





Abstract
Nonhomologous end joining (NHEJ) in yeast depends on eight different proteins in at least three different functional complexes: Yku70–Yku80 (Ku), Dnl4–Lif1–Nej1 (DNA ligase IV), and Mre11–Rad50–Xrs2 (MRX). Interactions between these complexes at DNA double-strand breaks (DSBs) are poorly understood but critical for the completion of repair. We previously identified two such contacts that are redundantly required for NHEJ, one between Dnl4 and the C terminus of Yku80 and one between the forkhead-associated (FHA) domain of Xrs2 and the C terminus of Lif1. Here, we first show that mutation of the Yku80 C terminus did not impair Ku binding to DSBs, supporting specificity of the mutant defect to the ligase interaction. We next show that the Xrs2–Lif1 interaction depends on Xrs2 FHA residues (R32, S47, R48, and K75) analogous to those known in other proteins to contact phosphorylated threonines. Two potential target threonines in Lif1 (T417 and T387) were inferred by identifying regions similar to a site in the human Lif1 homolog, XRCC4, known to be bound by the FHA domain of polynucleotide kinase. Mutating these threonines, especially T417, abolished the Xrs2–Lif1 interaction and impaired NHEJ epistatically with Xrs2 FHA mutation. Combining mutations that selectively disable the Yku80–Dnl4 and Xrs2–Lif1 interactions abrogated both NHEJ and DNA ligase IV recruitment to a DSB. The collected results indicate that the Xrs–Lif1 and Yku80–Dnl4 interactions are important for formation of a productive ligase–DSB intermediate.
NONHOMOLOGOUS end joining (NHEJ) is a form of DNA double-strand break (DSB) repair that entails direct religation of DSB termini (Daley et al. 2005). During NHEJ, broken DNA ends must first be recognized and bound, which is the function of the Ku heterodimer required for NHEJ in all organisms (Walker et al. 2001; Weller et al. 2002; Daley et al. 2005). Ends must next be brought together. In mammals, the DNA-dependent protein kinase catalytic subunit (DNA-PKcs) binds to Ku and can synapse DSB termini (Burma and Chen 2004). Yeast lack DNA-PKcs and this function is likely supplied by the Mre11–Rad50–Xrs2 (MRX) complex required for NHEJ in this organism (Chen et al. 2001). Next, damaged nucleotides must be removed and gaps filled by enzymes including Artemis (Ma et al. 2002) and Pol X family DNA polymerases (Nick McElhinny and Ramsden 2004). Finally, the DNA strands must be ligated to restore the continuity of the chromosome, in eukaryotes by DNA ligase IV (Wilson et al. 1997; Grawunder et al. 1998).
Ku, which consists of intertwined Ku70 and Ku80 subunits (Yku70 and Yku80 in yeast), specifically binds DSBs by threading a free DNA end into its ring structure (Walker et al. 2001). Ku70 and Ku80 both harbor conserved Von Willebrand A (VWA) domains, a central β-barrel core that forms the ring, and C-terminal tails (Daley et al. 2005). DNA-PKcs binds the C-terminal tail of mammalian Ku80 via a putative α-helix that is very similar to a C-terminal Yku80 motif we previously implicated in Dnl4 binding (Gell and Jackson 1999; Palmbos et al. 2005). The dynamics of assembly of these proteins at a DSB are poorly understood, but include inward translocation of Ku on the DNA (Kysela et al. 2003). Ku function is not restricted to NHEJ, but includes roles in telomere maintenance (Fisher and Zakian 2005).
The MRX complex consists of three proteins: Mre11, Rad50, and Xrs2/NBS1. Mre11 harbors a conserved nuclease domain important for meiosis and recombination but not NHEJ (Moreau et al. 1999; Zhang and Paull 2005). Rad50 is similar to structural maintenance of chromosome proteins and forms a long-coiled coil with an ABC ATPase and a Zn hook that facilitates dimerization (Hopfner et al. 2002). The third MRX protein is known as Xrs2 in yeast and NBS1 in mammalian cells. Although significantly diverged, both Xrs2 and NBS1 harbor N-terminal forkhead-associated (FHA) and BRCA1 C-terminal (BRCT) domains and interact with Tel1/ATM via their C terminus (Daley et al. 2005; Falck et al. 2005; Becker et al. 2006). Xrs2 possesses intrinsic DNA affinity and is required for MRX binding to DNA and bridging of DSB ends (Trujillo et al. 2003). Similar to Ku, MRX has many functions beyond NHEJ, including telomere maintenance, activation of Tel1/ATM-mediated damage response checkpoints, and support of homologous recombination through stimulation of DSB resection (Assenmacher and Hopfner 2004).
