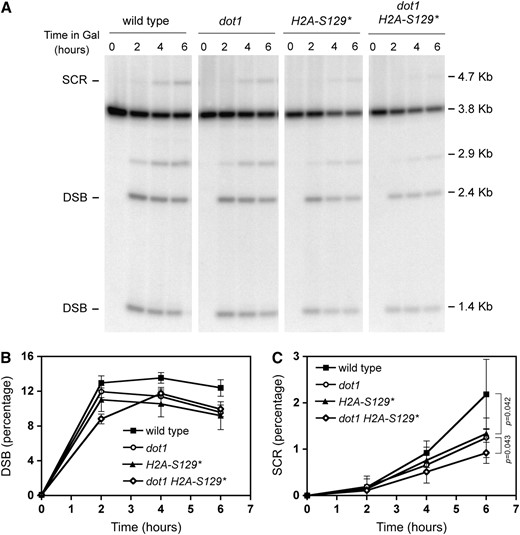
γH2A is important for SCR. Kinetics of HO-induced DSB formation and its repair by SCR in wild-type (MKOS-3C), dot1 (YP764), H2A-S129* (YP933), and dot1 H2A-S129* (YP934) cells incubated in galactose for the indicated times. (A) Representative Southern blot. (B and C) Quantification of DSBs and SCR, respectively, relative to the total DNA. Averages, standard deviations, and P-values of statistical comparisons are shown.
The defect in SCR was modestly increased in the dot1 H2A-S129* double mutant compared with the H2A-S129* single mutant (P < 0.05) (Figure 4C); however, the DSB levels also varied slightly among the strains analyzed (Figure 4B). Therefore, to determine whether the differences in SCR efficiency result from different DSB levels, we carried out the SCR assay following the same procedure described in Figure 3A; that is, turning off the expression of HO after 2 hr of induction and normalizing the SCR values to the initial DSB levels (Figure 5). Using this normalization, we confirmed that the H2A-S129* mutant is defective in SCR. In addition, the dot1 and H2A-S129* single mutants as well as the dot1 H2A-S129* double mutant displayed similar SCR defects (P = 0.901) (Figure 5C). Thus, both H3K79 methylation and γH2A contribute to DSB repair by SCR through the same pathway.
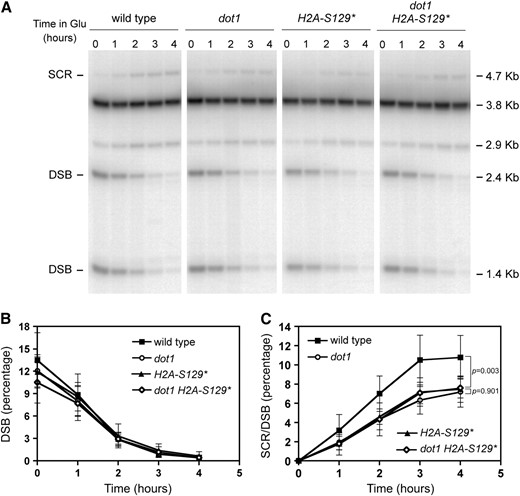
The dot1, H2A-S129*, and dot1 H2A-S129* mutants display similar defects in SCR. The repair by SCR of the HO-induced DSB was monitored and quantified as described in Figure 3 to normalize the SCR values to the initial DSB levels (A) Representative Southern blot of kinetics of DSB repair by SCR in wild-type (MKOS-3C), dot1 (YP764), H2A-S129* (YP933), and dot1 H2A-S129* (YP934). (B) Quantification of DSB disappearance. (C) Quantification of SCR levels relative to the initial DSB percentage for each strain at time zero. Averages, standard deviations, and P-values of statistical comparisons are shown.
Dot1 and Rad9 promote binding of cohesin to DSB sites:
To determine if, like γH2A, Dot1-dependent H3K79 methylation is also involved in loading cohesin in response to DSBs, we used chromatin immunoprecipitation (ChIP) to analyze binding of the Scc1 cohesin subunit to the chromosomal region flanking an irreparable HO-induced DSB at the MAT locus in G2/M-arrested cells. In the wild type, upon HO induction, the binding of cohesin to the regions flanking the DSB was significantly increased beyond the distance of ∼2 kb to both sites of the break (Figure 6A), as previously reported (Unal et al. 2004). In contrast, in the dot1 mutant, loading of cohesin in response to the presence of a DSB was dramatically impaired throughout the whole chromosomal region adjacent to the DSB (Figure 6A). FACS analysis confirmed that both wild type and dot1 remained arrested at G2/M (Figure S2).
