





Abstract
During meiosis, programmed DNA double-strand breaks (DSBs) are repaired to create at least one crossover per chromosome arm. Crossovers mature into chiasmata, which hold and orient the homologous chromosomes on the meiotic spindle to ensure proper segregation at meiosis I. This process is usually monitored by one or more checkpoints that ensure that DSBs are repaired prior to the meiotic divisions. We show here that mutations in Drosophila genes required to process DSBs into crossovers delay two important steps in meiotic progression: a chromatin-remodeling process associated with DSB formation and the final steps of oocyte selection. Consistent with the hypothesis that a checkpoint has been activated, the delays in meiotic progression are suppressed by a mutation in the Drosophila homolog of pch2. The PCH2-dependent delays also require proteins thought to regulate the number and distribution of crossovers, suggesting that this checkpoint monitors events leading to crossover formation. Surprisingly, two lines of evidence suggest that the PCH2-dependent checkpoint does not reflect the accumulation of unprocessed recombination intermediates: the delays in meiotic progression do not depend on DSB formation or on mei-41, the Drosophila ATR homolog, which is required for the checkpoint response to unrepaired DSBs. We propose that the sites and/or conditions required to promote crossovers are established independently of DSB formation early in meiotic prophase. Furthermore, the PCH2-dependent checkpoint is activated by these events and pachytene progression is delayed until the DSB repair complexes required to generate crossovers are assembled. Interestingly, PCH2-dependent delays in prophase may allow additional crossovers to form.
MEIOTIC crossovers promote genetic variation and mature into chiasmata, which hold the homologous chromosomes together at metaphase I and direct their segregation at anaphase I. In the absence of chiasmata, homologs may segregate randomly, resulting in aneuploidy, which can lead to infertility, severe developmental consequences, or lethality. Therefore, it is not surprising that crossover formation is a tightly regulated process. The formation of crossovers depends on the repair of programmed DNA double-strand breaks (DSBs) through homologous recombination (McKim and Hayashi-Hagihara 1998; Keeney 2001). DSBs are believed to be catalyzed by the Spo11 protein, a suspected paralog of a type II topoisomerase from archaebacteria. DSBs that do not become crossovers are repaired as noncrossovers, often referred to as “gene conversions.”
The mechanism for repairing DSBs to generate crossovers during meiotic prophase probably involves some kind of double Holliday junction intermediate (Stahl 1996; Heyer et al. 2003; Hollingsworth and Brill 2004; Whitby 2005). By contrast, noncrossovers can be generated by a combination of repair pathways such as synthesis-dependent strand annealing. The meiotic DSB repair program involves proteins specialized for the generation of crossovers as well as generic DSB repair proteins. In Drosophila, the former group of “crossover proteins” have been identified by mutations that cause reductions in the frequency of crossovers but not noncrossovers (reviewed in Mehrotra et al. 2007). The latter group includes proteins such as members of the Rad51 family, required to repair all DSBs (Hoffmann and Borts 2004; Kunz and Schar 2004).
Drosophila genes required for crossing over have been divided into two general classes: precondition and exchange genes (Sandler et al. 1968; Carpenter and Sandler 1974). The distinction between the precondition and exchange classes has been based mainly on the effects of mutations on the distribution of crossovers. The few crossovers observed in the progeny of females homozygous for precondition mutants show an altered distribution, while the few crossovers generated by mothers homozygous for exchange mutants show a relatively normal distribution. Therefore, precondition genes may have a role in establishing the crossover distribution, while exchange genes are required later to carry out the reaction that generates crossovers.
Meiotic DSB repair in Drosophila is monitored by at least one checkpoint. When there is a defect in repairing meiotic DSBs in Drosophila females, the ATR/MEI-41-dependent DSB repair checkpoint is activated (Jang et al. 2003), resulting in a variety of developmental defects, including the failure of the oocyte to establish dorsal–ventral polarity (Ghabrial and Schupbach 1999). This checkpoint pathway may also have a more direct role in DSB repair since mutations in the mei-41 gene cause a reduction in crossing over (Baker and Carpenter 1972). In budding yeast, checkpoint proteins may also have a role in determining whether repair occurs using the sister chromatid or the homolog (Grushcow et al. 1999).
We have found evidence for a new meiotic prophase checkpoint in Drosophila females. Mutations in DSB repair genes and exchange genes cause delays in two meiotic events: a chromatin-remodeling response to DSBs and oocyte selection. Both of these phenotypes may be a consequence of a general delay in pachytene progression, suggestive of an activated checkpoint. Surprisingly, the delay in pachytene progression in DSB repair and exchange mutants is independent of DSB formation but requires precondition genes like mei-218 and rec. This suggests that the checkpoint is not the canonical DSB-repair checkpoint that depends on ATR/MEI-41 (Ghabrial and Schupbach 1999). Instead, we propose that this delay is the result of a second checkpoint associated with the pathway leading to crossovers. We show that this DSB-independent checkpoint requires the Drosophila homolog of PCH2, an AAA–adenosine triphosphatase.
In Saccharomyces cerevisiae and Caenorhabditis elegans, it has been suggested that a PCH2-dependent checkpoint pathway responds to synapsis defects independent of DSBs (Bhalla and Dernburg 2005; Wu and Burgess 2006). However, some Drosophila mutants with PCH2-dependent delays in pachytene do not have obvious defects in synapsis. Thus, our results point to a defect in the pathway leading to crossover formation as the trigger that activates the checkpoint. Interestingly, the synapsis mutants analyzed in other organisms also have crossover defects, suggesting that there may be a common mechanism related to crossover specification for triggering the checkpoint in all three species.
MATERIALS AND METHODS
Fly stocks and genetic techniques:
The following mutations were used and have been previously described: hdmg7 (Liu et al. 2000; Joyce et al. 2009, accompanying article in this issue); mei-W684572 (Bhagat et al. 2004); mei-P22103 (Liu et al. 2002); okrWS, spn-A1, spn-BBU, and spn-D349 (Ghabrial et al. 1998; Abdu et al. 2003; Jang et al. 2003; Staeva-Vieira et al. 2003); mei-41D3 (Sibon et al. 1999); mei-2181 (Carpenter and Sandler 1974; McKim et al. 1996); rec1 and rec2 (Blanton et al. 2005); mei-9a, mei-9A2, mei-912, and mei-9RT1 (Yildiz et al. 2004); and mus312D1 (Yildiz et al. 2002). Experiments were done with both mei-9a and mei-9A2 since both are genetic null alleles. Experiments with rec were done with rec1/rec2 heterozygotes. The deficiency Df(3R)p-XT103 deletes cytological bands 85A2–85C2, which includes the pch2 locus. All crosses were raised at 25°. The frequency of X-chromosome nondisjunction is calculated as 2(Bar+ females + Bar males)/[2(Bar+ females + Bar males) + Bar females + Bar+ males].
Irradiation of oocytes:
Females were exposed to a dose of 10 Gy of X rays (at a dose rate of 1 Gy/min) and were dissected and fixed at 1, 5, or 24 hr after irradiation.
Cytology and immunofluorescence:
For immunolocalization experiments, females were aged at room temperature for ∼16 hr and ovaries were dissected and fixed using using Buffer A (Belmont et al. 1989; McKim et al. 2008). The antibody to γ-HIS2AV was described by Mehrotra and McKim (2006) and used at a 1:500 dilution. Additional primary antibodies included mouse anti-C(3)G antibody used at 1:500 (Page and Hawley 2001), rabbit anti-C(2)M antibody used at 1:400 (Manheim and McKim 2003), and a combination of two mouse anti-ORB antibodies (4H8 and 6H4) used at 1:100 (Lantz et al. 1994).
The secondary antibodies were Cy3-labeled goat anti-rabbit (Jackson Labs) used at 1:250 and FITC-labeled goat anti-mouse (Vector Labs) used at 1:125. Chromosomes were stained with Hoechst at 1:5000 (10 mg/ml solution) for 7 min at room temperature. Images were collected using a Leica TCS SP2 confocal microscope with a ×63, N.A. 1.3 lens. In most cases, whole germaria were imaged by collecting optical sections through the entire tissue. These data sets are shown as maximum-intensity projections. The analysis of the images, however, was performed by examining one section at a time.
Counting two oocytes and calculating P-values:
The oocytes were observed using an anti-C(3)G antibody. A cell was scored as an oocyte if complete SC filaments were clear and distinct. P-values were calculated using the Fisher's exact test. The P-value from the test compares the ratio of one-oocyte to two-oocyte cysts that were observed in two genotypes. In experiments where C(3)G staining was not visible [such as in the c(3)G null mutant], a concentration of ORB staining in the cytoplasm of a cell was used to identify the oocytes (Gonzalez-Reyes et al. 1997).