The catalytic subunit of DNA ligase IV (Dnl4 in yeast) has a typical ATP-dependent DNA ligase domain as well as two tandem BRCT domains that allow it to bind in a tight complex to its partner protein XRCC4 (Lif1 in yeast) (Grawunder et al. 1997; Teo and Jackson 2000). XRCC4/Lif1 has an N-terminal globular head, a central coiled-coil domain, which is the docking site for the ligase BRCT domains, and a structurally uncharacterized but functionally essential C terminus (Sibanda et al. 2001; Daley et al. 2005). XRCC4/Lif1 also binds to a third XRCC4-related protein known as Nej1 in yeast and XLF/Cernunnos in mammalian cells (Kegel et al. 2001; Ahnesorg et al. 2006; Callebaut et al. 2006). Unlike Ku and MRX, the only known role of DNA ligase IV is NHEJ.
In addition to the protein contacts that individually assemble the Ku, MRX, and DNA ligase IV complexes, NHEJ demands a coordinated series of interactions between them. In previous work, we used systematic two-hybrid analysis to identify three such putative interactions (Palmbos et al. 2005). One interaction, between Dnl4 and the C terminus of Yku80, could be selectively disabled by yku80Δ605 C-terminal truncation such that the telomere functions of Ku remained intact. The other interaction, between the FHA domain of Xrs2 and the C terminus of Lif1, could be selectively disabled by xrs2ΔFHA mutation such that the telomere, checkpoint, and recombination functions of MRX remained intact. Combining these two mutations severely impaired NHEJ while individually they had a much smaller functional effect, suggesting redundancy in support of Dnl4–Lif1 action. The importance of the Xrs2 FHA domain for in vitro and in vivo interaction with Lif1 was recently confirmed by others (Matsuzaki et al. 2008) who further made refined mutations of Lif1 and implicated S383 as a target residue for binding by the Xrs2 FHA domain.
Here, we analyze the Yku80–Dnl4 and Xrs–Lif1 interactions and their role in NHEJ in further detail. DNA binding studies confirm that Ku complex made with Yku80Δ605 binds to DSBs normally. Mutational analysis of the Xrs2 FHA domain shows that the Xrs2–Lif1 interaction depends on residues known in other FHA domains to contact phosphorylated threonine residues in peptide ligands. Accordingly, two Lif1 threonine residues, T417 and T387, are shown to be critical to the Xrs2–Lif1 interaction, although confirmation of their phosphorylation has not been possible. Finally, we demonstrate failed recruitment of Dnl4 to a DSB in vivo when both the Yku80–Dnl4 and Xrs2–Lif1 interactions are selectively disrupted. These results are discussed relative to the recent findings of Matsuzaki et al. (2008) and the multiple interactions required to stabilize DNA ligase IV at a DSB during NHEJ.
MATERIALS AND METHODS
Yeast strains and growth:
Yeast strain PJ694α (MATα, GAL2-ADE2, gal4Δ, gal80Δ, his3Δ200, leu2-3,112, LYS2∷GAL1-HIS3, met2∷GAL7-lacZ, trp1-901, ura3-52), which harbors Gal4 responsive HIS3, ADE2, and LacZ markers, was used for two hybrid studies (Uetz et al. 2000). Yeast strains BY4741 (MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0) and BY4742 (MATα, his3Δ1, leu2Δ0, lys2Δ0, ura3Δ0) were used for Ku purification (Brachmann et al. 1998). Diploids of these a/α strain pairs were made by mating to combine various expression constructs. The HO(+2) suicide deletion strain YW1276 [MATα-inc, ade2∷HOSD(+1)∷STE3-MET15, his3Δ1, leu2Δ, met15Δ, ura3Δ] was previously described (Palmbos et al. 2005). The chromatin immunoprecipitation (ChIP) strain YW1752 (MATa-inc∷ChIP-QPCR∷URA3, can1Δ∷repair-QPCR, DNL4- Myc13∷HisMX6, gal1∷HOcs∷HOcod, his3Δ1, leu2Δ0, met15Δ0, ura3Δ) was previously described (Wu et al. 2008). Both YW1276 and YW1752 are direct descendants of BY4741/BY4742. All strains were grown at 30° in a rich medium containing 1% yeast extract, 2% peptone, 2% dextrose, and 40 μg/ml adenine (YPAD) or a synthetic defined (SD) medium with either 2% glucose or galactose as needed.
Oligonucleotides:
The sequences of oligonucleotides used to generate the various mutations and expression constructs are available upon request.
Site-directed mutagenesis:
Single and multiple amino acid substitutions were generated in YW1276 at the chromosomal gene locus with expression driven by the native promoter using a previously described PCR-based “pop-in/pop-out” method (Palmbos et al. 2005). All alleles were sequenced to verify the presence of the expected and absence of unexpected mutations. The coding sequence was subsequently amplified from these strains for construction of mutant expression and two-hybrid plasmids.
