





Abstract
The Rad51 paralogs Rad55 and Rad57 form a heterodimer required to mediate the formation and/or stabilization of the Rad51 filament. To further characterize the function of Rad55-Rad57, we used a combination of rad57 partial suppressors to determine whether the DNA repair and recombination defects of the rad57 mutant could be completely suppressed. The combination of all suppressors, elevated temperature, srs2, rad51-I345T, and mating-type (MAT) heterozygosity resulted in almost complete suppression of the rad57 mutant defect in the recruitment of Rad51 to DNA-damaged sites, as well as survival in response to ionizing radiation and camptothecin. In a physical assay to monitor the kinetics of double-strand-break (DSB)-induced gene conversion, the rad57 mutant defect was effectively suppressed by srs2 and MAT heterozygosity, but these same suppressors failed to suppress the spontaneous recombination defect. Thus the Rad55-Rad57 heterodimer appears to have a unique function in spontaneous recombination that is not essential for DSB repair. Furthermore, we investigated the currently unknown mechanism of rad57 suppression by MAT heterozygosity and found that it is independent of DNL4.
HOMOLOGOUS recombination is required for the faithful repair of DNA double-strand breaks (DSBs) that arise during normal cellular processes or from exposure of cells to DNA-damaging agents. Central to the process of homologous recombination is the Rad51 protein, which facilitates synapsis and strand invasion into homologous duplex DNA (San Filippo et al. 2008). Rad51 belongs to the RecA family of homologous pairing proteins (Aboussekhra et al. 1992; Basile et al. 1992; Shinohara et al. 1992). Yeast and humans have two RecA homologs: Rad51 and the meiosis-specific Dmc1 (Bishop et al. 1992; San Filippo et al. 2008). In addition, the Saccharomyces cerevisiae RAD55 and RAD57 genes encode proteins with sequence similarity to RecA and Rad51 and are considered to be Rad51 paralogs (Kans and Mortimer 1991; Lovett 1994). Mutation of RAD51, RAD55, or RAD57 confers sensitivity of ionizing radiation (IR) and defects in mitotic and meiotic recombination, indicating that their functions are not redundant (Symington 2002). rad51 mutants generally exhibit more severe defects than rad55 or rad57 mutants in DSB-induced recombination assays; however, rad55 and rad57 mutants are more defective than rad51 in some assays that measure spontaneous recombination between repeated sequences (Rattray and Symington 1995; Mozlin et al. 2008).
The molecular details of homologous recombination are largely based on genetic, physical, and cytological studies of DSB repair (DSBR) and on biochemical characterization of purified proteins (Paques and Haber 1999; Sugawara et al. 2003; Lisby et al. 2004; San Filippo et al. 2008). The single-stranded DNA (ssDNA)-binding protein, replication protein A (RPA), initially binds ssDNA that forms by nucleolytic processing of DNA ends at DSBs. In vitro, RPA has been shown to be inhibitory to Rad51 binding to ssDNA, but this inhibition can be overcome by addition of Rad52 or the Rad55-Rad57 heterodimer to the reaction (Sung 1997a,b; New et al. 1998; Shinohara and Ogawa 1998). Rad52, via its interaction with both RPA and Rad51, recruits Rad51 to the DNA and stimulates the removal of RPA (Sugiyama et al. 1998; Song and Sung 2000; Seong et al. 2008). Once Rad51 has been recruited to the ssDNA, a nucleoprotein filament forms in an ATP-dependent manner. A competent Rad51 filament is then able to interact with another DNA molecule to search for homologous sequences and initiate strand exchange (Krogh and Symington 2004). Consistent with the in vitro assays, rad52 mutants are completely defective in the recruitment of Rad51 to meiotic and mitotic DSBs in vivo (Gasior et al. 1998; Sugawara et al. 2003; Lisby et al. 2004; Miyazaki et al. 2004).
Cytological and chromatin immunoprecipitation studies suggest that Rad55 and Rad57 mediate filament formation by facilitating nucleation of Rad51 onto ssDNA or by stabilizing the filament once it is assembled. In rad55 mutants, Rad51 is recruited to DSBs with slower kinetics and forms dimmer IR-induced foci compared to wild-type cells (Sugawara et al. 2003; Lisby et al. 2004; Fung et al. 2006). The IR sensitivity of rad55 and rad57 mutants is partially bypassed by overexpression of Rad51, which increases the availability of the protein for filament formation, or by RAD51 gain-of-function alleles, such as rad51-I345T, that encode proteins with a higher affinity for DNA than wild-type Rad51 (Hays et al. 1995; Johnson and Symington 1995; Fortin and Symington 2002; Malik and Symington 2008). Deletion of SRS2, which encodes a helicase that disrupts Rad51-ssDNA complexes in vitro, also suppresses the IR sensitivity of rad55 and rad57 mutants (Krejci et al. 2003; Veaute et al. 2003; Fung et al. 2006). The expression of both mating-type alleles in haploid or diploid cells suppresses the IR sensitivity and interhomolog recombination defects of rad55 and rad57 mutants through an unknown mechanism (Lovett and Mortimer 1987; Mozlin et al. 2008). Its target of action is presumed to be the Rad51 nucleoprotein filament since mating-type heterozygosity suppresses other mutations that result in Rad51 filament defects such as rad51-K191R and rad52-20, and these mutations are also suppressed by deletion of SRS2 or by overexpression of Rad51 (Schild 1995; Morgan et al. 2002; Fung et al. 2006). The DSBR defect of rad55 and rad57 mutants is cold sensitive (Lovett and Mortimer 1987; Symington 2002). Cold sensitivity is a property often associated with proteins composed of multiple subunits or large multi-protein complexes (Scheraga et al. 1962), consistent with a role for the Rad51 paralogs in stabilizing Rad51 nucleoprotein filaments. Together, these data support the proposed role for Rad55 and Rad57 as accessory factors for Rad51 during the initiation of recombination.
Vertebrates encode five Rad51 paralogs: Rad51B, Rad51C, Rad51D, Xrcc2, and Xrcc3. Mutations in genes encoding the Rad51 paralogs in chicken DT40 cells confer defects in DSB-induced homologous recombination and result in spontaneous chromosomal aberrations, high sensitivity to DNA crosslinking agents, and decreased Rad51 focus formation upon exposure to IR (Takata et al. 2001). Overexpression of human Rad51 suppresses the sensitivity of the DT40 Rad51 paralog-defective cell lines to DNA crosslinking agents, consistent with their function as accessory proteins for Rad51 (Takata et al. 2001). However, studies in mammalian cells suggest that there could be a later function for the Rad51 paralogs, possibly involving Holliday junction resolution. The human Rad51B-Rad51C-Rad51D-Xrcc2 complex preferentially binds to branched DNA substrates, including a synthetic Holliday junction substrate, over other DNA substrates (Yokoyama et al. 2004). XRCC3- and RAD51C-defective cell lines exhibit longer-than-normal gene conversion tract lengths, which could result from defects in the resolution of recombination intermediates or be due to a different mode of recombination by the Rad51-independent pathway (Brenneman et al. 2002; Nagaraju et al. 2006; Pohl and Nickoloff 2008). Extracts made from XRCC3- or RAD51C-defective Chinese hamster ovary cells show reduced levels of Holliday junction resolvase activity (Liu et al. 2004). Furthermore, Rad51C colocalizes with the mismatch repair protein Mlh1, which serves as a marker for the later pachytene/diplotene stages during meiosis, and both Rad51C and Xrcc3 associate with the pseudoautosomal region, a crossover hotspot during meiosis (Liu et al. 2007).