Counting γ-HIS2AV foci:
The γ-HIS2AV foci were counted from germaria where the foci were clear and distinct. Foci numbers in wild type were at a maximum in region 2a (early pachytene) and few foci were visible by region 2b (mid-pachytene). Therefore, to compare foci numbers in different genotypes, we used a method that includes all cysts with γ-HIS2AV foci, averaging the number in each pair of pro-oocytes. We compared the average number of foci in all the pro-oocytes or oocytes of each germarium, starting with the youngest cysts at the anterior end, by examining a full series of optical sections.
Plotting γ-HIS2AV foci as a function of relative cyst age:
Since the position of a cyst in the germarium is only a rough estimate of its meiotic stage, the foci were first counted in all the pro-oocytes/oocytes [identified by C(3)G staining] in the germarium. The meiotic stage of each pro-oocyte was then normalized according to the relative position of the entire cyst within the germarium since the relative position is more important than the absolute position. The pro-oocytes from 13 wild-type germaria, 18 hdmg7, 6 mei-2181, 5 hdmg7 mei-2181, 5 spn-D349, 4 mei-2181; spn-D349, 5 pch2EY01788a, 6 hdmg7; pch2EY01788a, 5 mei-9A2, and 5 mei-9A2; pch2EY01788a were arranged according to their relative age. The average number of γ-HIS2AV foci per pro-oocyte at each stage was then calculated and plotted as a function of relative cyst age.
Isolation of a pch2 insertion allele:
We used an allele of pch2 in which the coding region was disrupted by 473 bp of a partially deleted P element (P{EPgy2}CG31453[EY01788a]) inserted toward the end of the first exon (Bellen et al. 2004). This pch2EY01788a mutation causes a frameshift early in the protein and is therefore likely a null allele. The original stock contained two third-chromosome insertions (P{EPgy2}CG31453EY01788a P{EPgy2}EY01788b). One insertion, EY01788a, was located inside the coding region of CG31453 at 85A3. The other insertion, EY01788b, was located at 95F1 near CG5524. To isolate the pch2 (CG31453) insertion, we crossed y1 w67c23; P{EPgy2}CG31453EY01788a P{EPgy2}EY01788b males to cu e ca/TM6B females. The Tb+ female progeny were then crossed to cu e Pr ca/TM6B males, and cu+ e ca recombinants were collected and crossed individually to yw; Dr/TM3 females. Five stocks were made from crossing Pr+ Sb females to their Pr+ Sb brothers. We confirmed the presence of EY01788a by isolating DNA from homozygote pch2EY01788a e ca females. In all five lines, PCR revealed an ∼500-bp insertion in the pch2 locus. Sequence analysis showed that there was 473 bp of a deleted P{Epgy2} element inserted into the coding region of pch2, which is expected to cause a frameshift mutation.
RESULTS
Many of the exchange genes have been shown to encode proteins with informative biochemical functions. MEI-9 and ERCC1 form an endonuclease, supporting the hypothesis that exchange proteins play direct roles in the recombination process. In contrast, the function of precondition genes has been ambiguous and confusing because their biochemical functions are not known. Furthermore, the distinction between the precondition and exchange type genes is based mostly on the distribution of crossing over in the mutants although the mechanistic basis for this difference is not known.
To establish meaningful parameters to distinguish precondition from exchange genes and gain insights into how these genes promote crossover formation, we used cytological tools to characterize synaptonemal complex (SC) formation and DSB repair in mutations required for crossing over. This was done by comparing the progression through meiotic pachytene in wild-type and mutant females. Whole-mounted ovaries were stained with antibodies recognizing the SC components C(3)G and C(2)M to observe synapsis, identify the pro-oocytes, and estimate the meiotic stage of each nucleus. DSB formation and repair was monitored using an antibody to γ-HIS2AV. Phosphorylation of HIS2AV (or H2AX in mammals) is a rapid chromatin-remodeling response to DSBs that appears during the pachytene stage in Drosophila female meiosis (Rogakou et al. 1999; Madigan et al. 2002; Jang et al. 2003).
Meiotic progression in wild-type females:
During wild-type oogenesis, oocytes differentiate within a 16-cell germline cyst (see Figure 1 for description of oogenesis). Several cysts are contained in each germarium and are arranged in temporal order, with the earliest cysts located in the most anterior positions. These germarium cysts can be separated into three stages on the basis of their morphology. First, within region 2a cysts, two pro-oocytes initially appear equivalent as both enter meiosis and reach early pachytene. At this time the SC forms and is observed as complete filaments of C(3)G or C(2)M staining in each pair of pro-oocytes. Second, within region 2b cysts, one of the two pro-oocytes begins to exit meiosis, converts to a nurse cell fate, and loses staining of SC proteins. Third, within region 3 cysts, located in the most posterior position of the germarium, oocyte selection has occurred. This is characterized by the presence of only a single oocyte with SC staining (Figure 1).
![SC formation and the two-oocyte phenotype in hdm mutants. (A and B) Maximum-intensity projections showing a merge of all the confocal optical sections through a wild-type and hdm mutant germarium stained for SC [C(3)G] in green and DNA in blue. A cartoon of oocyte development in wild-type and hdm mutant germaria is next to its corresponding image. Oocyte development begins in the germarium, where four incomplete divisions form a 16-cell cyst (region 1, not shown). Two of the cells in each cyst have four interconnections, or ring canals, and become the pro-oocytes. Changes in cyst morphology differentiate regions 2a, 2b, and 3. In region 2a, both pro-oocytes enter meiosis, including zygotene and early pachytene, where the SC assembles between homologs and meiotic recombination initiates. Region 2a cysts are round, region 2b cysts flatten out, and region 3 cysts become round again before leaving the germarium into the vitellarium (stages 2–14). (A) In some region 2b and most region 3 wild-type cysts, one cell is identifiable as the oocyte by robust localization of the SC component C(3)G protein. The white arrow indicates the “loser” pro-oocyte in region 2b, which shows trace amounts of C(3)G staining, and the white arrowhead indicates a region 3 oocyte with robust C(3)G staining. (B) In hdm mutants, there are two late pachytene cells (oocytes) with robust C(3)G staining in most region 3 cysts (white arrowheads). Bars, 10 μm.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/genetics/181/1/10.1534_genetics.108.093112/7/m_39fig1.jpeg?Expires=1716542197&Signature=MiktOHaMv8NQ6YfKr1KMjrICrWyLwO9iIMRM14eLmVqvp7~wwDmSfOxytmzkvc1t7TZvEiR1ZrJxCujFQWa2~e8B~uxXpHO6LXFA4-K-0S6QFpdFhFFkOvJkuQaRxnzs4qfEPuTEBhWI9qDs64bHmeJzuwQ9LPVpvSFavBuJ3rxYTV6iaPXpmxoIcHufOcADI8OqHaYc9WxYJZpY2rEiupYuy01m19N1ZN9XMNwhZbnWnlPbmCTixcA0pB9E0oBkRoCEWpbd6TZU90oA~PQr50k16U-lIuXYylwgEgj47PnAabCvEWUSGq3xhxVHwDDut1rQFdpxYnIjo-ZhBCV7Rg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
SC formation and the two-oocyte phenotype in hdm mutants. (A and B) Maximum-intensity projections showing a merge of all the confocal optical sections through a wild-type and hdm mutant germarium stained for SC [C(3)G] in green and DNA in blue. A cartoon of oocyte development in wild-type and hdm mutant germaria is next to its corresponding image. Oocyte development begins in the germarium, where four incomplete divisions form a 16-cell cyst (region 1, not shown). Two of the cells in each cyst have four interconnections, or ring canals, and become the pro-oocytes. Changes in cyst morphology differentiate regions 2a, 2b, and 3. In region 2a, both pro-oocytes enter meiosis, including zygotene and early pachytene, where the SC assembles between homologs and meiotic recombination initiates. Region 2a cysts are round, region 2b cysts flatten out, and region 3 cysts become round again before leaving the germarium into the vitellarium (stages 2–14). (A) In some region 2b and most region 3 wild-type cysts, one cell is identifiable as the oocyte by robust localization of the SC component C(3)G protein. The white arrow indicates the “loser” pro-oocyte in region 2b, which shows trace amounts of C(3)G staining, and the white arrowhead indicates a region 3 oocyte with robust C(3)G staining. (B) In hdm mutants, there are two late pachytene cells (oocytes) with robust C(3)G staining in most region 3 cysts (white arrowheads). Bars, 10 μm.