Plasmids:
Yeast two-hybrid Gal4 DNA binding domain (bait, pOBD2) and transcriptional activating domain (prey, pOAD) vectors were previously described (Uetz et al. 2000). Vectors for tagged expression for purification were previously described (Deshpande and Wilson 2007). Briefly, they include the tag pairs FLAG–His6 or calmodulin binding peptide (CBP)-His6 on URA3- and LEU2-marked 2μ-backbones, respectively. Insertion of wild-type or mutant Ku sequences into these vectors was accomplished by gap repair to create FlagHis6–Yku70 and CBP–Yku80. Correct constructs were identified by expression of the expected protein and subsequent sequence verification.
Protein purification:
Diploid yeast strains were configured to coexpress FlagHis6–Yku70 and CBP–Yku80 to allow sequential purification using His6 and CBP tags as follows. Protein expression was induced by overnight growth in 600 ml galactose SD medium selective for the plasmids. Next, ∼10 g of cells were harvested by centrifugation, resuspended in buffer A (10 mm Tris pH 7.5, 20 mm imidazole, 0.5 m KCl, 5 mm MgCl2, 10% glycerol, 10 mm 2-mercaptoethanol, 0.1% NP40, and a protease inhibitor cocktail consisting of 1 mm PMSF, 2 μg/ml aprotinin, 2 μg/ml leupeptin, 1 μg/ml pepstatin), and lysed using zirconium beads (Biospec) and a Turbomix vortexer (Scientific Instruments). Lysates were cleared by centrifugation at 15,000 g and proteins bound in batch to 0.5 ml packed volume Ni-NTA agarose (QIAGEN) for 1 hr at 4°. The resin was transferred to a disposable column (BioRad), washed with 10 column volumes of buffer A, and bound proteins eluted into buffer B (10 mm Tris pH 7.5, 250 mm imidazole, 0.5 m KCl, 1 mm MgCl2, 2 mm CaCl2, 10% glycerol, 10 mm 2-mercaptoethanol, 0.1% NP40, and protease inhibitor cocktail). Eluates were immediately incubated with 50 μl calmodulin affinity resin (Stratagene) for 1.5 hr at 4°, washed with buffer C (50 mm Tris pH 7.5, 0.5 m KCl, 1 mm MgCl2, 2 mm CaCl2, 10% glycerol, 10 mm 2-mercaptoethanol, 0.1% NP40, and protease inhibitor cocktail) and bound proteins eluted with 5 × 100 μl buffer C containing 2 mm EGTA instead of CaCl2. These final fractions were dialyzed against protein storage buffer (50 mm KCl, 10 mm Tris pH 7.5, 0.1 mm EDTA, 50% glycerol, 1 mm DTT) and stored at −20°.
Electrophoretic mobility shift assay:
Oligonucleotides OW1543 (5′-GTC TTT GGT TCA TGA TCT TCC CAT ACA ATT GCC TCA ATG TCT CTT GTT TTC AAA GCT GAT AAT GA) and OW2564 (5′-ATT ATC AGC TTT GAA AAC AAG AGA CAT TGA GGC AAT TGT ATG GGA AGA TCA TGA ACC AAA G) were annealed by heating to 100° followed by slow cooling, electrophoresed on a 8% polyacrylamide gel, and purified by excising the duplex DNA followed by passive elution and ethanol precipitation. This purified duplex probe was then end-labeled with γ-32P using polynucleotide kinase (NEB) and diluted to 50 fmol/μl in electrophoretic mobility shift assay (EMSA) buffer (25 mm Tris pH 7.5, 100 mm NaCl, 30 mm KCl, 0.1 mm EDTA, 0.05% Triton-X, 50 μg/mL BSA, 5% glycerol, 2 mm DTT). Five microliters were added to 5 μl of purified protein in protein storage buffer and incubated for 30 min at 4°. Binding reactions were run on a 4% native polyacrylamide gel (29:1) in TE buffer (45 mm Tris pH 8.0, EDTA 1 mm) for 45 min at 60 V. The gel was dried and imaged using a phosphorimager.
Yeast two hybrid:
Yeast two-hybrid assays were performed in PJ694α as previously described (Palmbos et al. 2005). Briefly, wild-type or mutant Xrs2(1–125) coding sequences were introduced into the bait vector pOBD2 by gap repair as described above. Strains were then transformed with the Lif1(245–421) prey. Alternatively, wild-type or mutant Lif1(245–421) coding sequences were introduced into the prey vector pOAD by gap repair, with subsequent transformation with Xrs2(1–125) bait constructs. Strains were grown for 2 days in SD medium lacking leucine and tryptophan and spotted to plates lacking histidine or adenine followed by 3- or 5-days growth, respectively. β-galactosidase activity was determined as previously described (Rupp 2002).