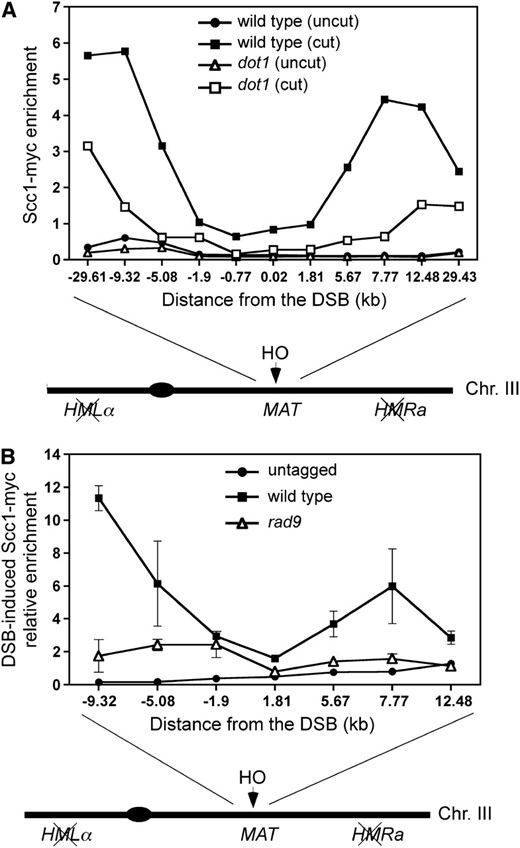
Dot1 and Rad9 contribute to DSB-induced cohesin loading. (A) ChIP analysis of Scc1 binding to the region flanking the HO site in the MAT locus in wild-type (CCG2876) and dot1 (YP960) strains lacking HMLα and HMRa, without (uncut) or with a DSB (cut) at MAT. HO expression was induced for 4 hr in nocodazole-arrested cells before processing for ChIP. Input DNA and DNA immunoprecipitated with anti-myc antibody were analyzed by quantitative real-time PCR using primer pairs located at different positions flanking the HO site in chromosome III. The scheme of chromosome III is not drawn to scale. (B) ChIP analysis of Scc1 binding to the region flanking the HO site in the MAT locus in Scc1-myc tagged wild-type (CCG2876) and rad9 (YP1150) strains lacking HMLα and HMRa. The results from untagged cells (JKM179) are also shown as control. The increment of Scc1 binding to the indicated locations flanking the HO site after DSB induction (4 hr in galactose) relative to the binding when there is no DSB (no galactose added) is represented. Averages and standard deviations are shown.
Since we have found that Rad9 is required for efficient DSB repair by SCR, we asked whether Rad9 function was also involved in cohesin recruitment. Using ChIP, we found that the DSB-promoted binding of Scc1 to the regions flanking the break was reduced in the rad9 mutant, especially in the regions where cohesin binding is higher in the wild type (−9.32 kb and +7.77 kb from the break site) (Figure 6B). To rule out the possibility that the reduced DSB-promoted cohesin deposition in the G2/M DNA-damage checkpoint-defective rad9 mutant results from cells that escaped from the nocodazole-induced arrest, we monitored the cell cycle stage by FACS analysis and microscopic observation. After 2 hr of DSB induction in nocodazole-treated cells, the majority of wild-type, dot1, and rad9 cells displayed a 2C DNA content and possessed a large bud indicative of a G2/M cell cycle arrest (Figure 7A). However, DSB-induced cohesin binding was impaired in the dot1 and rad9 mutants (Figure 7B), confirming that the reduced cohesin levels in the mutants do not result from inappropriate cell cycle progression. Taken together, these observations suggest that Dot1-mediated recruitment of Rad9 to DSB sites is required for efficient cohesin loading to promote SCR.