In the progression from early (region 2a) to late (region 3) pachytene, there is also a characteristic pattern of γ-HIS2AV staining in the pro-oocytes and oocyte (Mehrotra and McKim 2006). γ-HIS2AV foci are most abundant at early pachytene (region 2a) and by late pachytene (region 3) no γ-HIS2AV foci are observed. We used cyst morphology, SC, and γ-HIS2AV staining to compare the relative timing of progression through pachytene in wild-type and recombination-defective mutants (summarized in Table 1).
Comparison of crossover-defective mutant phenotypes
Pachytene progression defects | ||||||||
|---|---|---|---|---|---|---|---|---|
| Class | Mutant | SC formation | Crossover levels | Crossover distribution | Delayed γ-HIS2AV | Persistence of γ-HIS2AV focia | Two oocytes | Suppresses pachytene delay phenotypes |
| Synapsis | c(3)G | No | None | Noneb | ND | 0.0 | No | ND |
| DSB formation | mei-W68 | Yes | None | Noneb | Noneb | Noneb | No | No |
| mei-P22 | Yes | None | Noneb | Noneb | Noneb | No | No | |
| Precondition | mei-218 | Yes | Reduction | Abnormal | No | 0.0 | No | Yes |
| rec | Yes | Reduction | Abnormal | No | 0.0 | No | Yes | |
| DSB repair | spn-A | Yes | NDc | NDc | Yes | 21.0 | Yes | NAd |
| spn-De | Yes | Reduction | Abnormal | Yes | 20.9 | Yes | NAd | |
| okra | Yes | Reductionc | Abnormalc | Yes | 22.0 | Yes | NAd | |
| Exchange | mei-9e | Yes | Reduction | Normal | Yes | 1.3 | Yes | NAd |
| hdm | Yes | Reduction | Normal | Yes | 1.4 | Yes | NAd | |
| Surveillance mechanisms | mei-41 | Yes | Reductionc | Abnormalc | Yes | 21.0 | Yes | NAd |
| pch2 | Yes | Normal | Normal | No | 0.0 | No | Yes | |
Pachytene progression defects | ||||||||
|---|---|---|---|---|---|---|---|---|
| Class | Mutant | SC formation | Crossover levels | Crossover distribution | Delayed γ-HIS2AV | Persistence of γ-HIS2AV focia | Two oocytes | Suppresses pachytene delay phenotypes |
| Synapsis | c(3)G | No | None | Noneb | ND | 0.0 | No | ND |
| DSB formation | mei-W68 | Yes | None | Noneb | Noneb | Noneb | No | No |
| mei-P22 | Yes | None | Noneb | Noneb | Noneb | No | No | |
| Precondition | mei-218 | Yes | Reduction | Abnormal | No | 0.0 | No | Yes |
| rec | Yes | Reduction | Abnormal | No | 0.0 | No | Yes | |
| DSB repair | spn-A | Yes | NDc | NDc | Yes | 21.0 | Yes | NAd |
| spn-De | Yes | Reduction | Abnormal | Yes | 20.9 | Yes | NAd | |
| okra | Yes | Reductionc | Abnormalc | Yes | 22.0 | Yes | NAd | |
| Exchange | mei-9e | Yes | Reduction | Normal | Yes | 1.3 | Yes | NAd |
| hdm | Yes | Reduction | Normal | Yes | 1.4 | Yes | NAd | |
| Surveillance mechanisms | mei-41 | Yes | Reductionc | Abnormalc | Yes | 21.0 | Yes | NAd |
| pch2 | Yes | Normal | Normal | No | 0.0 | No | Yes | |
ND, not determined.
Average no. of foci that persist into late pachytene (region 3) oocytes.
Crossing over is eliminated by null alleles but the crossover distribution is altered by hypomorphic alleles (Liu et al. 2002; Bhagat et al. 2004).
Crossover distribution in null mutants is not known because they are sterile. However, mei-41 and okr hypomorphs are fertile and have reduced crossing over with an altered distribution (Baker and Carpenter 1972; Bhagat et al. 2004).
These mutants exhibit delay phenotypes.
Results with spn-B were similar to spn-D and with mus312 were similar to mei-9.
Comparison of crossover-defective mutant phenotypes
Pachytene progression defects | ||||||||
|---|---|---|---|---|---|---|---|---|
| Class | Mutant | SC formation | Crossover levels | Crossover distribution | Delayed γ-HIS2AV | Persistence of γ-HIS2AV focia | Two oocytes | Suppresses pachytene delay phenotypes |
| Synapsis | c(3)G | No | None | Noneb | ND | 0.0 | No | ND |
| DSB formation | mei-W68 | Yes | None | Noneb | Noneb | Noneb | No | No |
| mei-P22 | Yes | None | Noneb | Noneb | Noneb | No | No | |
| Precondition | mei-218 | Yes | Reduction | Abnormal | No | 0.0 | No | Yes |
| rec | Yes | Reduction | Abnormal | No | 0.0 | No | Yes | |
| DSB repair | spn-A | Yes | NDc | NDc | Yes | 21.0 | Yes | NAd |
| spn-De | Yes | Reduction | Abnormal | Yes | 20.9 | Yes | NAd | |
| okra | Yes | Reductionc | Abnormalc | Yes | 22.0 | Yes | NAd | |
| Exchange | mei-9e | Yes | Reduction | Normal | Yes | 1.3 | Yes | NAd |
| hdm | Yes | Reduction | Normal | Yes | 1.4 | Yes | NAd | |
| Surveillance mechanisms | mei-41 | Yes | Reductionc | Abnormalc | Yes | 21.0 | Yes | NAd |
| pch2 | Yes | Normal | Normal | No | 0.0 | No | Yes | |
Pachytene progression defects | ||||||||
|---|---|---|---|---|---|---|---|---|
| Class | Mutant | SC formation | Crossover levels | Crossover distribution | Delayed γ-HIS2AV | Persistence of γ-HIS2AV focia | Two oocytes | Suppresses pachytene delay phenotypes |
| Synapsis | c(3)G | No | None | Noneb | ND | 0.0 | No | ND |
| DSB formation | mei-W68 | Yes | None | Noneb | Noneb | Noneb | No | No |
| mei-P22 | Yes | None | Noneb | Noneb | Noneb | No | No | |
| Precondition | mei-218 | Yes | Reduction | Abnormal | No | 0.0 | No | Yes |
| rec | Yes | Reduction | Abnormal | No | 0.0 | No | Yes | |
| DSB repair | spn-A | Yes | NDc | NDc | Yes | 21.0 | Yes | NAd |
| spn-De | Yes | Reduction | Abnormal | Yes | 20.9 | Yes | NAd | |
| okra | Yes | Reductionc | Abnormalc | Yes | 22.0 | Yes | NAd | |
| Exchange | mei-9e | Yes | Reduction | Normal | Yes | 1.3 | Yes | NAd |
| hdm | Yes | Reduction | Normal | Yes | 1.4 | Yes | NAd | |
| Surveillance mechanisms | mei-41 | Yes | Reductionc | Abnormalc | Yes | 21.0 | Yes | NAd |
| pch2 | Yes | Normal | Normal | No | 0.0 | No | Yes | |
ND, not determined.
Average no. of foci that persist into late pachytene (region 3) oocytes.
Crossing over is eliminated by null alleles but the crossover distribution is altered by hypomorphic alleles (Liu et al. 2002; Bhagat et al. 2004).
Crossover distribution in null mutants is not known because they are sterile. However, mei-41 and okr hypomorphs are fertile and have reduced crossing over with an altered distribution (Baker and Carpenter 1972; Bhagat et al. 2004).
These mutants exhibit delay phenotypes.
Results with spn-B were similar to spn-D and with mus312 were similar to mei-9.
Mutations in the exchange and DSB repair classes of genes result in a delay in oocyte selection during pachytene:
Meiotic progression was examined in females homozygous for mutations in each of three exchange class genes: two previously characterized genes, mei-9 and mus312, and hold'em (hdm), a new member of the exchange class that we recently identified (Joyce et al. 2009, accompanying article in this issue). Ovaries from these exchange mutant females were stained for C(3)G. As in wild-type, zygotene was an infrequently observed stage, and complete threads of C(3)G staining were observed in most region 2a pro-oocytes of each exchange mutant (Figure 1). This indicates that SC formation and synapsis occurred rapidly and without notable delay, as expected from previous electron microscopy studies of SC formation in mei-9 homozygotes (Carpenter 1979).