Suicide deletion:
The HO(+2) suicide deletion assay for monitoring precise and imprecise NHEJ has been previously described (Della et al. 2004) and was used according to published procedures (Palmbos et al. 2005). See results and the figure legends for a description of the method and its interpretation.
Plasmid recircularization NHEJ assay:
The LEU2-marked plasmid pRS315 was cut with the restriction enzyme AgeI (Roche) to create a DSB within LEU2 bearing compatible 4-base 5′ overhangs. The cut plasmid (100 ng) was cotransformed into the suicide deletion strains generated above with 10 ng of the supercoiled HIS3-marked plasmid pRS413 (Brachmann et al. 1998). Relative repair efficiency was measured as the ratio of Leu+ colonies to His+ colonies.
Chromatin immunoprecipitation:
ChIP methodology and the associated DSB repair assay have been described and validated in detail (Wu et al. 2008). Briefly, yeast strain YW1752 contains a recognition site for the HO endonuclease in the native GAL1 promoter, and the HO coding sequence under control of that same GAL1 promoter (gal1∷HOcs∷HOcod). yku80Δ605–629 and lif1-T387A/T417A alleles were added to this background by mating YW1752 to the otherwise isogenic suicide deletion strains generated above, followed by sporulation and selection for appropriate recombinants. A DSB in the GAL1 promoter was induced by growing these strains overnight in YPA with 3% glycerol and then adding 2% galactose for 60 min. HO expression was terminated by adding 2% glucose and cells harvested at various time points. For monitoring chromosome breakage and repair, PCR was performed on purified genomic DNA using primers that flank the HO cut site, so that only intact GAL1 alleles could be amplified. The fraction of intact sites was assessed by comparing to competitive amplification of a different unbroken chromosomal locus with the same primer pair (Wu et al. 2008). For ChIP, a Myc13 tag was inserted at the C terminus of the endogenous DNL4 gene (DNL4-Myc13∷HisMX6) as described (Longtine et al. 1998). Cell lysis and ChIP were performed essentially as described (Aparicio et al. 2005). PCR detection utilized a primer pair targeted to a sequence immediately adjacent to the GAL1 DSB site. We again utilized a competitive PCR approach to control for background amplification and reveal specific binding of Dnl4 to the DSB site as excess amplification of the GAL1 product (Wu et al. 2008).
RESULTS
The Yku80 C terminus is not required for DNA end binding:
Our previous work identified a yeast two-hybrid interaction between Dnl4 and Yku80 (Palmbos et al. 2005). Truncation of the C-terminal amino acids 605–629 of Yku80 blocked this interaction and impaired NHEJ in vivo. However, Ku must also bind to DSB ends, which could provide an alternative and nonexclusive explanation for the NHEJ defect caused by truncated Yku80. To address this issue, we performed EMSA using wild-type FlagHis6-Yku70/CBP-Yku80 Ku heterodimer and similarly tagged Yku80Δ605–629 mutant Ku heterodimer (KuΔ605) purified from yeast. Both the wild-type and KuΔ605 forms showed equivalent multiple slow-migrating bands with a 65-mer probe indicative of dsDNA binding by multiple Ku heterodimers (Figure 1). Thus, the extreme C terminus of Yku80 is not required for DSB binding by Ku.
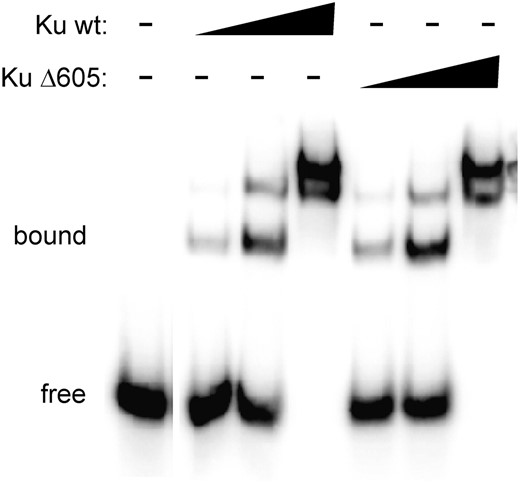
Deletion of the extreme Yku80 C terminus does not impair DSB binding. EMSA was performed using a 65-bp radiolabeled dsDNA probe (200 fmol) and increasing amounts (20 ng, 100 ng, 500 ng) of tagged wild-type or Yku80Δ605 Ku (purified as FlagHis6-Yku70/CBP-Yku80 Ku heterodimer). Each showed equivalent dsDNA binding.