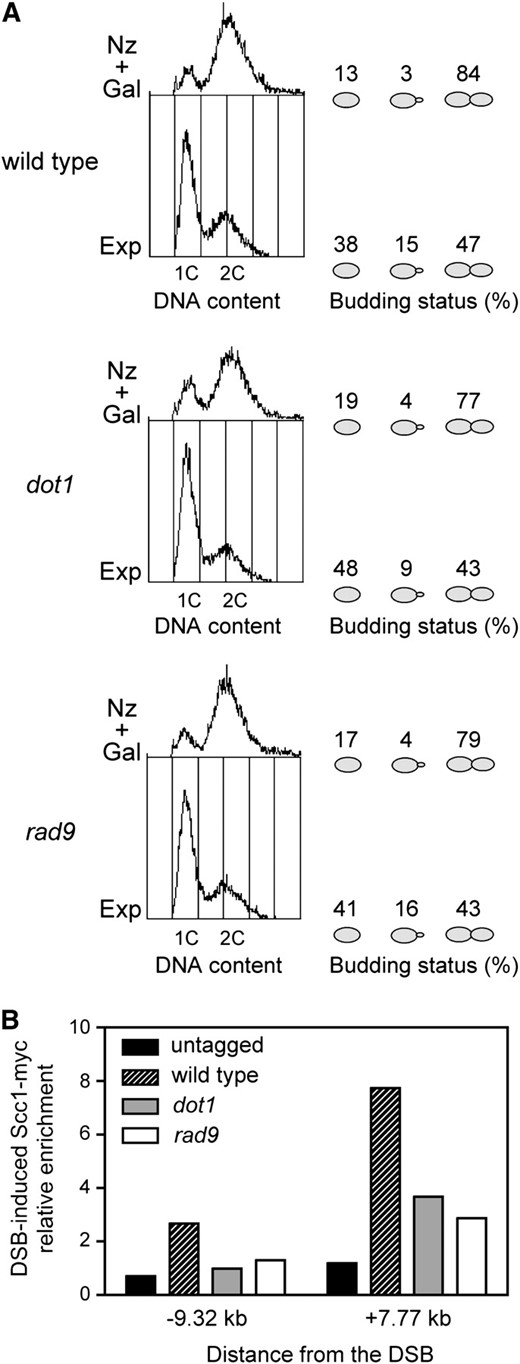
The reduced level of DSB-promoted cohesin binding in the checkpoint-defective rad9 mutant does not result from cell cycle progression. (A) Exponentially growing cells (Exp) of wild-type (CCG2876), dot1 (YP960), and rad9 (YP1150) strains were treated with nocodazole for 2 hr and then galactose was added. After 2 hr of DSB induction (Nz + Gal), cells were fixed with formaldehyde for ChIP analysis (B). The DNA content determined by FACS analysis and the budding status of the cells (no bud, small bud, or large bud) are presented to illustrate the cell cycle stage. The budding status of 300 cells was scored for each strain in each growth condition. (B) DSB-induced enrichment of Scc1-myc to the indicated locations flanking the HO site at MAT in the strains mentioned in A. The untagged strain (JKM179) is also shown as control.
DISCUSSION
To further understand how chromatin modulation regulates the DNA damage response, we have investigated the role of the H3K79 methyltransferase Dot1 in HR. Here, we demonstrate that, indeed, Dot1 functions in HR and, in particular, it contributes to SCR, which is the major mechanism of DSB repair in mitotic yeast cells. To our knowledge, we provide for the first time physical evidence for a role of Dot1 in recombinational repair. Therefore, our study confirms at the molecular level previous genetic and cytological studies that have suggested such a role for Dot1, though mostly based in indirect observations, such as sensitivity to damage (Game et al. 2006; Toh et al. 2006; Conde and San-Segundo 2008). Although we clearly prove that Dot1 is involved in SCR, it is likely that Dot1 function also impinges onto general HR because DSB-induced ectopic gene conversion events are reduced about twofold in the dot1 mutant (F. Conde and P. San-Segundo, unpublished data).