In all three exchange mutants, however, oocyte selection was delayed; the choice between the two pro-oocytes occurred later than in wild type. For example, in hdm mutant females, two pro-oocytes were visible by C(3)G staining in 62.5% of region 3 cysts (Figure 1; Figure 2), which was significantly greater than the frequency of 9.5% observed in wild type (P = 0.0005, Fisher's exact test). Similarly, both the mei-9 and mus312 mutants showed a high frequency of two oocytes in region 3 cysts (69.6%, P = 0.00006, and 75.0%, P = 0.00002, respectively; Figure 2). The presence of two oocytes was associated with the failure of either pro-oocyte to localize to the posterior end of the cyst and frequently showed weaker C(3)G staining in region 3 compared either to wild-type region 3 oocytes or to earlier-stage oocytes in the same mutant germarium (Figure 3A). These two observations indicate that the delay affects both pro-oocytes. The presence of two pro-oocytes in region 3 cysts will be referred to as the “two-oocyte” phenotype in this article.
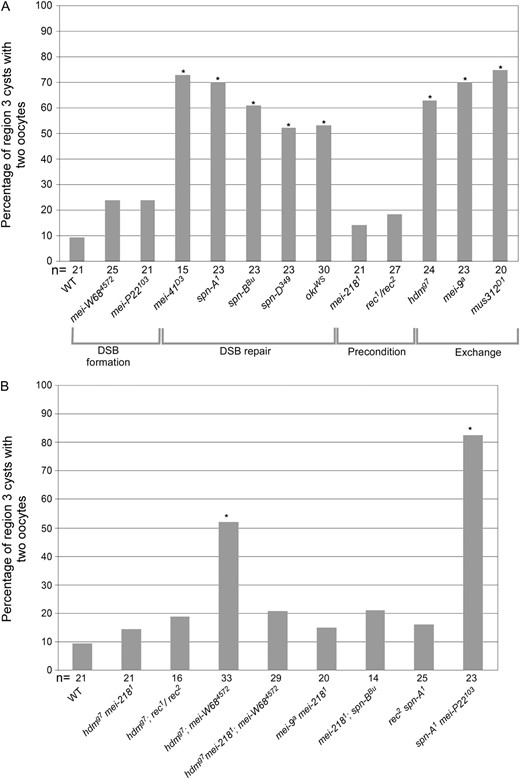
Two-oocyte phenotype in region 3 cysts of wild-type and crossover-defective single mutant females. The percentage of region 3 cysts with two oocytes is based on C(3)G staining. Asterisks located above each bar correspond to a genotype that gave a P-value <0.05 when compared to wild type. The number of cysts (which is equivalent to the number of germaria) counted is shown at the bottom of each bar. (A) Two-oocyte phenotype in single-mutant females. The mutants are grouped by their primary defect in recombination. A significantly high frequency of the two-oocyte phenotype was found in DSB repair and exchange class mutants, but not in mutants of the DSB formation and precondition groups. The two-oocyte phenotype appears to be robust and reproducible. The frequencies reported here are similar to those in Huynh and St. Johnston (2000). (B) Two-oocyte phenotype in double-mutant females. The high frequency of two oocytes observed in exchange mutants (hdmg7 or mei-9a) was suppressed by mei-2181 or rec1/2 but not by mei-W684572. Similarly, the two-oocyte phenotypes of DSB repair mutants (spn-A1 or spn-BBU) were suppressed by rec1/2 and mei-2181 but not by mei-P22103.
![Pattern of γ-HIS2AV staining in wild-type and crossover-defective mutants. (A) Representative examples of γ-HIS2AV staining (red) at different stages of pachytene in wild-type and hdmg7 mutants, with SC staining [C(3)G] in green and DNA in blue. Each image shows a projection of all confocal sections through the oocyte nucleus. The panels of each genotype were cropped from the same germarium and image stack. When two oocytes were observed, the C(3)G staining often had reduced intensity relative to other oocytes in the same cyst. Bar, 5 μm. (B) The average number of γ-HIS2AV foci is plotted relative to cyst age in hdmg7 and mei-2181 mutants. Cyst 1 is the first to have complete SC, cyst 8 is in late pachytene (region 3), and cysts 9–11 are in later-stage cysts (stages 2–4), which have left the germarium. The age difference between each cyst is ∼12 hr (King 1970). Because the oocytes are arranged in temporal order in the ovary, the lower number of γ-HIS2AV foci in early stage oocytes (cysts 2–4) of hdmg7 mutants indicates that there is a delay in their appearance. mei-2181 suppresses the delayed onset and persistence of γ-HIS2AV in hdmg7 mutants. (C) Same graph as B but for spn-DD349 and mei-2181 mutants. mei-2181 suppresses the delayed onset of γ-HIS2AV in spn-D349 mutants. Because spn-D349 has a DSB repair defect, the γ-HIS2AV foci persist in high numbers into late stages of pachytene. Error bars denote the standard error of the mean.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/genetics/181/1/10.1534_genetics.108.093112/7/m_39fig3.jpeg?Expires=1716542197&Signature=r74MPsJPA0rupHGhiKWnMvziwVF70LIVJic0JYcKS9uVGncW6ep4dGaIG5CrIKcnXkf82e6waPURRalFAPYFpt9pyhsvebsmGIDyY7Tk83GTD3-oe7Nwsm4Nz6P42iAxZiOzp7nge4XF8h6p2BtiBtwRZtpeFh6NaeopkAB-AKsVziZqhfFWLjG6WZdqHQ83q1SljEIUg-RRFcebP07BmvJ2sohKEcTa9YmF4Nq2TkIarvVXfwSXZ1QTrHJyuNXE3KkTIGkw8t2aisj8yKS5DA8nYXFWYJoO0v4w3519ou8HmqPo2ZIQV0P9GMgJHI1axmOUm13qmlKH7MaYAvhoRA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Pattern of γ-HIS2AV staining in wild-type and crossover-defective mutants. (A) Representative examples of γ-HIS2AV staining (red) at different stages of pachytene in wild-type and hdmg7 mutants, with SC staining [C(3)G] in green and DNA in blue. Each image shows a projection of all confocal sections through the oocyte nucleus. The panels of each genotype were cropped from the same germarium and image stack. When two oocytes were observed, the C(3)G staining often had reduced intensity relative to other oocytes in the same cyst. Bar, 5 μm. (B) The average number of γ-HIS2AV foci is plotted relative to cyst age in hdmg7 and mei-2181 mutants. Cyst 1 is the first to have complete SC, cyst 8 is in late pachytene (region 3), and cysts 9–11 are in later-stage cysts (stages 2–4), which have left the germarium. The age difference between each cyst is ∼12 hr (King 1970). Because the oocytes are arranged in temporal order in the ovary, the lower number of γ-HIS2AV foci in early stage oocytes (cysts 2–4) of hdmg7 mutants indicates that there is a delay in their appearance. mei-2181 suppresses the delayed onset and persistence of γ-HIS2AV in hdmg7 mutants. (C) Same graph as B but for spn-DD349 and mei-2181 mutants. mei-2181 suppresses the delayed onset of γ-HIS2AV in spn-D349 mutants. Because spn-D349 has a DSB repair defect, the γ-HIS2AV foci persist in high numbers into late stages of pachytene. Error bars denote the standard error of the mean.
All known exchange gene products have roles in both somatic DNA repair and meiotic crossover production. To examine whether the two-oocyte phenotype reflected the loss of the meiotic recombination functions, we tested two special mei-9 alleles (Yildiz et al. 2004). The mei-912 mutant, which is defective for meiotic crossover formation but proficient in somatic DNA repair, had a high frequency of the two-oocyte phenotype (52.9%, P = 0.005). The mei-9RT1 mutant, which is proficient in meiotic crossover formation but is defective in somatic DNA repair, had a low frequency of the two-oocyte phenotype (28.6%, P = 0.21). These results suggest that the delay in oocyte selection is due to a defect in a meiotic function of the exchange genes.
We next examined mutations in four genes required for meiotic DSB repair for the two-oocyte phenotype: the Rad51 ortholog spn-A, Rad51 paralogs spn-B and spn-D, and the Rad54 ortholog okr. In all of these DSB repair mutants, complete threads of C(3)G staining were observed in most region 2a pro-oocytes, indicating that SC formation and synapsis occurred normally. Like the exchange mutants, each DSB repair mutant exhibited a high frequency of the two-oocyte phenotype (52.5–69.6%, each P < 0.05 compared to wild type; Figure 2). Indeed, a two-oocyte phenotype has previously been described in some of these mutants using different markers for the oocyte, such as the cytoplasmic ORB protein (Gonzalez-Reyes et al. 1997; McCaffrey et al. 2006) or a different SC antibody (Huynh and St. Johnston 2000).
In all mutant females analyzed, SC staining was limited to one cell at stage 2 of oogenesis, which is shortly after a cyst leaves the germarium. Thus, one of the two pro-oocytes does eventually become a nurse cell. These results suggest that exchange and DSB repair mutations delay but do not block the pro-oocyte-to-oocyte decision, causing region 3 oocytes to be in mid-pachytene rather than late pachytene.