To determine whether the function of Rad55 and Rad57 is limited to the initiation of recombination, we used a combination of rad55 and rad57 suppressors that are thought to function by promoting Rad51 filament function to see if these suppressors can make a competent filament that can fully suppress rad55 and rad57 defects. We show almost complete suppression of the Rad51 recruitment and DSBR defects of the rad57 mutant by combining the suppressors, which suggests that the primary function of Rad55-Rad57 in DSBR is recruitment and/or stabilization of Rad51. However, the same combination of suppressors that suppresses the DSB-induced gene conversion defect of rad57 does not suppress the spontaneous recombination defect, suggesting that Rad55-Rad57 has a role in spontaneous recombination that is distinct from its role in DSBR.
MATERIALS AND METHODS
Media, growth conditions, and genetic methods:
Rich medium [yeast extract–peptone–dextrose (YPD)], synthetic complete medium (SC) lacking the appropriate amino acids or nucleic acid bases, sporulation medium, and genetic methods were as described previously (Sherman et al. 1986). Synthetic deficient medium containing 2% raffinose and supplemented with adenine, uracil, histidine, and leucine was used for the galactose induction of I-SCEI in the direct-repeat recombination assays.
Yeast strains and plasmids:
S. cerevisiae strains used in this study are listed in Table 1. All strains are in the W303 background (his3-11,15 leu2-3,112 trp1-1 ura3-1 ade2-1 can1-100) except those listed as BY4742 and LSY1786 (Zou and Rothstein 1997). The yellow fluorescent protein (YFP) fusion strains were made by crossing the appropriate haploid parents, sporulating the resulting diploids, and screening the haploid progeny for the correct phenotype; the expression of YFP was confirmed by epifluorescence microscopy. To construct LSY1957-1 and LSY2004-1, pRS406-rad51-I345T was cut with Bsu36I and transformed into the YFP-RAD51 strain W5857-2C or into the YFP-RAD51 rad57 strain LSY1956-8B, creating a repeat. The resulting Ura+ transformants were screened for the presence of the rad51-I345T allele fused to YFP by PCR and for restriction digestion to detect the novel HpaII restriction site introduced by the allele. The second RAD51 allele in the repeat formed by integration lacks a promoter and is not expressed (Fung et al. 2006). Haploid strains expressing both mating types were made by transforming haploids with the opposite mating-type allele on the pRS414 vector. SIR4, DNL4, and RAD55 deletion strains were made by a one-step gene replacement of the relevant locus with a linear PCR fragment containing homologous 5′ and 3′ flanking sequences and the KanMX4 selectable marker from the appropriate BY4742 deletion strain. LSY1786 was constructed by one-step gene replacement of RAD55 in BY4742 dnl4∷KanMX4 with a linear PCR fragment containing homologous 5′ and 3′ flanking sequences and LEU2 from YHK597-2B. Most other haploid strains were made by mating appropriate haploid strains, sporulating the resulting diploids, and screening the haploid segregants for the desired genotype. Unless otherwise indicated, null alleles of all genes were used in these studies.
Yeast strains
Strain | Genotype | Source or reference |
|---|---|---|
| W1588-4C | MATa | Zou and Rothstein (1997) |
| W1588-4A | MATα | Zou and Rothstein (1997) |
| W5857-2C | MATa ADE2 YFP-RAD51 | Lisby et al. (2004) |
| YHK597-2B | MATa rad55∷LEU2 | H. Klein |
| YHK598-8B | MATα rad57∷LEU2 | H. Klein |
| YHK1186-5C | MATα dnl4∷URA3 | H. Klein |
| LSY1421-2A | MATα srs2∷HIS3 ade2-n∷URA3∷ade2-a | This study |
| LSY1422-2A | MATα rad57∷LEU2 ade2-n∷URA3∷ade2-a rad5-535 | This study |
| LSY1422-6B | MATα rad57∷LEU2 srs2∷HIS3 ade2-n∷URA3∷ade2-a | This study |
| LSY1519-1D | MATα ade2-n∷TRP1∷ade2-I | Mozlin et al. (2008) |
| LSY1682-1B | MATα rad55∷LEU2 dnl4∷URA3 | This study |
| LSY1786 | MATα dnl4∷KanMX4 rad55∷LEU2 his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 | This study |
| LSY1827-1D | MATα ADE2 rad51-I345T srs2∷HIS3 | This study |
| LSY1867 | MATa ADE2 srs2∷HIS3 | This study |
| LSY1892 | MATα ade2-n∷URA3∷ade2-a | This study |
| LSY1894-3B | MATα rad57∷LEU2 ade2-n∷URA3∷ade2-a | This study |
| LSY1895 | MATa rad57∷LEU2 rad51-I345T ade2-n∷URA3∷ade2-a | This study |
| LSY1896-1A | MATα rad57∷LEU2 rad51-I345T srs2∷HIS3 ade2-n∷URA3∷ade2-a | This study |
| LSY1898 | MATα srs2∷HIS3 sir4∷KanMX4 ade2-n∷URA3∷ade2-a | This study |
| LSY1900 | MATα rad57∷LEU2 srs2∷HIS3 sir4∷KanMX4 ade2-n∷URA3∷ade2-a | This study |
| LSY1956-8B | MATa ADE2 YFP-RAD51 rad57∷LEU2 | This study |
| LSY1957-1 | MATa ADE2 YFP-rad51-I345T-URA3-RAD51 | This study |
| LSY2004-1 | MATα ADE2 YFP-rad51-I345T-URA3-RAD51 rad57∷LEU2 | This study |
| LSY2005-8A | MATα ADE2 YFP-rad51-I345T-URA3-RAD51 rad57∷LEU2 srs2∷HIS3 | This study |
| LSY2029 | MATα rad57∷URA3 ade2-I dnl4∷KanMX4 | This study |
| LSY2032-1C | MATa ade2-n∷TRP1∷ade2-I lys2∷GAL-I-SCEI rad57∷LEU2 srs2∷HphMX4 sir4∷KanMX4 | This study |
| LSY2032-10C | MATa ade2-n∷TRP1∷ade2-I lys2∷GAL-I-SCEI | This study |
| LSY2032-12A | MATa ade2-n∷TRP1∷ade2-I lys2∷GAL-I-SCEI rad57∷LEU2 | This study |
| LSY2086-2B | MATα ADE2 YFP-rad51-I345T-URA3-RAD51 srs2∷HIS3 | This study |
| LSY2113-4 | MATα sir4∷KanMX4 srs2∷HIS3 ade2∷TRP1∷ade2-I lys2∷GAL-I-SCEI | This study |
| BY4742 dnl4∷KanMX4 | MATα dnl4∷KanMX4 his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 | Winzeler et al. (1999) |
| BY4742 sir4∷KanMX4 | MATα sir4∷KanMX4 his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 | Winzeler et al. (1999) |
| BY4742 rad55∷KanMX4 | MATα rad55∷KanMX4 his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 | Winzeler et al. (1999) |
Strain | Genotype | Source or reference |
|---|---|---|
| W1588-4C | MATa | Zou and Rothstein (1997) |
| W1588-4A | MATα | Zou and Rothstein (1997) |
| W5857-2C | MATa ADE2 YFP-RAD51 | Lisby et al. (2004) |
| YHK597-2B | MATa rad55∷LEU2 | H. Klein |
| YHK598-8B | MATα rad57∷LEU2 | H. Klein |
| YHK1186-5C | MATα dnl4∷URA3 | H. Klein |