Identification of conserved Xrs2 FHA domain residues required for Lif1 binding:
Previous work also identified an interaction between Xrs2 and Lif1 (Chen et al. 2001), which we refined to the N-terminal 125 amino acids of Xrs2, a region encompassing its FHA domain, and the C terminus of Lif1 (Palmbos et al. 2005). Subsequent work by Matsuzaki et al. (2008) identified two mutations (S47A/H50A and G31E) that disrupt the FHA domain and correspondingly block an in vitro interaction with Lif1. We sought to determine more specifically which residues of the Xrs2 FHA domain mediate its interaction with Lif1 and explain its role in NHEJ. FHA domains are protein interaction motifs known to bind peptides with phosphorylated threonine residues (Durocher et al. 2000; Durocher and Jackson 2002). When aligned over many species, the Xrs2/NBS1 N terminus was seen to retain a cluster of highly conserved amino acids important for phosphothreonine contact in crystallized FHA domains (Durocher et al. 2000; Pike et al. 2001), specifically Xrs2 R32, S47, R48, and K75 (Figure 2A). These residues localized as expected to a putative binding surface in a modeled Xrs2 FHA structure (Figure 2B). We created alanine substitutions of these residues to examine their importance in Lif1 binding. Each was incorporated into Xrs2(1–125) two-hybrid baits and tested for interaction with the Lif1(245–421) prey. R32A, S47A, R48A, and K75A mutations all completely blocked growth on indicator plates lacking adenine (Figure 3A). Interestingly, the Xrs2(1–125) bait does show some auto-activation, i.e., with prey vector controls, evident on indicator plates lacking histidine. S47A and K75A mutants reduced the two-hybrid readout only to this background, but R32A and R48A mutants abrogated even the auto-activation (Figure 3A). This may indicate that the latter mutations destabilize the protein while S47A and K75A mutants are stable enough to support the nonspecific activation. Despite this, we cannot be certain that any of the mutant FHA domains studied by us or others (Matsuzaki et al. 2008) are folded properly. The consistency of the phenotype over a large series of mutations that typify FHA domain function in other proteins is nonetheless consistent with the hypothesis that the Xrs2 FHA domain binds a Lif1 phosphopeptide.
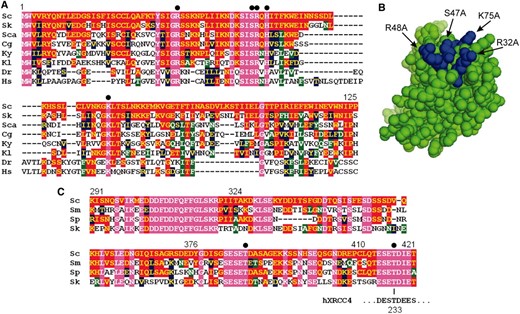
Alignments of the Xrs2/NBS1 FHA domain and the Lif1 C terminus. (A) Alignment of Xrs2 and NBS1 FHA domains. Solid circles identify residues mutated in this study. (B) Predicted structure of the Saccharomyces cerevisiae Xrs2 FHA domain, generated by mapping multiple sequence alignments onto the N-terminal Rad53 FHA domain crystal structure (PDB 1G6G) (Durocher et al. 2000) with the Phyre domain prediction software (Kelley et al. 2000). Residues mutated in this study are labeled and highlighted in blue. Their side chains cluster on the surface at which phosphopeptides bind to Rad53 other crystallized FHA domains. (C) Alignment of Lif1 from Saccharomyces species. Also shown is the human XRCC4 sequence surrounding threonine 233 that is phosphorylated and bound by the PNKP FHA domain. Solid circles identify the Lif1 threonine residues shown here to be recognized by Xrs2. For each alignment, residues conserved in all sequences are shaded in magenta. Residues conserved with S. cerevisiae are shaded in red. More or less conservative substitutions are shown in blue or green, respectively. Species designations are: Sc, S. cerevisiae; Sk, S. kudriavzevii; Sca, S. castelli; Sm, S. mikatae; Sp, S. paradoxus; Cg, Candida glabrata; Ky, Kluyveromyces yarrowii; Kl, K. lactis; Dr, Danio rerio; and Hs, Homo sapiens.