In contrast to exchange and DSB repair mutants, the two precondition class mutants that we examined, mei-218 and rec, showed a frequency of two oocytes in region 3 that was not significantly greater than wild type (14.3%, P = 1, and 18.5%, P = 0.44, respectively; Figure 2). Similarly, mutants with defects in DSB formation, mei-W68 and mei-P22, also showed a low frequency of two oocytes in region 3 (24.0%, P = 0.26, and 23.8%, P = 0.41, respectively). Taken together, our results indicate that only mutations in DSB repair and exchange class genes induce a significant delay in oocyte selection. In contrast, neither defects in DSB formation or precondition genes cause such a delay.
Delayed oocyte selection in crossover mutants is DSB independent:
The delay in oocyte selection caused by mutations in exchange and DSB repair genes could be caused by the activation of a checkpoint sensitive to the accumulation of repair intermediates. This hypothesis was tested by eliminating meiotic DSBs using mutants defective in DSB formation. In Drosophila, the programmed DSBs that initiate meiotic recombination require the genes mei-W68 (encoding a Spo11 ortholog) and mei-P22 (McKim et al. 1998; Liu et al. 2002). A high frequency of the two-oocyte phenotype was still observed in hdm; mei-W68 (51.5%, P = 0.420 compared to hdm) and mei-P22 spn-A (82.6%, P = 0.491 compared to spn-A) double mutants (Figure 2). A similar result was previously observed in a mei-W68; mus301 double mutant (mus301 is also required for meiotic DSB repair; McCaffrey et al. 2006). These results demonstrate that the delay in oocyte selection does not depend on the induction of DSBs or persistent repair intermediates.
Exchange and DSB repair mutants exhibit a delay in the chromatin-remodeling response to DSBs:
To determine if exchange and DSB-repair mutations, which caused a delay in oocyte selection, delayed other aspects of meiotic progression, we examined the dynamics of DSB formation and repair by staining for γ-HIS2AV. In wild-type oocytes, γ-HIS2AV foci were most abundant at early pachytene (region 2a, cyst 3 in Figure 3) and absent by late pachytene (region 3, cyst 8 in Figure 3). Interestingly, in the exchange mutants, hdm (Figure 3B), mei-9 (see supplemental Figure S1), and mus312 (data not shown), γ-HIS2AV foci did not reach maximum numbers until cyst 5 (approximately the last cyst in region 2a). These mutants did not affect the total number of γ-HIS2AV foci (Joyce et al. 2009, accompanying article in this issue), only the timing of their appearance. Thus, exchange mutant females exhibited a delay in the appearance of γ-HIS2AV foci in region 2a.
Mehrotra and McKim (2006) previously reported a delay in the appearance of γ-HIS2AV staining in DSB repair mutants. To examine this in more detail and compare to the exchange mutants, we examined the effect on γ-HIS2AV staining of mutations in spn-D (Figure 3C) and spn-B (data not shown). These and other mutations in DSB repair genes, such as spn-A and okr, have two effects on γ-HIS2AV foci. First, they cause a delay in the appearance of γ-HIS2AV foci, much like that observed in exchange mutants. Second, they cause large numbers of γ-HIS2AV foci to accumulate into late stages of pachytene because DSBs are not repaired.
Exchange mutants show only the first phenotype observed in DSB repair mutants: the delay in the appearance of γ-HIS2AV foci. In contrast to what we observed in DSB repair mutants, only a few γ-HIS2AV foci (1.4 foci/region 3 oocyte, n = 26) persisted into late pachytene (region 3) oocytes in hdm mutant cysts (Table 1). mei-9 mutants also showed the persistence of only a few γ-HIS2AV foci (1.3 foci/region 3 oocyte, n = 10). In wild type, the persistence of γ-HIS2AV foci in region 3 oocytes was extremely rare (0.04 foci/region 3 oocyte, n = 26). These observations are consistent with the conclusion that mutants like spn-A and spn-B have a block in DSB repair while exchange mutants like hdm and mei-9 have a delay only in DSB repair.
Unlike the DSB repair and the exchange mutants, precondition mutants mei-218 and rec did not exhibit a delay in either the appearance or disappearance of γ-HIS2AV foci staining, with the exception of a reproducibly tighter curve from cyst 2 through 4 (Figure 3 and data not shown).
The delayed appearance of γ-HIS2AV foci in exchange and DSB repair mutants could represent either a delay in DSB formation or a delay in the response to DSBs. To test for a delayed response to DSB formation, we compared γ-HIS2AV staining in hdm; mei-W68 double mutants to mei-W68 single mutants following irradiation. Because mei-W68 mutants do not generate meiotic DSBs, the only DSBs in this experiment were induced by irradiation. Furthermore, the X-ray treatment would induce the same average number of DSBs in early pachytene (region 2a) cells as in late pachytene (region 3) cells of the germarium, allowing for a direct comparison of the γ-HIS2AV response to DSBs at different stages of meiotic prophase. Specifically, we examined the effects of exchange mutations on X-ray-induced γ-HIS2AV foci in the first pachytene pro-oocytes (same as cyst 1 in Figure 3). mei-W68 single-mutant pro-oocytes in early pachytene (region 2a) showed a low number of γ-HIS2AV foci at 1 hr after irradiation and did not reach maximum numbers until ∼5 hr after irradiation (Figure 4; see supplemental Figure S2). This is in agreement with Mehrotra and McKim's observation (2006) that early pachytene oocytes respond to X-ray-induced DSBs more slowly than late pachytene oocytes or somatic cells (Madigan et al. 2002).
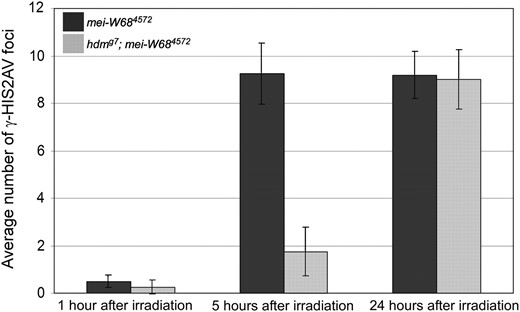
hdm delays the response to X-ray-induced DSBs. The average number of γ-HIS2AV foci in mei-W684572 (solid) and hdmg7; mei-W684572 (shaded) females at 1, 5, and 24 hr after irradiation. The data at the 1- and 5-hr time points are from the first cyst with pro-oocytes in pachytene (the first pair of region 2a pro-oocytes with SC staining). At 5 hr after irradiation, hdmg7; mei-W684572 pro-oocytes had 18.9% of the γ-HIS2AV foci found in mei-W684572. The data at the 24-hr time point are from the third cyst with pro-oocytes in pachytene, which, due to movement down the germarium, was approximately the first cyst at the time of irradiation. Error bars denote the standard error of the mean.
If hdm mutations delayed only the formation of DSBs, we would have expected the response time to X-ray-induced breaks to be the same in hdm; mei-W68 and mei-W68 single mutants at all time points. At 1 hr after irradiation, the hdm; mei-W68 mutant had the same low number of γ-HIS2AV foci in early pachytene oocytes as the mei-W68 mutant (Figure 4). At 5 hr after irradiation, however, the number of γ-HIS2AV foci in hdm; mei-W68 mutants was significantly reduced compared to mei-W68 mutants. At 24 hr after irradiation, the number of γ-HIS2AV foci in both mei-W68 and hdm; mei-W68 mutants was again similar throughout the germarium, indicating that the same number of γ-HIS2AV foci develop in hdm mutants as in wild type, but take longer to appear. A strikingly similar delayed response time to X-ray-induced DSBs was previously found in the DSB repair mutants spn-B and okr (Mehrotra and McKim 2006). Thus, along with the delay in oocyte selection, mutations in exchange genes such as hdm and mei-9 and DSB repair genes such as spn-B and spn-D cause a delay in the γ-HIS2AV response to DSBs. It remains possible that these mutations also cause delays in DSB formation, but this cannot currently be tested.