Our study reveals that Dot1, as well as the Rad9 DNA damage checkpoint adaptor, and phosphorylation of histone H2A (γH2A) are required for efficient SCR. We show that γH2A and H3K79 methylation contribute to SCR through the same mechanism, consistent with previous studies reporting overlapping functions for these histone modifications in the DNA damage response (Javaheri et al. 2006; Toh et al. 2006). We also demostrate that, like γH2A, Dot1 and Rad9 promote DSB-induced loading of cohesin onto chromatin. Therefore, we propose that the role of γH2A and H3K79 methylation in SCR is mediated, at least in part, through Rad9-dependent recruitment of cohesin to chromatin at the sites of damage (Figure 8). In addition to histone modifications, chromatin remodeling also functions in the DNA damage response. This is the case of the RSC (remodel the structure of chromatin) ATP-dependent chromatin remodeler, which is required for activation of the Rad53-dependent checkpoint and for association of cohesin with DSBs (Liang et al. 2007). Consistent with the observations that H3K79 is constitutively methylated (van Leeuwen et al. 2002) and the extent of methylation does not change upon DNA damage (Figure S3), it has been proposed that DSB-induced nucleosome remodeling may expose the methylated H3K79 allowing the binding of Rad9 (Huyen et al. 2004). The RSC complex may carry out this remodeling function.
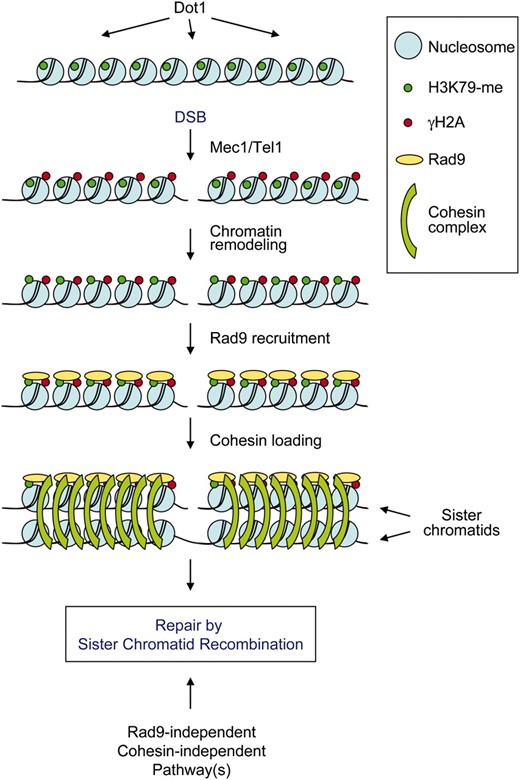
A model for the role of γH2A and H3K79 methylation in SCR. The lysine 79 positioned in the histone H3 globular domain is constitutively methylated by Dot1 (H3K79-me; green dots). When a DSB occurs, the Mec1/Tel1 kinases phosphorylate histone H2AS129 (γH2A) in the chromosomal region surrounding the break (red dots) and H3K79-methylated residues are exposed as a consequence of chromatin remodeling. Both γH2A and exposed H3K79-me recruit Rad9 (yellow ovals) to the DSB site, which in turn promotes cohesin binding (green arcs) facilitating repair of the DSB by SCR. Since neither the rad9 mutant nor the H2A-S129* mutant, which is defective in cohesin loading, is completely deficient in SCR, another pathway(s) contributing to SCR must exist. See text for details.
The only known direct target of Dot1 is H3K79 (van Leeuwen et al. 2002), whose methylation contributes to Rad53 activation by promoting Rad9 recruitment to sites of damage (Figure S1) (Giannattasio et al. 2005; Wysocki et al. 2005; Grenon et al. 2007). As the Scc1 cohesin subunit undergoes DNA damage-induced phosphorylation, which is partially dependent on the Rad53 checkpoint kinase (Sidorova and Breeden 2003) and the rad53 mutant is partially defective in cohesin recruitment to a DSB (Unal et al. 2004), it is possible that the effect of Dot1 and Rad9 in SCR is exerted by Rad53 activation, which in turn would promote cohesin loading. Indeed, it has been reported that Rad53 is required for DSB-induced SCR (Fasullo et al. 2005). However, a “late” role for Rad9 in DSB repair by HR, distinct from its “early” checkpoint function on Rad53 activation, has been proposed because Rad9 chromatin retention and colocalization with a subset of Rad52 foci correlate with checkpoint signaling downregulation and reconstitution of intact chromosomes after IR treatment (Toh et al. 2006). Moreover, the impaired G2/M checkpoint arrest of the rad9 mutant cannot solely explain the defect in SCR, because the dot1 and H2A-S129 mutants are quite proficient at DNA-damage-induced G2/M arrest (Wysocki et al. 2005; Javaheri et al. 2006), but they display similar SCR defects.