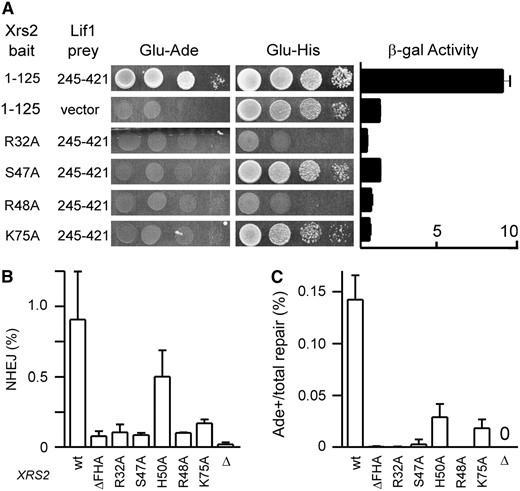
Mutational analysis of the Xrs2 FHA domain. (A) Wild-type and mutant Xrs2(1–125) two-hybrid baits were tested for interaction with the Lif1(245–421) prey by spotting 10-fold serial dilutions of strains to plates lacking adenine (Glu −Ade) or histidine (Glu −His), as well as by measuring β-galactosidase activity. Empty prey vector reveals the degree of Xrs2(1–125) bait auto-activation, most evident on Glu −His. Mutation of conserved FHA domain residues blocked yeast two-hybrid interaction with Lif1, and in some cases also reduced the auto-activation. (B) yku80Δ605–629 yeast strains additionally bearing the indicated XRS2 mutations were analyzed by HO(+2) suicide deletion analysis by plotting the fraction of cells that repaired the DSB and survived, which primarily reveals defects in precise NHEJ. Results are the mean ± standard deviation of three independent experiments. xrs2-R32A, S47A, and R48A mutations impaired NHEJ similarly to xrs2ΔkanMX4 complete gene deletion (Δ) or FHA domain deletion (ΔFHA). (C) Similar to B, except plotting Ade+ suicide deletion repair events as a fraction of the total repair, which in this system reveals defects in the highly NHEJ-dependent HO(+2) processed repair event.
Mutation of conserved Xrs2 FHA domain residues specifically impairs NHEJ:
We next examined whether these conserved FHA residues are also essential for NHEJ. To monitor NHEJ, we primarily used the HO(+2) suicide deletion assay we have described in detail (Della et al. 2004; Palmbos et al. 2005). Briefly, an HO endonuclease-mediated DSB is created at ADE2. Most cells die, but ∼1% repair the DSB by precise NHEJ, which is monitored by counting the fraction of cells that survive HO induction. Among the survivors, ∼1% (i.e., ∼0.01% of all cells) repair the DSB by the HO(+2) processed NHEJ event. HO(+2) places ADE2 in frame and so is monitored by counting the Ade+ fraction among survivors. HO(+2) is entirely dependent on Ku-dependent NHEJ and provides a more sensitive and specific assay. R32A, S47A, R48A, and K75A mutations were introduced at the XRS2 locus in the suicide deletion strain, in addition to H50A mutation. This histidine does not directly contact the peptide ligand in known FHA structures but has been used as a defining residue in previous analyses, for example of Xrs2 (Matsuzaki et al. 2008). Similar to complete deletion of the FHA domain (xrs2ΔFHA) (Palmbos et al. 2005), these alanine replacements alone had no effect on total suicide deletion NHEJ and caused at most a twofold reduction of HO(+2) NHEJ (Table 1). To increase sensitivity for FHA NHEJ defects, we also introduced the alanine replacements into yeast bearing the yku80Δ605–629 allele, since a synergistic decrease in NHEJ efficiency is observed in yku80Δ605–629 xrs2ΔFHA yeast (Palmbos et al. 2005) (Table 1). Xrs2 R32A, S47A, and R48A mutations each nearly completely blocked total and HO(+2) NHEJ in combination with yku80Δ605–629 (Figure 3, B and C). This NHEJ defect was equivalent to xrs2ΔFHA and demonstrates the importance of the FHA domain itself in NHEJ. Xrs2 H50A and K75A showed intermediate NHEJ defects, suggesting residual FHA domain function in these mutants.
NHEJ phenotype of Xrs2 FHA domain mutants
YKU80 | yku80Δ605–629 | |||
|---|---|---|---|---|
| XRS2 | Total | HO(+2) | Total | HO(+2) |
| wt | 1a | 1 | 2 | 8 |
| Δ∷kanMX4 | 8 | 100 | 125 | ∞ |
| ΔFHA | 1 | 1.7 | 30 | >1000 |
| R32A | 1 | 1.8 | 22 | >1000 |
| S47A | 1 | 2 | 27 | >1000 |
| R48A | 1 | 1.6 | 24 | >1000 |
| H50A | 1 | 1 | 5 | 37 |
| K75A | 1 | 1.2 | 14 | 60 |
YKU80 | yku80Δ605–629 | |||
|---|---|---|---|---|
| XRS2 | Total | HO(+2) | Total | HO(+2) |
| wt | 1a | 1 | 2 | 8 |
| Δ∷kanMX4 | 8 | 100 | 125 | ∞ |
| ΔFHA | 1 | 1.7 | 30 | >1000 |
| R32A | 1 | 1.8 | 22 | >1000 |
| S47A | 1 | 2 | 27 | >1000 |
| R48A | 1 | 1.6 | 24 | >1000 |
| H50A | 1 | 1 | 5 | 37 |
| K75A | 1 | 1.2 | 14 | 60 |
wt, wild type.