Delays in meiotic progression require the precondition class of crossover genes:
Mutations in the precondition genes mei-218 and rec did not lead to a significant increase in the frequency of the two-oocyte phenotype or delayed γ-HIS2AV response to DSBs. Therefore, we examined whether precondition mutations could suppress these delays in DSB repair and exchange mutants. In all cases, mei-218 and rec mutations suppressed the delay phenotypes of DSB repair or exchange mutants. For example, a mei-218 mutation reduced the frequency of the two-oocyte phenotype in mei-9 and hdm mutants to 15.0% (P = 0.0006 compared to mei-9) and 14.3% (P = 0.002 compared to hdm), respectively, which is not significantly different from wild type (Figure 2). Similarly, we observed a reduced frequency of the two-oocyte phenotype in the hdm; rec mutant (18.8%, P = 0.0097 compared to hdm). Furthermore, while hdm; mei-W68 mutant females had a high frequency of the two-oocyte phenotype, hdm mei-218; mei-W68 mutant females did not (Figure 2; 20.7%, P = 0.033 compared to hdm; mei-W68), indicating that the suppression of the two-oocyte phenotype by a mei-218 mutation did not depend on DSBs. Precondition mutations also reduced the frequency of the two-oocyte phenotype in DSB repair mutants, such as in rec spn-A (16.0%, P = 0.0003 compared to spn-A) and mei-218; spn-B double mutants (21.4%, P = 0.039 compared to spn-B) (Figure 2). Similar results were observed using γ-HIS2AV staining. A mei-218 mutation suppressed the delayed onset of γ-HIS2AV foci in hdm and spn-D mutants (Figure 3, B and C). We have not determined why the high numbers of persistent foci typical of DSB repair mutants disappear in the mei-218; spn-D double mutant.
Precondition mutations could suppress the delay phenotypes indirectly by accelerating progression through pachytene. This hypothesis was tested by ascertaining the frequency of region 2b (mid-pachytene) cysts with two oocytes in mei-218 mutants and wild type. If progression through pachytene was accelerated in a mei-218 mutant, we would expect a lower frequency of region 2b cysts with two oocytes compared to wild type. Instead, we found two oocytes in 75 and 87.5% of the wild-type and mei-218 mutant region 2b cysts, respectively, indicating no significant change in progression through pachytene in mei-218 mutant females (P = 0.63). Furthermore, the γ-HIS2AV response in these mutants was not faster than wild type (Figure 3). Therefore, precondition genes are required for the mechanism, which delays pachytene progression in exchange and DSB repair mutants.
pch2 is required for the pachytene delay observed in DSB repair and exchange mutants:
Mutations in DSB repair but not the exchange genes activate a DSB repair checkpoint pathway that requires the Drosophila ATR homolog mei-41 (Ghabrial and Schupbach 1999). However, the mei-41-dependent DSB repair checkpoint pathway is not responsible for the delays that we observed in pachytene progression because this phenotype occurs independently of DSBs. In support of this conclusion, mei-41 mutants showed a high frequency of the two-oocyte phenotype (73% region 3 cysts with two oocytes; Figure 2) and also showed a pattern of γ-HIS2AV staining in oocytes similar to that seen in DSB repair mutants, including delayed onset and persistence of foci into late pachytene (see supplemental Figure S3). These results suggest that MEI-41 is required for the repair of DSBs during meiosis in addition to its role in the DSB repair checkpoint (see also Larocque et al. 2007).
A DSB-independent surveillance mechanism has been proposed to monitor pachytene events in S. cerevisiae and C. elegans. The conserved pch2 gene, which encodes an AAA–adenosine triphosphatase, is essential for a pachytene arrest in response to mutants with synapsis defects in S. cerevisiae (Wu and Burgess 2006). In C. elegans, pch-2 is required for a DSB-independent checkpoint pathway that induces apoptosis in response to mutations that cause synapsis defects (Bhalla and Dernburg 2005). The Drosophila ortholog of pch2 is CG31453, which encodes a predicted protein that is 22.4% identical and 35.2% similar to S. cerevisiae Pch2, and 34.4% identical and 48.3% similar to C. elegans PCH-2 (see supplemental Figure S4).
We determined whether a mutation in pch2 could suppress the delay in oocyte selection and the delayed γ-HIS2AV response to DSBs in DSB repair and exchange mutants. Females homozygous for a pch2 null allele (see materials and methods) were fully viable and fertile with a normal distribution and frequency of X-chromosome crossing over (Table 2), showing that pch2 is not required for meiotic recombination. Furthermore, the pch2 mutant did not exhibit a delay in oocyte selection or the γ-HIS2AV response to DSBs (Figure 5). However, the frequency of the two-oocyte phenotype in the hdm; pch2 double mutant was reduced to 23.3% (P = 0.003 compared to hdm and P = 0.31 compared to wild type) (Figure 5A). Consistent with this result, hdm; pch2 mutants did not show the delayed onset of γ-HIS2AV staining observed in hdm single mutants (Figure 5B). Similar results were found with mei-9; pch2 and okr; pch2 double mutants in which the pachytene delay phenotypes observed in the mei-9 and okr single mutants were suppressed (Figure 5A; see supplemental Figure S1). These results show that pch2 is required for both of the pachytene delay phenotypes observed in hdm, mei-9, and okr mutants. In the course of these experiments, we also found that okr; pch2 mutants had the same number of γ-HIS2AV foci (23.3 ± 6.9) in region 3 oocytes as okr mutants (22.0 ± 2.2), suggesting that pch2 mutations do not affect the number of DSBs.
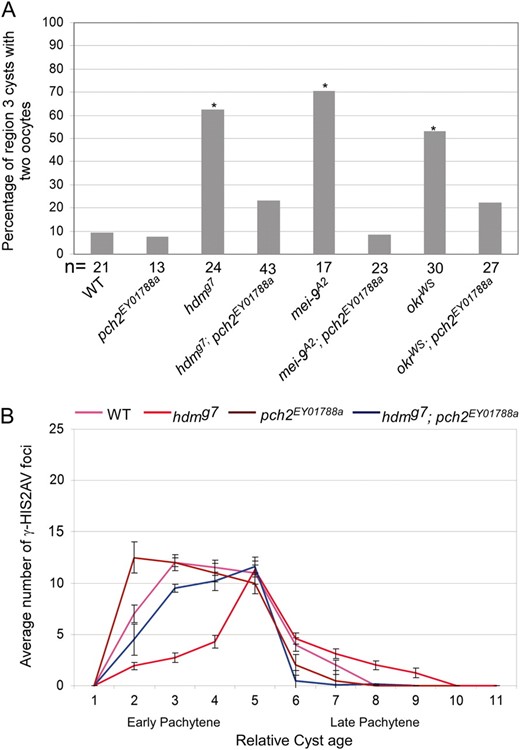
A pch2 mutation suppresses the pachytene delay phenotypes in hdm, mei-9, and okr mutants. (A) For each genotype, the percentage of region 3 cysts with two oocytes is given on the basis of C(3)G staining. pch2EY01788a mutations have no effect on the frequency of the two-oocyte phenotype and suppress the high frequency of two oocytes observed in hdmg7, mei-9A2, and okrWS mutants. Asterisks above each bar correspond to a mutant giving a P-value <0.05 compared to wild type and the number of cysts counted is at the bottom of each bar. (B) The average number of γ-HIS2AV foci relative to oocyte age in hdmg7 and pch2EY01788a mutants (see Figure 3B for details). pch2EY01788a mutations do not alter the wild-type γ-HIS2AV staining pattern and suppress the delayed onset and persistence of γ-HIS2AV in hdmg7 mutants. pch2EY01788a also suppressed the delayed onset of γ-HIS2AV in mei-9A2 mutants (supplemental Figure S4). Error bars denote the standard error of the mean.
Crossing over in hdm and pch2 mutants
Nondisjunction and crossing over on the X chromosome | |||||||
|---|---|---|---|---|---|---|---|
| Genotype | % X-chromosome NDa | pn-cv b | cv-m | m-f | f-y+ | Total pn-y+ | Total ♂ progenya |
| Wild type | 0.1 (2440) | 15.3 (100) | 21.6 (100) | 19.1 (100) | 6.9 (100) | 62.9 (100) | 1479 |
| hdmg7 c | 7.2 (3524) | 3.9 (25.5) | 15.0 (69.2) | 6.8 (35.3) | 4.2 (60.1) | 29.9 (47.5) | 1656 |
| pch2EY01788a/Df | 0.3 (2098) | 12.6 (82.4) | 25.2 (116.7) | 19.5 (102.1) | 7.8 (113.0) | 65.1 (103.5) | 960 |
| hdmg7; pch2EY01788a/Df | 15.0 (2305) | 0.7 (4.6) | 9.6 (44.4) | 5.1 (26.7) | 3.6 (52.2) | 19.0 (30.2) | 1107 |
Nondisjunction and crossing over on the X chromosome | |||||||
|---|---|---|---|---|---|---|---|
| Genotype | % X-chromosome NDa | pn-cv b | cv-m | m-f | f-y+ | Total pn-y+ | Total ♂ progenya |
| Wild type | 0.1 (2440) | 15.3 (100) | 21.6 (100) | 19.1 (100) | 6.9 (100) | 62.9 (100) | 1479 |
| hdmg7 c | 7.2 (3524) | 3.9 (25.5) | 15.0 (69.2) | 6.8 (35.3) | 4.2 (60.1) | 29.9 (47.5) | 1656 |
| pch2EY01788a/Df | 0.3 (2098) | 12.6 (82.4) | 25.2 (116.7) | 19.5 (102.1) | 7.8 (113.0) | 65.1 (103.5) | 960 |
| hdmg7; pch2EY01788a/Df | 15.0 (2305) | 0.7 (4.6) | 9.6 (44.4) | 5.1 (26.7) | 3.6 (52.2) | 19.0 (30.2) | 1107 |
For wild-type or pch2EY01788a/Df, y/y pn cv m f · y+ females were crossed to C(1:Y)1, v f B; C(4)RM, ci ey males. For hdm and hdmg7; pch2EY01788a/Df, y pn cv hdmg7/y hdmg7 m f y+ females were crossed to C(1:Y)1, v f B; C(4)RM, ci ey males. Only the male progeny counts were used to calculate the crossover frequency.