| LSY1421-2A | MATα srs2∷HIS3 ade2-n∷URA3∷ade2-a | This study |
| LSY1422-2A | MATα rad57∷LEU2 ade2-n∷URA3∷ade2-a rad5-535 | This study |
| LSY1422-6B | MATα rad57∷LEU2 srs2∷HIS3 ade2-n∷URA3∷ade2-a | This study |
| LSY1519-1D | MATα ade2-n∷TRP1∷ade2-I | Mozlin et al. (2008) |
| LSY1682-1B | MATα rad55∷LEU2 dnl4∷URA3 | This study |
| LSY1786 | MATα dnl4∷KanMX4 rad55∷LEU2 his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 | This study |
| LSY1827-1D | MATα ADE2 rad51-I345T srs2∷HIS3 | This study |
| LSY1867 | MATa ADE2 srs2∷HIS3 | This study |
| LSY1892 | MATα ade2-n∷URA3∷ade2-a | This study |
| LSY1894-3B | MATα rad57∷LEU2 ade2-n∷URA3∷ade2-a | This study |
| LSY1895 | MATa rad57∷LEU2 rad51-I345T ade2-n∷URA3∷ade2-a | This study |
| LSY1896-1A | MATα rad57∷LEU2 rad51-I345T srs2∷HIS3 ade2-n∷URA3∷ade2-a | This study |
| LSY1898 | MATα srs2∷HIS3 sir4∷KanMX4 ade2-n∷URA3∷ade2-a | This study |
| LSY1900 | MATα rad57∷LEU2 srs2∷HIS3 sir4∷KanMX4 ade2-n∷URA3∷ade2-a | This study |
| LSY1956-8B | MATa ADE2 YFP-RAD51 rad57∷LEU2 | This study |
| LSY1957-1 | MATa ADE2 YFP-rad51-I345T-URA3-RAD51 | This study |
| LSY2004-1 | MATα ADE2 YFP-rad51-I345T-URA3-RAD51 rad57∷LEU2 | This study |
| LSY2005-8A | MATα ADE2 YFP-rad51-I345T-URA3-RAD51 rad57∷LEU2 srs2∷HIS3 | This study |
| LSY2029 | MATα rad57∷URA3 ade2-I dnl4∷KanMX4 | This study |
| LSY2032-1C | MATa ade2-n∷TRP1∷ade2-I lys2∷GAL-I-SCEI rad57∷LEU2 srs2∷HphMX4 sir4∷KanMX4 | This study |
| LSY2032-10C | MATa ade2-n∷TRP1∷ade2-I lys2∷GAL-I-SCEI | This study |
| LSY2032-12A | MATa ade2-n∷TRP1∷ade2-I lys2∷GAL-I-SCEI rad57∷LEU2 | This study |
| LSY2086-2B | MATα ADE2 YFP-rad51-I345T-URA3-RAD51 srs2∷HIS3 | This study |
| LSY2113-4 | MATα sir4∷KanMX4 srs2∷HIS3 ade2∷TRP1∷ade2-I lys2∷GAL-I-SCEI | This study |
| BY4742 dnl4∷KanMX4 | MATα dnl4∷KanMX4 his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 | Winzeler et al. (1999) |
| BY4742 sir4∷KanMX4 | MATα sir4∷KanMX4 his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 | Winzeler et al. (1999) |
| BY4742 rad55∷KanMX4 | MATα rad55∷KanMX4 his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 | Winzeler et al. (1999) |
Yeast strains
Strain | Genotype | Source or reference |
|---|---|---|
| W1588-4C | MATa | Zou and Rothstein (1997) |
| W1588-4A | MATα | Zou and Rothstein (1997) |
| W5857-2C | MATa ADE2 YFP-RAD51 | Lisby et al. (2004) |
| YHK597-2B | MATa rad55∷LEU2 | H. Klein |
| YHK598-8B | MATα rad57∷LEU2 | H. Klein |
| YHK1186-5C | MATα dnl4∷URA3 | H. Klein |
| LSY1421-2A | MATα srs2∷HIS3 ade2-n∷URA3∷ade2-a | This study |
| LSY1422-2A | MATα rad57∷LEU2 ade2-n∷URA3∷ade2-a rad5-535 | This study |
| LSY1422-6B | MATα rad57∷LEU2 srs2∷HIS3 ade2-n∷URA3∷ade2-a | This study |
| LSY1519-1D | MATα ade2-n∷TRP1∷ade2-I | Mozlin et al. (2008) |
| LSY1682-1B | MATα rad55∷LEU2 dnl4∷URA3 | This study |
| LSY1786 | MATα dnl4∷KanMX4 rad55∷LEU2 his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 | This study |
| LSY1827-1D | MATα ADE2 rad51-I345T srs2∷HIS3 | This study |
| LSY1867 | MATa ADE2 srs2∷HIS3 | This study |
| LSY1892 | MATα ade2-n∷URA3∷ade2-a | This study |
| LSY1894-3B | MATα rad57∷LEU2 ade2-n∷URA3∷ade2-a | This study |
| LSY1895 | MATa rad57∷LEU2 rad51-I345T ade2-n∷URA3∷ade2-a | This study |
| LSY1896-1A | MATα rad57∷LEU2 rad51-I345T srs2∷HIS3 ade2-n∷URA3∷ade2-a | This study |
| LSY1898 | MATα srs2∷HIS3 sir4∷KanMX4 ade2-n∷URA3∷ade2-a | This study |
| LSY1900 | MATα rad57∷LEU2 srs2∷HIS3 sir4∷KanMX4 ade2-n∷URA3∷ade2-a | This study |
| LSY1956-8B | MATa ADE2 YFP-RAD51 rad57∷LEU2 | This study |
| LSY1957-1 | MATa ADE2 YFP-rad51-I345T-URA3-RAD51 | This study |
| LSY2004-1 | MATα ADE2 YFP-rad51-I345T-URA3-RAD51 rad57∷LEU2 | This study |
| LSY2005-8A | MATα ADE2 YFP-rad51-I345T-URA3-RAD51 rad57∷LEU2 srs2∷HIS3 | This study |
| LSY2029 | MATα rad57∷URA3 ade2-I dnl4∷KanMX4 | This study |
| LSY2032-1C | MATa ade2-n∷TRP1∷ade2-I lys2∷GAL-I-SCEI rad57∷LEU2 srs2∷HphMX4 sir4∷KanMX4 | This study |
| LSY2032-10C | MATa ade2-n∷TRP1∷ade2-I lys2∷GAL-I-SCEI | This study |
| LSY2032-12A | MATa ade2-n∷TRP1∷ade2-I lys2∷GAL-I-SCEI rad57∷LEU2 | This study |
| LSY2086-2B | MATα ADE2 YFP-rad51-I345T-URA3-RAD51 srs2∷HIS3 | This study |
| LSY2113-4 | MATα sir4∷KanMX4 srs2∷HIS3 ade2∷TRP1∷ade2-I lys2∷GAL-I-SCEI | This study |
| BY4742 dnl4∷KanMX4 | MATα dnl4∷KanMX4 his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 | Winzeler et al. (1999) |
| BY4742 sir4∷KanMX4 | MATα sir4∷KanMX4 his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 | Winzeler et al. (1999) |
| BY4742 rad55∷KanMX4 | MATα rad55∷KanMX4 his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 | Winzeler et al. (1999) |
Strain | Genotype | Source or reference |
|---|---|---|
| W1588-4C | MATa | Zou and Rothstein (1997) |
| W1588-4A | MATα | Zou and Rothstein (1997) |
| W5857-2C | MATa ADE2 YFP-RAD51 | Lisby et al. (2004) |
| YHK597-2B | MATa rad55∷LEU2 | H. Klein |
| YHK598-8B | MATα rad57∷LEU2 | H. Klein |
| YHK1186-5C | MATα dnl4∷URA3 | H. Klein |
| LSY1421-2A | MATα srs2∷HIS3 ade2-n∷URA3∷ade2-a | This study |
| LSY1422-2A | MATα rad57∷LEU2 ade2-n∷URA3∷ade2-a rad5-535 | This study |
| LSY1422-6B | MATα rad57∷LEU2 srs2∷HIS3 ade2-n∷URA3∷ade2-a | This study |
| LSY1519-1D | MATα ade2-n∷TRP1∷ade2-I | Mozlin et al. (2008) |
| LSY1682-1B | MATα rad55∷LEU2 dnl4∷URA3 | This study |
| LSY1786 | MATα dnl4∷KanMX4 rad55∷LEU2 his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 | This study |
| LSY1827-1D | MATα ADE2 rad51-I345T srs2∷HIS3 | This study |
| LSY1867 | MATa ADE2 srs2∷HIS3 | This study |
| LSY1892 | MATα ade2-n∷URA3∷ade2-a | This study |
| LSY1894-3B | MATα rad57∷LEU2 ade2-n∷URA3∷ade2-a | This study |
| LSY1895 | MATa rad57∷LEU2 rad51-I345T ade2-n∷URA3∷ade2-a | This study |
| LSY1896-1A | MATα rad57∷LEU2 rad51-I345T srs2∷HIS3 ade2-n∷URA3∷ade2-a | This study |
| LSY1898 | MATα srs2∷HIS3 sir4∷KanMX4 ade2-n∷URA3∷ade2-a | This study |
| LSY1900 | MATα rad57∷LEU2 srs2∷HIS3 sir4∷KanMX4 ade2-n∷URA3∷ade2-a | This study |
| LSY1956-8B | MATa ADE2 YFP-RAD51 rad57∷LEU2 | This study |