Numbers indicate the average fold decrease as compared to YKU80 XRS2 yeast for total and HO(+2) NHEJ in the suicide deletion assay.
NHEJ phenotype of Xrs2 FHA domain mutants
YKU80 | yku80Δ605–629 | |||
|---|---|---|---|---|
| XRS2 | Total | HO(+2) | Total | HO(+2) |
| wt | 1a | 1 | 2 | 8 |
| Δ∷kanMX4 | 8 | 100 | 125 | ∞ |
| ΔFHA | 1 | 1.7 | 30 | >1000 |
| R32A | 1 | 1.8 | 22 | >1000 |
| S47A | 1 | 2 | 27 | >1000 |
| R48A | 1 | 1.6 | 24 | >1000 |
| H50A | 1 | 1 | 5 | 37 |
| K75A | 1 | 1.2 | 14 | 60 |
YKU80 | yku80Δ605–629 | |||
|---|---|---|---|---|
| XRS2 | Total | HO(+2) | Total | HO(+2) |
| wt | 1a | 1 | 2 | 8 |
| Δ∷kanMX4 | 8 | 100 | 125 | ∞ |
| ΔFHA | 1 | 1.7 | 30 | >1000 |
| R32A | 1 | 1.8 | 22 | >1000 |
| S47A | 1 | 2 | 27 | >1000 |
| R48A | 1 | 1.6 | 24 | >1000 |
| H50A | 1 | 1 | 5 | 37 |
| K75A | 1 | 1.2 | 14 | 60 |
wt, wild type.
Numbers indicate the average fold decrease as compared to YKU80 XRS2 yeast for total and HO(+2) NHEJ in the suicide deletion assay.
xrs2ΔFHA mutation was previously shown to preserve the checkpoint and recombination functions of MRX (Palmbos et al. 2005; Shima et al. 2005). To verify that this was also true of our alanine mutants, their survival on media containing camptothecin or hydroxyurea was determined. In contrast to xrs2Δ yeast, none of the alanine replacement mutants, whether alone or in combination with yku80-Δ605–629, was hypersensitive to camptothecin or hydroxyurea, confirming their specificity to NHEJ (data not shown).
Identification of conserved Lif1 threonine residues required for Xrs2 binding:
Our previous two-hybrid screen identified the Xrs2-interacting region of Lif1 as amino acids 245–421 (construct A in Figure 4A) (Palmbos et al. 2005). Because no known protein domains are evident in this region, we generated deletions of the Lif1(245–421) prey (constructs B–G in Figure 4A) and monitored their interaction with the Xrs2(1–125) bait using the β-galactosidase reporter (Figure 4B). Lif1(291–421) (construct D) was the minimal domain necessary to support the strongest interaction with Xrs2. Lif1(324–421) (E) and Lif1(376–421) (F) also supported the two-hybrid interaction, albeit more weakly. Interestingly, Lif1(245–410) (C), which lacks only the last 11 amino acids, did not interact with Xrs2. These results pointed to residues 376–421, and more specifically 410–421, as the region of Lif1 recognized by Xrs2. Lif1(410–421) (G) did not interact with Xrs2, but this construct contained only 11 amino acids and may not fold properly. Interestingly, this interpretation differs from that of a recent similar two-hybrid experiment where Lif1 residues 375–402 were suggested to be most essential (Matsuzaki et al. 2008), but examination of that data reveals an appreciable defect for the construct lacking residues 402–421.
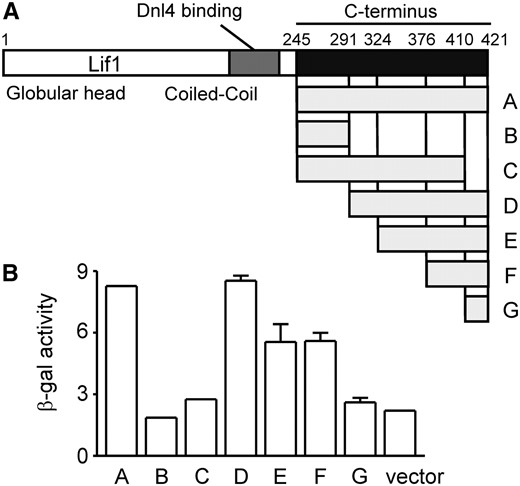
Mapping the Lif1 C-terminal region that interacts with Xrs2. (A) Lif1 is shown in schematic form, with the span of various yeast two-hybrid prey constructs indicated below. Specific amino acid endpoints were 245–421 (A), 245–291 (B), 245–410 (C), 291–421 (D), 324–421 (E), 376–421 (F), and 410–421 (G). (B) Yeast two-hybrid interaction with wild-type Xrs2(1–125) bait was assessed by β-galactosidase activity, which identified the extreme C-terminal Lif1 region as most important.