Nondisjunction and crossing over were measured in the same experiment. Total progeny for nondisjunction are shown in parentheses. Total progeny for crossing over are only the male progeny from this experiment (see materials and methods).
Crossing over is expressed as map units across the intervals shown. Numbers in parentheses denote the percentage of wild-type crossover frequency.
Data on hdm single mutant are from Joyce et al. (2009, accompanying article in this issue).
Crossing over in hdm and pch2 mutants
Nondisjunction and crossing over on the X chromosome | |||||||
|---|---|---|---|---|---|---|---|
| Genotype | % X-chromosome NDa | pn-cv b | cv-m | m-f | f-y+ | Total pn-y+ | Total ♂ progenya |
| Wild type | 0.1 (2440) | 15.3 (100) | 21.6 (100) | 19.1 (100) | 6.9 (100) | 62.9 (100) | 1479 |
| hdmg7 c | 7.2 (3524) | 3.9 (25.5) | 15.0 (69.2) | 6.8 (35.3) | 4.2 (60.1) | 29.9 (47.5) | 1656 |
| pch2EY01788a/Df | 0.3 (2098) | 12.6 (82.4) | 25.2 (116.7) | 19.5 (102.1) | 7.8 (113.0) | 65.1 (103.5) | 960 |
| hdmg7; pch2EY01788a/Df | 15.0 (2305) | 0.7 (4.6) | 9.6 (44.4) | 5.1 (26.7) | 3.6 (52.2) | 19.0 (30.2) | 1107 |
Nondisjunction and crossing over on the X chromosome | |||||||
|---|---|---|---|---|---|---|---|
| Genotype | % X-chromosome NDa | pn-cv b | cv-m | m-f | f-y+ | Total pn-y+ | Total ♂ progenya |
| Wild type | 0.1 (2440) | 15.3 (100) | 21.6 (100) | 19.1 (100) | 6.9 (100) | 62.9 (100) | 1479 |
| hdmg7 c | 7.2 (3524) | 3.9 (25.5) | 15.0 (69.2) | 6.8 (35.3) | 4.2 (60.1) | 29.9 (47.5) | 1656 |
| pch2EY01788a/Df | 0.3 (2098) | 12.6 (82.4) | 25.2 (116.7) | 19.5 (102.1) | 7.8 (113.0) | 65.1 (103.5) | 960 |
| hdmg7; pch2EY01788a/Df | 15.0 (2305) | 0.7 (4.6) | 9.6 (44.4) | 5.1 (26.7) | 3.6 (52.2) | 19.0 (30.2) | 1107 |
For wild-type or pch2EY01788a/Df, y/y pn cv m f · y+ females were crossed to C(1:Y)1, v f B; C(4)RM, ci ey males. For hdm and hdmg7; pch2EY01788a/Df, y pn cv hdmg7/y hdmg7 m f y+ females were crossed to C(1:Y)1, v f B; C(4)RM, ci ey males. Only the male progeny counts were used to calculate the crossover frequency.
Nondisjunction and crossing over were measured in the same experiment. Total progeny for nondisjunction are shown in parentheses. Total progeny for crossing over are only the male progeny from this experiment (see materials and methods).
Crossing over is expressed as map units across the intervals shown. Numbers in parentheses denote the percentage of wild-type crossover frequency.
Data on hdm single mutant are from Joyce et al. (2009, accompanying article in this issue).
The pch2 mutation exacerbates the crossover recombination defect in hdm mutants:
Checkpoints often function to slow progression through the cell cycle so that a problem can be corrected. Thus, a defective checkpoint can increase the severity of a mutation that would normally activate the checkpoint. This situation was observed in the hdm; pch2 double mutant, in which X-chromosome nondisjunction was increased from 7.2 to 15.0% and crossing over was reduced a further 36% relative to the hdm single mutant (Table 2). Although pch2 is not required for crossing over in a wild-type background, this result demonstrates that it is required for some of the crossovers that occur in hdm mutants.
Asynapsis does not activate the pch2-dependent checkpoint:
The pch2–dependent checkpoint in C. elegans and S. cerevisiae has been proposed to respond to defects in synapsis (Bhalla and Dernburg 2005; Wu and Burgess 2006). We tested whether asynapsis could cause delays in Drosophila meiotic prophase by examining a c(3)G mutant. C(3)G encodes a transverse filament protein that may be the functional homolog of S. cerevisiae Zip1 (Page and Hawley 2004). Because synapsis is abolished in c(3)G68 mutant females, we used ORB staining to mark the oocytes (Lantz et al. 1994). The oocyte is identified by a concentration of cytoplasmic ORB protein around 1 cell in the 16-cell cyst. To confirm the efficacy of ORB staining at identifying the two-oocyte phenotype, we assayed how often ORB protein concentrated in the cytoplasm of 2 cells rather than 1 cell in late-pachytene hdm mutant cysts. We found a two-oocyte phenotype in 20% (n = 30) of region 3 cysts in hdm mutants, comparable to the frequencies previously reported in some DSB repair mutants (Gonzalez-Reyes et al. 1997; McCaffrey et al. 2006). In contrast, we did not observe any region 3 cysts with two oocytes in c(3)G68 mutant (0%, n = 71) or wild-type control females (0%, n = 43), indicating that asynapsis does not cause pachytene delay phenotypes in Drosophila.
DISCUSSION
Mutations in exchange class and DSB repair genes cause delays in pachytene progression:
As in most other cell types, there is a checkpoint response to unrepaired DSBs in Drosophila female meiosis (Ghabrial and Schupbach 1999). Our results define a second and distinct DSB-independent checkpoint that operates during pachytene. Mutations in exchange class (e.g., hdm, mei-9, and mus312) and DSB repair genes (e.g., okr, spn-A, spn-B, spn-D, and mei-41) cause a delay in the timing of at least two events: the chromatin-remodeling response to DSBs (phosphorylation of HIS2AV) and, through a process that is DSB independent, the selection of a single oocyte. Both of the delay phenotypes that we have observed can be explained if their timing is linked to the progression through pachytene (Figure 6). A delay in pachytene has also been proposed to explain why Rad51 foci, DSB response markers, persist into late pachytene in synapsis-defective C. elegans mutants (Carlton et al. 2006).
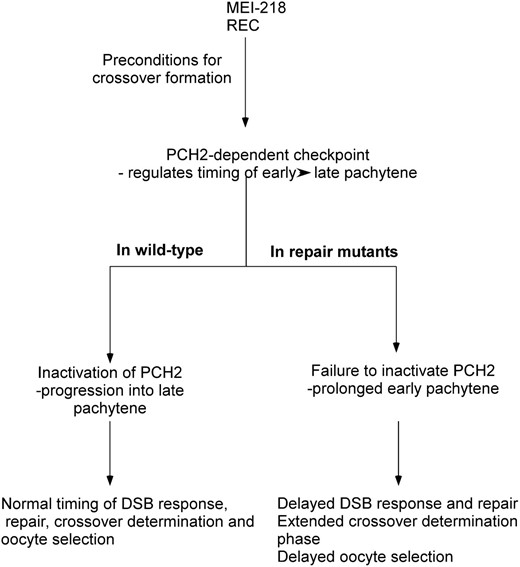
Model for how precondition genes, the PCH2-dependent checkpoint, and the exchange genes interact to affect pachytene progression. Crossover determination requires the precondition gene products MEI-218 and REC. Crossover determination generates a substrate, which activates the PCH2-dependent checkpoint and delays pachytene progression until the early functions of DSB repair and exchange proteins alleviate the activator of PCH2. This process—from crossover determination to the early function of the DSB repair and exchange genes—is DSB independent, but the timing relative to DSB formation is not known. Since the consequence of a DSB repair or exchange mutant is observed in early pachytene (the γ-HIS2AV response to DSBs), their activity may turn off the checkpoint close to this time. In DSB repair and exchange mutants, the PCH2-dependent checkpoint remains active, which has several effects on prophase events. Activation of the PCH2-dependent checkpoint early in pachytene leads to the delayed appearance of γ-HIS2AV foci observed in DSB repair and exchange mutants. In the case of the two-oocyte phenotype, the transition into late pachytene could be required for one of the two pro-oocytes to convert to a nurse cell fate. When PCH2 remains active, the transition into late pachytene is delayed. Finally, the active PCH2-dependent checkpoint may prolong the “crossover determination” phase, which in some cases can result in additional crossovers.