| LSY1957-1 | MATa ADE2 YFP-rad51-I345T-URA3-RAD51 | This study |
| LSY2004-1 | MATα ADE2 YFP-rad51-I345T-URA3-RAD51 rad57∷LEU2 | This study |
| LSY2005-8A | MATα ADE2 YFP-rad51-I345T-URA3-RAD51 rad57∷LEU2 srs2∷HIS3 | This study |
| LSY2029 | MATα rad57∷URA3 ade2-I dnl4∷KanMX4 | This study |
| LSY2032-1C | MATa ade2-n∷TRP1∷ade2-I lys2∷GAL-I-SCEI rad57∷LEU2 srs2∷HphMX4 sir4∷KanMX4 | This study |
| LSY2032-10C | MATa ade2-n∷TRP1∷ade2-I lys2∷GAL-I-SCEI | This study |
| LSY2032-12A | MATa ade2-n∷TRP1∷ade2-I lys2∷GAL-I-SCEI rad57∷LEU2 | This study |
| LSY2086-2B | MATα ADE2 YFP-rad51-I345T-URA3-RAD51 srs2∷HIS3 | This study |
| LSY2113-4 | MATα sir4∷KanMX4 srs2∷HIS3 ade2∷TRP1∷ade2-I lys2∷GAL-I-SCEI | This study |
| BY4742 dnl4∷KanMX4 | MATα dnl4∷KanMX4 his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 | Winzeler et al. (1999) |
| BY4742 sir4∷KanMX4 | MATα sir4∷KanMX4 his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 | Winzeler et al. (1999) |
| BY4742 rad55∷KanMX4 | MATα rad55∷KanMX4 his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 | Winzeler et al. (1999) |
To construct pRS406-rad51-I345T, a PCR fragment containing the rad51-I345T complete open reading frame was made using pRS413-rad51-I345T (Fortin and Symington 2002) as the PCR template. The PCR fragment was cloned into the pGEM-T Easy vector (Promega, Madison, WI) and subsequently cloned into the pRS406 vector using the SacI and NotI restriction sites. pRS414-MATa and pRS414-MATα were gifts from R. Rothstein.
Microscopy:
Cells were grown in SC or SC–TRP liquid medium until an optical density at 600 nm (OD600) of 0.2, at which time the liquid cultures were exposed to the defined doses of radiation in a Gammacell-220 irradiator containing 60Co, or treated with camptothecin (CPT) at a concentration of 7 μg/ml for 3 hr. Aliquots of the cultures were processed immediately for imaging as described by Lisby et al. (2001). Live cell images were captured as described (Lisby et al. 2004). YFP fluorescence was acquired using Openlab software (Improvision) and quantified using Volocity software (Improvision). For each set of strains shown in Figures 3 and 6, samples were processed at the same time because of day-to-day variation in focus brightness.
Clastogen sensitivity tests:
For IR sensitivity tests, cells were grown in liquid medium to mid-log phase at 23° or 30°. The cultures were serially diluted, and aliquots of each 10-fold dilution were spotted onto YPD or SC–TRP plates. The plates were left unirradiated or irradiated in a Gammacell-220 irradiator containing 60Co for the designated dose and then incubated for 3 or 5 days at 30° or 23°, respectively. For CPT sensitivity tests, cells were grown in SC–TRP overnight at 30° or 23°. Strains were diluted to a concentration of 7 × 106 cells/ml and then 10-fold serially diluted and spotted onto SC–TRP plates or onto SC–TRP plates containing the specified concentration of CPT buffered with 0.25% dimethyl sulfoxide (DMSO). A stock solution was made by dissolving CPT in DMSO at 1 mg/ml. Control plates contained 0.25% DMSO. The plates were incubated for 3 or 5 days at 30° or 23°, respectively.
DSB-induced gene conversion assay:
SC–TRP glucose cultures (5 ml) were grown overnight at 30°. Cells were diluted to a concentration of 1 × 105 cells/ml in 300 ml SC–TRP with raffinose replacing glucose. Cultures were grown overnight to a concentration of 3 × 106 cells/ml, and galactose was added to the cultures for a final concentration of 2%. Fifty milliliters of cells were harvested at each indicated time point after galactose induction. DNA was isolated from each time point and used as template for PCR with the following primers amplifying the 2.5-kb region encompassing the ade2-I allele: 5GCA 5′-GTTGTGTGGAATTGTGAGCG-3′ and 3GCA 5′-CGCCATACTGGAGGCAATAA-3′. PCR in the linear range was performed using 25 cycles and 5 ng of genomic DNA. Control primers amplifying 1.8 kb of chromosome IV (TRP1 locus) were used in a PCR reaction with the ade2 reporter primers to normalize the amount of PCR product. Control primers used were 5QTrp1 5′-CACGGCAGAGACCAATCAGTA-3′ and 3QTrp1 5′-GCACTCCTGATTCCGCTAATA-3′. PCR products were analyzed on a 1.5% agarose gel. To amplify repaired products, PCR was performed for 35 cycles and with 500 ng template DNA. PCR products were digested with AatII and analyzed on a 1.8% agarose gel.
Determination of spontaneous mitotic recombination rates:
Mitotic recombination rates between ade2 direct repeats were determined as described previously by Mozlin et al. (2008).
RESULTS
YFP-rad51-I345T is functional:
Previous studies have shown a defect in the formation and/or brightness of DSB-induced Rad51 foci in rad55 or rad57 mutants (Gasior et al. 1998, 2001; Mozlin et al. 2008). To determine whether the defect in Rad51 recruitment to DSBs observed in the rad57 mutant could be rescued by factors thought to act at the level of the Rad51 filament—namely elevated temperature, deletion of SRS2, rad51-I345T and expression of both mating-type alleles—we monitored Rad51 foci formation by epifluorescence microscopy. To study the suppression conferred by the rad51-I345T allele, the endogenous Rad51-I345T protein was tagged with YFP. First, a strain expressing this fusion protein in a RAD57 background was tested for IR sensitivity and foci formation. Several groups have noted previously that terminally tagged versions of Rad51 are not fully functional (Lisby et al. 2004; Kojic et al. 2005). Surprisingly, the YFP-rad51-I345T strain was almost as IR resistant as the untagged RAD51 strain, in contrast to the YFP-RAD51 strain (Figure 1A). Although the number of cells with IR-induced Rad51 foci was comparable for the YFP-RAD51 and YFP-rad51-I345T strains, the foci formed by the YFP-Rad51 fusion protein were on average 1.5 times brighter than the IR-induced foci formed by the more functional YFP-Rad51-I345T protein (Figure 1B). The brighter YFP-Rad51 foci could be a result of inappropriate recruitment, retention, aggregation, or turnover of YFP-Rad51 fusion protein.