Recent findings indicate that phosphorylation of Scc1 at serine 83 by the Chk1 kinase promotes DSB-induced cohesion at G2/M. However, Chk1 is not required for recruitment of cohesin to DSBs; it has been proposed that phosphorylation of Scc1 by Chk1 promotes chromatin-bound cohesin to become cohesive (Heidinger-Pauli et al. 2008). Although we cannot rule out the possibility that Dot1 and Rad9 may also be required for Chk1-dependent cohesin activation in response to DNA damage, they must play additional functions because, unlike chk1, we found that the dot1 and rad9 mutants are defective in cohesin recruitment to DSB sites.
It has been recently shown that Dot1 and Rad9 limit the formation of single-stranded DNA during DSB resection (Lazzaro et al. 2008); thus, in contrast to our observations, one might expect that a faster resection in the dot1 or rad9 mutants could result in increased SCR. However, it is unlikely that this function is influencing SCR repair, because γH2A performs the opposite function, that is, promotes DSB resection (van Attikum et al. 2004), but it is also required for wild-type levels of SCR (Figure 5). Our SCR assay relies on the formation of a DSB during replication, but it does not preclude that the break can be repaired outside S phase; that is, during G2, when the sister chromatid is still available. Moreover, the induction of cohesin binding surrounding a DSB is similar in S phase and G2 cells (Unal et al. 2004). Therefore, the defect in DSB-promoted cohesin loading of the dot1, H2A-S129*, and rad9 G2 cells can account for the reduced SCR observed in these mutants.
We found that damage-induced loading of cohesin at DSBs is reduced, but not abolished, in dot1 and rad9 mutants; however, Rad9 is not required for genomewide DSB-induced cohesion (Strom et al. 2007), implying that the residual amount of cohesin observed in dot1 and rad9 might be sufficient to support sister chromatid cohesion but not enough for promoting wild-type levels of SCR. These results open the possibility that local cohesion at DSB sites, but not genomewide damage-induced cohesion, could be impaired in dot1 and rad9 mutants. Consistent with different requirements for the recombination and the cohesion function of cohesin, it has been recently shown that the Rec8 cohesin subunit, which is the meiotic counterpart of Scc1, promotes meiotic recombination independently of its role in sister chromatid cohesion (Brar et al. 2009). On the other hand, we found that SCR is impaired but not absolutely eliminated in the H2AS129* mutant, whereas cohesin deposition is completely dependent on γH2A (Unal et al. 2004). Therefore, a cohesin-independent SCR pathway must exist (Figure 8). It is tempting to speculate that this SCR function independent of cohesin might be carried out by another SMC-family complex, such as Smc5/6, which also participates in SCR (De Piccoli et al. 2006).
In summary, our study permits us to conclude that recruitment of Rad9 to DSB sites mediated by γH2A and H3K79 methylation contributes to DSB repair via SCR by regulating cohesin binding to these sites of damage. This result uncovers a new level in the control of sister-chromatid recombinational repair, providing new clues for our understanding of the mechanisms of DSB repair and its role in maintaining genome integrity.
Footnotes
Supporting information is available online at http://www.genetics.org/cgi/content/full/genetics.109.101899/DC1.
Footnotes
Communicating editor: S. E. Bickel
Acknowledgements
We thank Jessica Downs, Jim Haber, and Rodney Rothstein for providing strains; Félix Prado for comments on the manuscript; Purificación Galindo-Villardón for statistical advice; and Rocío Vizcaíno and Isabel Acosta for technical help. This work was supported by grants from the Ministry of Education and Science of Spain (BFU2006-05260 to A.A. and BFU2005-00955 and BFU2008-01014 to P.S.-S.); from the Junta de Andalucía (CVI624 to A.A.), from Junta de Castilla y León (CSI05A07 to P.S.-S.), and from the Medical Research Council of the United Kingdom (to L.A.). F.C. and E.R. were supported by predoctoral fellowships from the Ministry of Education and Science of Spain.
References
Javaheri, A., R. Wysocki, O. Jobin-Robitaille, M. Altaf, J. Cote et al.,
Lazzaro, F., V. Sapountzi, M. Granata, A. Pellicioli, M. Vaze et al.,

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}