pch2 is required for a pachytene checkpoint that is independent of DSBs:
Our results show that the proposed pachytene checkpoint depends on the Drosophila pch2 ortholog. In C. elegans and S. cerevisiae, pch2 is required for a DSB-independent checkpoint pathway that responds to synapsis defects (Bhalla and Dernburg 2005; Wu and Burgess 2006). In Drosophila, however, two sets of observations suggest that synapsis defects may not be the trigger of the PCH2-dependent checkpoint. First, our immunofluorescent studies using the SC components C(2)M (data not shown) and C(3)G suggest that exchange mutants (e.g., hdm and mei-9) and DSB repair mutants (e.g., okr and spn-D) are able to form SC. Indeed, complete reconstructions from electron micrographs have shown that mei-9 mutants synapse their chromosomes normally (Carpenter 1979). Second, c(3)G mutations, which abolish synapsis in Drosophila, do not trigger pachytene delays.
Because the exchange mutants have reduced crossover formation but no detectable synapsis defects, our results point to a defect in the pathway that leads to crossovers as the mechanism that triggers pachytene delays. Interestingly, the synapsis mutants analyzed in C. elegans and S. cerevisiae also have defects in crossover production, suggesting there may be a common mechanism to activate the PCH2-dependent checkpoint in all three species. In fact, a non-null crossover-defective zip1 allele in budding yeast was reported to exhibit normal synapsis by immunofluorescence but still activated the PCH2-dependent checkpoint (Mitra and Roeder 2007). In C. elegans and S. cerevisiae, it could be a secondary consequence of the synapsis defects on the crossover pathway that triggers the pch2-dependent checkpoint pathway.
A model for the determination and monitoring of crossover formation:
As with most checkpoints, there are two components to the PCH2-dependent checkpoint. First, activation of the checkpoint signal must depend on a specific substrate in the cell (such as a DSB in the canonical DNA repair checkpoint). Second, there must be a process that turns off the checkpoint signal. The first component of the PCH2-dependent checkpoint depends on the precondition genes, but not on DSB formation. This is based on the observation that mutations in the precondition genes mei-218 and rec suppress the pachytene delay phenotypes while mutations in the DSB formation genes mei-P22 and mei-W68 do not (Table 1). Similar to the original proposal by Carpenter and Sandler (1974), precondition genes such as mei-218 and rec may be required for establishing the pattern of crossovers, such as their distribution and frequency. Both MEI-218 and REC have homology to MCM proteins (Blanton et al. 2005) and recently a hypomorphic allele of the Drosophila mcm5 gene has been found to have a precondition mutant phenotype (Lake et al. 2007). In addition to their role in DNA replication, MCM proteins affect chromosome structure in as yet poorly defined ways (Bailis and Forsburg 2004) and may interact with checkpoint and recombination proteins (Bailis et al. 2008). Thus, the function of precondition gene products could include modifying the meiotic chromosome structure (see Zickler and Kleckner 1999), which in turn interacts with and is required to activate the PCH2-dependent checkpoint signal.
These data are also consistent with previous models that place mei-218 function upstream of exchange genes in the generation of crossovers (Sekelsky et al. 1995; Bhagat et al. 2004). However, mei-218 and rec also suppressed the pachytene delay phenotypes observed in the DSB repair mutants, spn-A and spn-D, which provides evidence for precondition gene products functioning early, during, or prior to the first steps of DSB repair. While it is possible that the effects of precondition mutations on pachytene progression and crossover formation are not related, the simplest model is that precondition genes function early in the repair process, close to the time of DSB formation, to commit a subset of DSBs to the crossover pathway (Figure 6). Such an early time for crossover decision has also been proposed in budding yeast (Bishop and Zickler 2004; Fung et al. 2004) and in C. elegans (Couteau and Zetka 2005).
The second component, which turns off the checkpoint signal, depends on a previously undescribed DSB-independent function of the DSB repair and exchange genes. If the initial activation of the checkpoint involves precondition gene-dependent changes in chromosome structure, then the DSB repair and exchange genes may function to reverse these changes or block how they interact with the checkpoint. Importantly, a defect in any one of the DSB repair or exchange proteins can trigger the checkpoint. One possibility is that all the proteins required for meiotic recombination preassemble for a “dry run” prior to the actual repair of DSBs. The absence of a functional DSB repair or exchange protein would result in a reduction or impairment of DSB repair complexes capable of generating crossovers and modifying the activity of the PCH2 checkpoint. In support of this hypothesis, exchange gene products are known to form a complex (Sekelsky et al. 1995; Yildiz et al. 2002; Joyce et al. 2009, accompanying article in this issue). Whether the exchange and DSB repair proteins form one or multiple complexes has yet to be determined. Preassembling repair complexes before programmed DSB formation occurs could suppress alternative repair pathways as well as provide a mechanism to ensure the proper number of crossovers.
Another implication of the delay phenotypes is that the exchange and DSB repair genes are required in early pachytene before the phosphorylation of HIS2AV. A function at this time is not surprising for the DSB repair proteins, which are presumably recruited shortly after the break is formed. It is surprisingly early, however, for the exchange genes, considering they are thought to function in the resolution step, relatively late in the repair process (Yildiz et al. 2004; Joyce et al. 2009, accompanying article in this issue). However, if all the proteins required for crossover repair preassemble as proposed above, relatively early mutant phenotypes could be the result.
Although Drosophila pch2 single mutants had no significant change in crossover distribution or frequency, the hdm; pch2 double mutant had fewer crossovers than the hdm single mutant, establishing a functional link between the checkpoint and generating crossovers. The activated PCH2-dependent checkpoint may promote crossover formation in some situations, such as when crossover formation is compromised. Additional crossovers may form due to an extended “window of opportunity” to generate crossovers (Lucchesi and Suzuki 1968; Carlton et al. 2006) (Figure 6) or the activation of additional crossover-promoting gene products.
Despite the sequence conservation of PCH2 in many organisms, a conservation of function is not clear. For example, the PCH2-dependent checkpoint causes a pachytene delay in flies and budding yeast (San-Segundo and Roeder 1999) but apoptosis in nematodes (Bhalla and Dernburg 2005). Most surprising, the mouse PCH2 homolog is required to complete recombination events but may not have a checkpoint function (Li and Schimenti 2007). One way to reconcile these differences may be that PCH2 has a role in regulating the timing of important transitions during pachytene. PCH2 may be constitutively active in early pachytene until turned off by activities of proteins involved in crossover formation. This is supported by the observation that mutations in budding yeast pch2 cause delays in the progression of both the crossover and noncrossover pathways but do not affect the final frequency of these events (Borner et al. 2008). In the future, it will be important to identify what the PCH2-dependent checkpoint responds to. In Drosophila, for example, it will be interesting to know if the proposed interaction of the precondition or repair gene products with the PCH2-dependent checkpoint is restricted to DSB sites or more generally dispersed along the chromosome.
Footnotes
Communicating editor: G. P. Copenhaver
Acknowledgements
We are grateful to Janet Jang and Li Nguyen for technical assistance and Scott Hawley, Sarah Radford, and Ruth Steward for comments on the manuscript. We also thank Jay Tischfield and members of his laboratory for use of the X-ray irradiator and R. Scott Hawley for antibodies. Some stocks used in this study were received from the Bloomington Stock Center and the ORB antibodies were obtained from the Developmental Studies Hybridoma Bank at the University of Iowa, developed under the auspices of the National Institute of Child Health and Human Development. A fellowship from the Busch Foundation to E.F.J. and a grant from the National Science Foundation to K.S.M. supported this work.
References
Borner, G. V., A. Barot and N. Kleckner,
Couteau, F., and M. Zetka,
Lucchesi, J. C., and D. T. Suzuki,
McKim, K., E. F. Joyce and J. K. Jang (Editors),
Mehrotra, S., R. S. Hawley and K. S. McKim,

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}