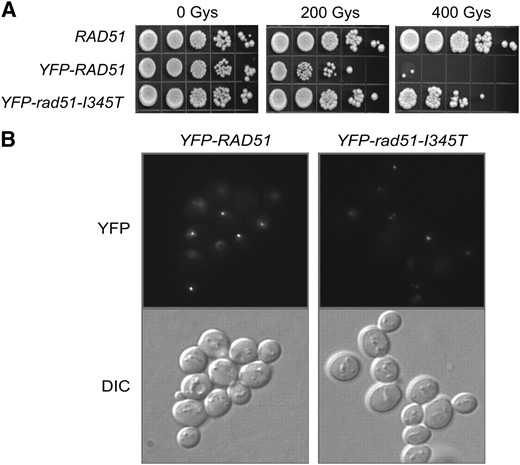
YFP-rad51-I345T is more functional in vivo than YFP-Rad51. (A) Serial dilutions of log-phase cultures of LSY1519-1D (RAD), W5857-2C (YFP-RAD51), and LSY1957-1 (YFP-rad51-I345T) were spotted onto YPD plates and left unirradiated or irradiated at 200 and 400 Gy. Survival was assessed following growth for 3 days at 30°. (B) Log-phase cultures of strains were exposed to 200 Gy of γ-irradiation, followed by microscopy to monitor focus formation.
The Rad51 foci formation defect of the rad57 mutant strain can be suppressed:
Rad51-I345T focus formation was monitored following treatment of cells with CPT (7 μg/ml) for 3 hr. CPT stabilizes the covalent DNA-Top1 intermediate that forms during the catalytic DNA nicking–closing cycle of Top1, and these stable nicks can then be converted into recombinogenic DSBs during replication (Hsiang et al. 1989). CPT was used instead of IR to maintain cells at a constant temperature throughout the experiment. For this analysis, we measured two parameters: the number of CPT-induced foci and the foci brightness. At 30° in the wild-type RAD57 background, YFP-Rad51-I345T formed at least one focus in 91% of the cells, whereas, in the rad57 mutant strain, only 26% of the cells had at least one YFP-Rad51-I345T focus (Figure 2B). In addition, the brightness of the YFP-Rad51-I345T foci in the rad57 mutant strain was only 31% of the brightness of the foci in the YFP-rad51-I345T RAD57 strain (Figure 2C). When the suppressors srs2 and MAT heterozygosity were combined with the rad51-I345T allele in the rad57 strain, 90% of cells had at least one focus at 30°, which is comparable to wild type. Although the brightness of the foci in the rad57-suppressed strain was increased to 90% of the RAD57 YFP-rad51-I345T strain, suppression was not complete (P = 0.0012). The degree of suppression of foci brightness was temperature dependent; at 23°, the brightness of the YFP-Rad51-I345T foci in the rad57 suppressor strain was only 73% of the brightness of the foci in the YFP-rad51-I345T strain, in contrast to the 90% brightness of YFP-Rad51-I345T foci in the rad57 suppressor strain at 30° (P = 0.0001) (Figure 2C). In the wild-type RAD57 background, YFP-Rad51-I345T formed at least one focus in 89% of the cells at 23°, whereas the rad57-suppressed strain had no defect with 86% of the cells having at least one YFP-Rad51-I345T focus (P = 0.08), suggesting that suppression of the number of foci formed is not temperature dependent (Figure 2B).
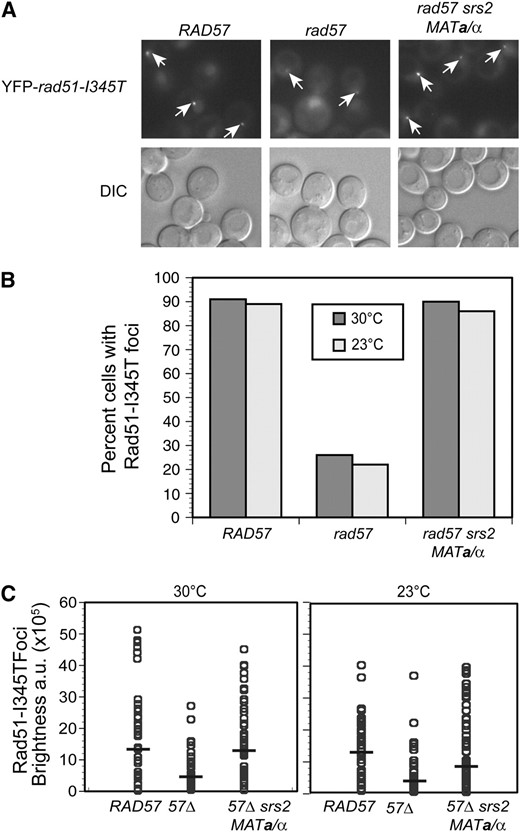
The combination of the suppressors srs2 and mating-type heterozygosity almost fully suppresses the Rad51 foci formation defect of rad57 in response to treatment with camptothecin. (A) To express both mating types, LSY2005-8A (MATα YFP-rad51-I345T rad57∷LEU2 srs2∷HIS3) was transformed with pRS414-MATa whereas LSY1957-1 (RAD57) and LSY2004-1 (rad57∷LEU2) were transformed with the pRS414 empty vector. Log-phase SC–TRP cultures of strains were treated with camptothecin at a concentration of 7 μg/ml for 3 hr at 30° or 23° followed by microscopy to monitor focus formation. (B) The percentage of cells with at least one focus was calculated; at least 100 cells were analyzed for each strain. (C) The mean focus brightness was normalized for each strain relative to LSY1957-1 (RAD57 YFP-rad51-I345T). The brightness of each focus was quantified and plotted; at least 80 foci were analyzed for each strain. A solid bar represents the mean focus brightness for each strain; a.u. represents arbitrary units.
The IR and CPT sensitivity of the rad57 null strain can be fully rescued by combining partial suppressors of the rad57 mutant:
A prediction from the above finding is that, if Rad55 and Rad57 function solely in recruitment and/or stabilization of Rad51 at damaged sites, then combinations of partial suppressors of rad55 and rad57 that suppress the Rad51 foci defect should additively suppress the sensitivity of rad55 and rad57 mutants to genotoxic agents. If, however, Rad55-Rad57 has a function independent of Rad51 filament formation, then suppressing the Rad51 recruitment defect of a rad57 strain would not be expected to suppress cell survival after DNA damage. The DNA repair defect of rad57 strains with all combinations of suppressors was assessed by the plating efficiency at 30° and 23° after exposure to IR or on medium containing CPT. DSBs are the toxic lesion generated by IR or CPT exposure.
Of the single suppressors, MAT heterozygosity showed the greatest suppression of the IR sensitivity of the rad57 mutant, and, when combined with srs2 or rad51-I345T, there was additive suppression (Figure 3). We found that there was almost full suppression of the IR and CPT sensitivity of the rad57 mutant when at least three of the four suppressors were combined. Because the srs2 mutation also confers sensitivity to IR and CPT, it appears that the suppression of the rad57 strain with all of the suppressors is complete. At both 23° and 30°, the rad57 srs2 rad51-I345T MATa/α strain showed equal or higher resistance than the srs2 single-mutant strain. Fewer than three suppressors resulted in incomplete suppression, especially at 23° (Figure 3).
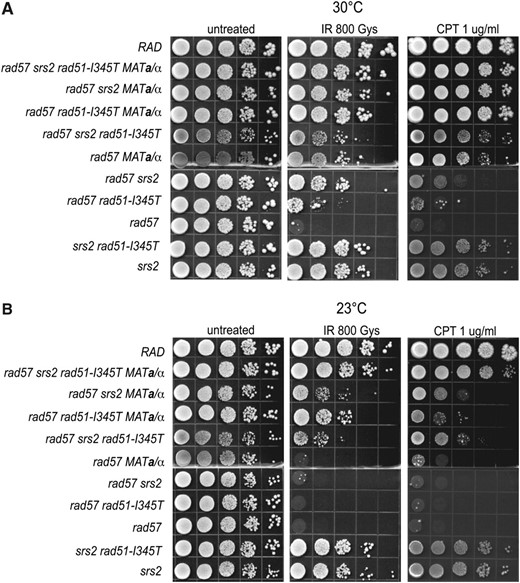
Combining known suppressors of rad57 fully suppresses the sensitivity of rad57 to DSB-inducing genotoxic agents. (A) LSY1519-1D (RAD), LSY1896-1A (rad57∷LEU2 srs2∷HIS3 rad51-I345T), LSY1422-6B (rad57∷LEU2 srs2∷HIS3), LSY1895 (rad57∷LEU2 rad51-I345T), LSY 1894-3B (rad57∷LEU2), LSY1827-1D (srs2∷HIS3 rad51-I345T), and LSY1867 (srs2∷HIS3) were transformed with either pRS414-MATα or pRS414-MATa to express both mating-type alleles or pRS414 empty vector as a control. Ten-fold serial dilutions of log-phase SC–TRP cultures grown at 30° were spotted onto SC–TRP plates and left unirradiated or exposed to 800 Gy of γ-irradiation. Dilutions were also spotted onto SC–TRP plates containing 1 μg/ml camptothecin buffered with 0.25% DMSO. Survival was assessed following 3 days of growth at 30°. (B) The same experiment as in A done at 23°.
Suppression of the DSB-induced gene conversion defect of the rad57 mutant:
Although we observed suppression of the IR and CPT sensitivity of the rad57 mutant, the survival assays offer insight only into the end result of repair: the ability to complete repair and form a colony or cell death. Because the survival assays are uninformative in regards to the timing of recombination, we used a DSB-induced gene conversion assay to monitor the kinetics of repair. The substrate used contains a direct repeat of alleles of the ade2 gene separated by vector sequences and a copy of the TRP1 gene integrated at the endogenous ADE2 locus (Mozlin et al. 2008) (Figure 4A). One allele contains a 2-bp fill-in mutation of the NdeI site (Huang and Symington 1994); the other allele contains an insertion of the I-SceI nuclease recognition site disrupting the AatII site. The strains also contain a fusion of the I-SCEI gene to the GAL1 promoter to provide regulated expression of the nuclease. After inducing I-SceI expression at 30°, collecting cells at appointed time points, and isolating genomic DNA, gene conversion events can be detected by PCR with primers that anneal to the vector sequence upstream of the ade2-I allele and to the ade2 sequence downstream of the I-SceI cut site. Uncut DNA or gene conversion using the ade2-n as the donor will produce a PCR product, whereas events repaired by single-strand annealing or unrepaired DNA will not (Figure 4, A and B). By quantitative PCR, the suppressed rad57 strain displayed increased PCR product at the later time points, comparable to wild type, but the rad57 single mutant did not (Figure 4B). If the I-SceI-induced DSB is repaired to the wild-type AatII sequence, the PCR product should be digested with AatII and the percentage of gene conversion calculated from the ratio of AatII− to AatII+ DNA (Figure 4, D and E). Nonlinear PCR was performed to obtain sufficient product for restriction digestion (see materials and methods). The percentage of gene conversion was then normalized to the amount of PCR product from the quantitative PCR. In a wild-type strain, 45% of the amplified DNA was AatII+ by 6 hr post-induction, and gene conversion plateaued after 8 hr at 80% (Figure 4, D and E). Compared to wild type, the rad57 single mutant was strongly defective in gene conversion with only 0.8% repair after 24 hr (Figure 4, D and E). The rad57 mutant combined with srs2 and sir4 [to eliminate silencing of the HMRa and HMLα loci (Rine and Herskowitz 1987)] displayed increased gene conversion and faster kinetics compared to the rad57 single mutant, but the timing of repair was slower compared to wild type. The rad57 suppressor strain had only 30% of repair at 6 hr post-induction but approached the wild-type level of repair at the 24-hr time point with 72% gene conversion (Figure 4, D and E). Consistent with previous results (Aylon et al. 2003), the srs2 sir4 mutant showed reduced efficiency of repair compared with wild type and was even slightly lower than the rad57 srs2 sir4 strain. Thus, the DSB-induced gene conversion defect of the rad57 mutant can be fully suppressed by srs2 and MAT heterozygosity.
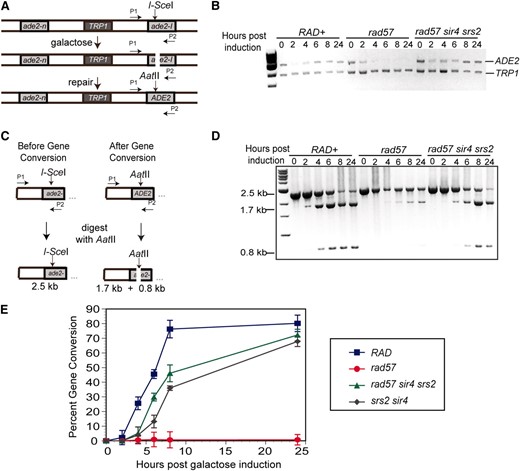
The DSB-induced gene conversion defect of rad57 is suppressed by srs2 and MAT heterozygosity. (A) The direct-repeat recombination substrate contains 3.6-kb repeats with different ade2 alleles integrated at the endogenous locus on chromosome XV separated by plasmid sequences and the TRP1 gene. Upon galactose induction, I-SceI is expressed and makes a DSB within the ade2-I allele. Primers were designed to anneal upstream of the ade2-I allele within vector sequences as well as downstream of the I-SceI cut site. (B) Quantitative PCR. Uncut DNA or gene conversion using ade2-n as the donor will result in a PCR product, whereas single-strand annealing or unrepaired DNA will not. The top band is the ADE2 PCR product and the bottom band is the control TRP1 PCR product. Strains used were LSY2032-10C (RAD), LSY2032-12A (rad57∷LEU2), LSY2113-4 (srs2∷HIS3 sir4∷KanMX), and LSY2032-1C (rad57∷LEU2 srs2∷HphMX sir4∷KanMX). (C) Gene conversion will restore the wild-type AatII site in the ade2-I allele. After PCR, products can be digested with AatII to monitor the kinetics of gene conversion within each strain. DNA uncut by I-SceI will result in a 2.5-kb fragment whereas gene conversion will result in 1.7- and 0.8-kb fragments. (D) PCR was performed to saturation (35 cycles), and the AatII-digested PCR products were analyzed by agarose gel electrophoresis. (E) The percentage of gene conversion was calculated as the ratio of AatII cut to uncut DNA and normalized to the amount of PCR product from the quantitative PCR (B).
The spontaneous recombination defect of rad57 is not suppressed by combining srs2 and mating-type heterozygosity:
Because the rad57 suppressed strain shows close to wild-type levels of DSB-induced gene conversion, we were interested to see if the same was true for spontaneous gene conversion between direct repeats. Previous studies showed a severe defect in spontaneous recombination between ade2 direct repeats in the rad57 mutant (Mozlin et al. 2008). Spontaneous recombination rates were determined using a ade2 repeat construct similar to the one described above, except the second allele contained a fill-in mutation of the AatII site and a URA3 marker was present between the repeats (Huang and Symington 1994). Unequal sister-chromatid or intrachromatid gene conversion between the two ade2 repeats can generate Ade+ Ura+ recombinants that retain the duplication (Figure 5A). The rate of recombination in the rad57 mutant was 1000-fold lower than in the wild-type strain (Figure 5B). As shown previously, the sir4 mutation resulted in a small, but significant, suppression of the rad57 spontaneous recombination defect (P < 0.05) (Mozlin et al. 2008). Although srs2 did not cause a significant increase in the rate of spontaneous recombination in the rad57 background, it did result in a small, but significant, suppression of the rad57 sir4 recombination defect (P < 0.05). Despite these small increases, the recombination rate of the rad57 srs2 sir4 strain was only 6-fold higher than that of rad57 and 170-fold lower than that of wild type (Figure 5B). This is the same combination of suppressors that resulted in an almost wild-type frequency of DSB-induced gene conversion in the rad57 background. As expected, the srs2 mutant showed an increased rate of spontaneous recombination compared with wild type (Aguilera and Klein 1988).
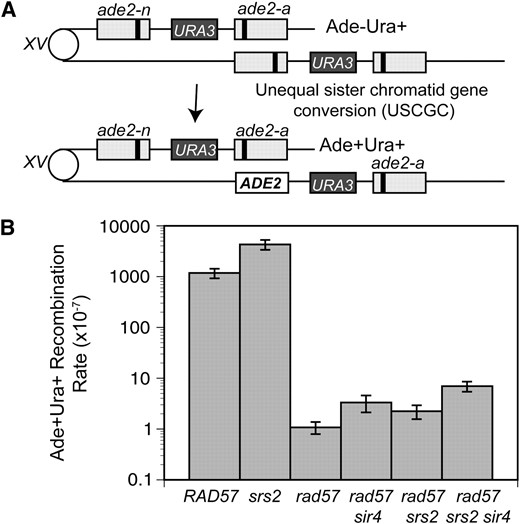
The spontaneous recombination repair defect of rad57 is weakly suppressed by combining srs2 and mating-type heterozygosity. (A) The direct-repeat recombination substrate contains 3.6-kb repeats with different ade2 alleles integrated at the endogenous locus on chromosome XV separated by plasmid sequences and the URA3 gene. Unequal sister-chromatid or intrachromatid gene conversion between the two ade2 repeats can generate Ade+Ura+ recombinants that retain the duplication. Either allele could be converted; only one type of conversion is shown here. (B) Spontaneous sister-chromatid recombination rates at 30°. Strains used were LSY1892 (RAD), LSY1894-3B (rad57∷LEU2), LSY1898 (rad57:LEU2 sir4∷kanMX), LSY1422-6B (rad57∷LEU2 srs2∷HIS3), LSY1421-2A (srs2∷HIS3), and LSY1900 (rad57∷LEU2 srs2∷HIS3 sir4∷KanMX).
The mechanism of suppression of rad55 and rad57 mutants by MAT heterozygosity is not through downregulation of SRS2 or nonhomologous end joining:
The mechanism for suppression of the rad57 mutant by srs2, overexpression of RAD51 or by rad51-I345T is most likely by increased recruitment or stabilization of Rad51 nucleoprotein filaments (Hays et al. 1995; Johnson and Symington 1995; Fortin and Symington 2002; Fung et al. 2006). However, the mechanism behind the suppression of rad55 and rad57 mutants by MAT heterozygosity has yet to be fully elucidated. We have previously shown that steady-state Rad51 protein levels are unchanged by MAT heterozygosity, suggesting that transcriptional induction of RAD51 is not the cause (Morgan et al. 2002). Furthermore, rad51-I345T fails to show greater suppression when present in high copy compared with single copy (Fortin and Symington 2002), but is additive with MAT heterozygosity in suppression of rad57 (Figure 3). Another possibility is that MAT heterozygosity decreases expression and/or functionality of SRS2 (Galitski et al. 1999). If MAT heterozygosity functioned via Srs2 regulation, then we would not have expected to see additive suppression of the IR sensitivity of the rad57 mutant by srs2 and MATa/α, contrary to our findings (Figure 3). As a further test of whether srs2 and MAT heterozygosity have the same effect on Rad51 recruitment to damaged sites, we examined IR-induced YFP-Rad51-I345T foci in both genetic backgrounds (Figure 6). Both the srs2 mutant and mating-type heterozygous strain showed an increase in brightness of YFP-Rad51-I345T foci over the MATa YFP-rad51-I345T SRS2 strain. However, the foci in the srs2 strain appeared larger and brighter compared to those in the mating-type heterozygous strain, indicating that the two suppressors have qualitatively different effects on the Rad51 recruitment or filament dynamics.
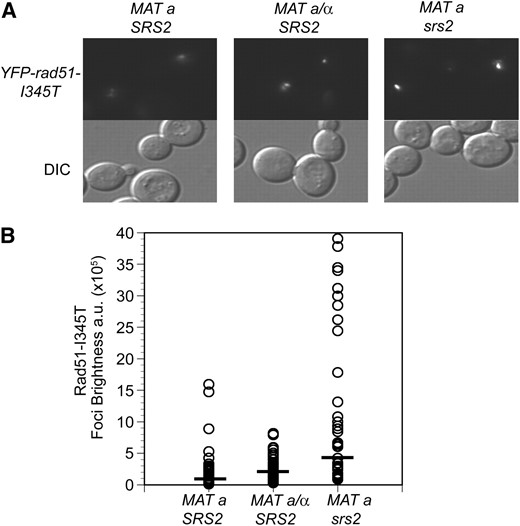
Mating-type heterozygosity and srs2 have different effects on rad51-I345T foci. (A) LSY1957-1 (MATa YFP-rad51-I345T) was transformed with pRS414 empty vector or pRS414-MATα to express both mating types, and LSY2086-2B (YFP-rad51-I345T srs2∷HIS3) was transformed with pRS414 empty vector. Log-phase cultures of the strains were exposed to 200 Gy of γ-irradiation, followed by microscopy to monitor focus formation. (B) The brightness of each focus was quantified and plotted; at least 50 foci were analyzed for each strain. A solid bar represents the mean focus brightness for each strain.
A prior report suggested that MAT heterozygosity suppresses rad55 and rad57 mutants solely through downregulation of the competing repair pathway, nonhomologous end joining (NHEJ) (Valencia-Burton et al. 2006). The haploid-specific NEJ1 gene, which is required for the NHEJ pathway of DSBR, is one of the genes repressed by the Mata1-α2 repressor (Frank-Vaillant and Marcand 2001; Kegel et al. 2001; Ooi et al. 2001; Valencia et al. 2001). However, we found that abrogation of NHEJ by deletion of DNL4, which encodes a DNA ligase essential for NHEJ, did not suppress the IR sensitivity of rad55 or rad57 in the W303 strain background (Figure 7). Because the previous study was performed with S288C derivatives, we constructed a dnl4 rad55 double mutant in the S288C background, but still failed to see suppression of the IR sensitivity conferred by rad55. Interestingly, suppression of the CPT sensitivity of the rad55 mutant by dnl4 is observed in the S288C but not the W303 background (Figure 7). This difference in suppression is probably due to strain background differences. Regardless of the strain background, it is evident that suppression of the IR sensitivity of rad55 and rad57 mutants by MAT heterozygosity is not due to downregulation of NHEJ. We tested mutations in other genes regulated by the Mata1-α2 transcriptional repressor (Galgoczy et al. 2004), but none of these was able to suppress the IR sensitivity conferred by rad57 (data not shown).
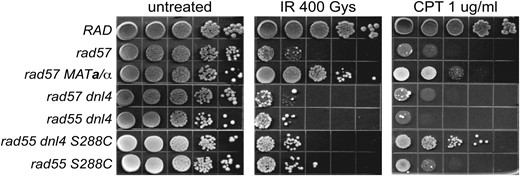
MAT heterozygosity does not suppress the IR sensitivity of rad57 by downregulating NHEJ. Strains used were W1588-4A (RAD), YHK598-8B (rad57∷LEU2), LSY2029 (rad57∷LEU2 dnl4∷KanMX), LSY1682-1B (rad55∷LEU2 dnl4∷URA3), LSY1786 (rad55∷LEU2 dnl4∷KanMX), and BY4742 rad55∷KanMX. To express both mating types, YHK598-8B (MATα rad57∷LEU2) was transformed with pRS414-MATa while the rest of the strains were transformed with the pRS414 empty vector. Ten-fold serial dilutions of log-phase cultures were spotted onto either SC–TRP-only plates or onto SC–TRP plates containing 1 μg/ml camptothecin buffered with 0.25% DMSO. SC–TRP-only plates either were left unirradiated or were exposed to 400 Gy of γ-irradiation.
DISCUSSION
Suppression of the DSBR defect of the rad57 mutant:
Several lines of evidence support a role for Rad55 and Rad57 as accessory proteins to promote nucleation or stabilization of the Rad51 nucleoprotein filament (Hays et al. 1995; Johnson and Symington 1995; Sung 1997b; Gasior et al. 1998; Fortin and Symington 2002; Sugawara et al. 2003). One important question in the field is whether the Rad51 paralogs have other roles in recombination in addition to Rad51 presynaptic filament formation. We investigated this possibility by combining partial suppressors of rad55 and rad57 that are thought to function by improving the stability or activity of the Rad51 nucleoprotein filament. The defect in the number of CPT-induced Rad51 foci (YFP-Rad51-I345T) observed in the rad57 strain was suppressed by elevated temperature, srs2, and MAT heterozygosity. Even though in the most suppressed strain the focus brightness was slightly reduced compared with the RAD57 YFP-rad51-I345T strain, the focus brightness was far greater than observed in the rad57 mutant (P = 0.0001) (Figure 2). We found an almost complete rescue of the IR and CPT sensitivity of the rad57 mutant when at least three of the suppressors were combined and the residual sensitivity reflected the innate DNA damage sensitivity conferred by the srs2 mutation (Figure 3). Furthermore, by monitoring the kinetics of DSB-induced gene conversion between direct repeats, the rad57 srs2 sir4 strain showed close to wild-type levels of recombination (72% vs. 80%), whereas the efficiency of repair in the rad57 mutant was <1%. These results show that the DSBR defect of rad57 mutants can be effectively suppressed by combining the partial suppressors. Because these suppressors are thought to act at the level of the Rad51 filament, we conclude that the main function of Rad57 in DSBR repair is in the assembly or maintenance of the Rad51 filament. However, we cannot rule out the possibility of a late function that is redundant with another protein(s).
Srs2 has been shown to dismantle Rad51-ssDNA filaments in vitro (Krejci et al. 2003; Veaute et al. 2003). This function of Srs2 is thought to be important in the context of replication fork stalling in the presence of DNA damage; removal of Rad51 by Srs2 allows repair to proceed by the error-free template-switching branch of post-replication repair instead of by recombination (Pfander et al. 2005). Consequently, srs2 suppresses the methyl methanesulfonate (MMS) sensitivity of rad6 and rad18 mutants by channeling lesions to the recombination pathway and srs2 mutants show elevated rates of spontaneous recombination (Schiestl et al. 1990; Rong et al. 1991; Robert et al. 2006). However, srs2 mutants show decreased efficiency of DSB-induced recombination and an altered outcome of these events in favor of crossovers (Aylon et al. 2003; Ira et al. 2003). The efficiency of DSB-induced recombination was lower in the sir4 srs2 strain than in wild type or the rad57 srs2 sir4 strain. Thus srs2 and rad57 show mutual suppression. Semidominant mutations of RAD51 are known to suppress the methyl methanesulfonate sensitivity of srs2 homozygous diploids (Chanet et al. 1996). One possible explanation is that Rad51 is recruited to inappropriate DNA sites in srs2 mutants and less Rad51 is available to bind to induced DNA damage, resulting in the reduced efficiency of DSBR in srs2 mutants (Osman et al. 2005). If Rad55-Rad57 mediates recruitment of Rad51 at these inappropriate sites, then in the absence of Rad57 more Rad51 would be available to bind at induced DSBs and would form a stable filament in the absence of Srs2, resulting in the observed suppression.
The spontaneous recombination defect of rad57 is poorly suppressed by srs2 and MAT heterozygosity:
We previously demonstrated a severe defect in the rate of spontaneous gene conversion between ade2 alleles oriented as direct repeats in rad55 and rad57 mutants. Surprisingly, this recombination defect was not suppressed by temperature and was only weakly (3-fold) suppressed by MAT heterozygosity or by overexpression of Rad51 (Mozlin et al. 2008). Here we show additive suppression of the rad57 recombination defect by combining MAT heterozygosity and srs2 at 30°, but the rate is still 170-fold less than that observed in wild type (Figure 5); these same suppressors fully suppress the DSB-induced gene conversion defect of the rad57 mutant.
We assume that most spontaneous recombination events occur during S-phase. Because the phenotype of the suppressed rad57 mutant in the spontaneous recombination assay is different from that observed for DSB-induced recombination at the same locus, we suggest that spontaneous recombination initiates at ssDNA gaps formed during DNA synthesis. The observation that srs2 and MATa/α strongly suppress the sensitivity of the rad57 mutant to CPT, which makes DSBs in the context of the replication fork, further supports the idea that collapsed replication forks are not the primary initiating lesion for spontaneous recombination. A block to leading-strand synthesis is thought to result in uncoupling of leading and lagging strands and in formation of ssDNA at the replication fork. Srs2 may act to prevent Rad51 binding to these structures, thereby promoting fork reversal and lesion repair or bypass. Single-stranded gaps that form on the lagging strand or following restart of the leading strand downstream of the lesion are bypassed by template switching, homologous recombination, or error-prone translesion DNA synthesis. These pathways appear to compete for the same substrate because rad57 mutants have an elevated spontaneous mutation rate that is dependent on REV3 (Rattray et al. 2002). Although recombination and template switching had previously been considered as separate mechanisms, recent studies suggest that Rad51 and Rad18 have overlapping functions in the formation of sister-chromatid joint molecules after MMS treatment (Branzei et al. 2008). Rad55-Rad57 may have a specific role in formation or maintenance of Rad51 at ssDNA gaps, perhaps by preventing filament extension into adjacent dsDNA. Alternatively, the pairing of the Rad51-bound ssDNA gapped substrate with the intact sister chromatid may be more dependent on Rad55-Rad57 than DSBR in which the ends are not torsionally constrained. Another possibility is that Rad55-Rad57 acts indirectly in spontaneous recombination by antagonizing the post-replication repair pathway, a hypothesis that we are currently testing.
Footnotes
These authors contributed equally to this work.
Present address: Department of Genetics, Harvard Medical School, 77 Ave. Louis Pasteur, Boston, MA 02115.
Footnotes
Communicating editor: S. Keeney
Acknowledgements
The authors thank R. Rothstein and members of the Rothstein lab for assistance with microscopy and helpful discussions. We thank H. Klein and R. Rothstein for yeast strains and plasmids and W. K. Holloman for comments on the manuscript. This research was supported by Public Health Service grants GM054099 (L.S.S.) and T32 AI07161 (A.M.M.) from the National Institutes of Health and by a National Science Foundation F31 predoctoral fellowship to A.M.M.
References
Aboussekhra, A., R. Chanet, A. Adjiri and F. Fabre,
Basile, G., M. Aker and R. K. Mortimer,
Galgoczy, D. J., A. Cassidy-Stone, M. Llinas, S. M. O'Rourke, I. Herskowitz et al.,
Gasior, S. L., H. Olivares, U. Ear, D. M. Hari, R. Weichselbaum et al.,
Hays, S. L., A. A. Firmenich and P. Berg,
Hsiang, Y. H., M. G. Lihou and L. F. Liu,
Huang, K. N., and L. S. Symington,
Lisby, M., R. Rothstein and U. H. Mortensen,
Sherman, F., G. Fink and J. Hicks,
Sugiyama, T., J. H. New and S. C. Kowalczykowski,

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}