We next examined alignments of Lif1 amino acids 291–421 to find conserved residues that might be recognized by the Xrs2 FHA domain. This analysis focused on Saccharomyces yeasts because of the high degree of divergence in the XRCC4/Lif1 C terminus, even among these closely related species (Figure 2C). Nevertheless, two regions showed a high degree of conservation, each containing threonine residues within acidic patches. Remarkably, these regions showed a great similarity to the phosphothreonine site of XRCC4 that is recognized by the polynucleotide kinase (PNKP) FHA domain (Figure 2C) (Koch et al. 2004). Thus, although the C termini of Lif1 and XRCC4 cannot be easily aligned, it seemed likely that Lif1 T387 and T417, by virtue of their analogous positions in this motif, might be the binding targets of the Xrs2 FHA domain. We therefore replaced these threonines with alanine, both alone and in combination, and tested for interaction with the Xrs2 FHA domain (Figure 5A). T417A mutation reduced β-galactosidase activity almost to background levels, although some residual growth was observed on the plates lacking adenine (just visible in Figure 5A). T387A mutants substantially supported the interaction with Xrs2(1–125), but did cause a two- to threefold reduction in β-galactosidase activity and reduced growth on plates without adenine. Combined Lif1 T417A/T387A mutation abolished the interaction with Xrs2. These amino acids together thus appear to account for the Xrs2 interaction, but with T417 having the greatest role.
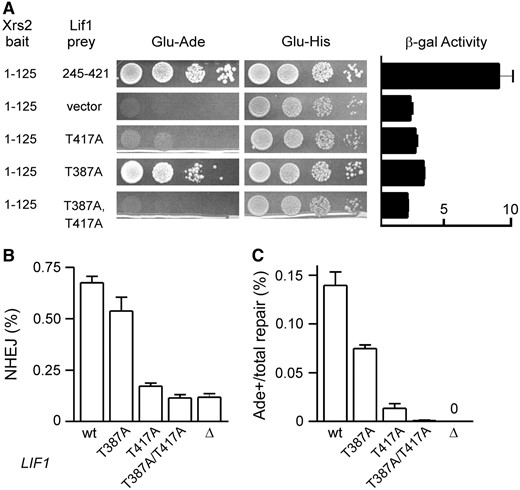
Mutational analysis of the C terminus of Lif1. (A) Two-hybrid analysis similar to Figure 3A, now mutating Lif1 residues in the context of the Lif1(245–421) prey. Combined Lif1 T387A/T417A mutation blocked its interaction with Xrs2(1–125) bait more than each mutation separately, with T417A having the greatest individual effect. (B and C) Lif1 T387A and T417A mutations were tested by suicide deletion analysis in the yku80Δ605–629 background as in Figure 3, B and C. Combined yku80Δ605–629 lif1-T387A/T417A mutations blocked NHEJ as completely as lif1Δ mutation in this assay.
Mutation of Lif1 T387A and T417A impairs NHEJ epistatically to Xrs2 FHA mutation:
To determine if these Lif1 threonines were also necessary for efficient NHEJ, we introduced lif1-T387A, T417A, and T387A/T417A alleles into YKU80 and yku80Δ605–629 suicide deletion strains. As with XRS2 FHA mutants, these LIF1 mutants showed wild-type levels of total NHEJ and less than twofold reduction in HO(+2) NHEJ when tested alone (Table 2) but strongly impaired NHEJ when combined with yku80Δ605–629 (Table 2 and Figure 5, B and C). As predicted by the two-hybrid results, the effect was greater for lif1-T417A, but even lif1-T387A measurably impaired NHEJ in the yku80Δ605–629 background. The lif1-T387A/T417A yku80Δ605–629 combination strongly and synergistically reduced NHEJ to levels comparable to complete LIF1 deletion (lif1Δ). These results demonstrate that these threonines, like the Xrs2 FHA domain, are required for an NHEJ function of Lif1 that is only fully unmasked by yku80Δ605–629 mutation. Interestingly, unlike these alanine mutants, deletion of the Lif1 C terminus (lif1Δ245–421) had a severe effect on NHEJ similar to lif1Δ, suggesting that the role of this protein region extends beyond just Xrs2 binding (data not shown). Moreover, the markedly different phenotype of Lif1 C-terminal deletion as compared to the point mutants strongly suggests that the latter are not grossly destabilizing the Lif1 C terminus. Similar conclusions were reached by Matsuzaki et al. (2008) studying mutants that included T387 alteration.
NHEJ phenotype of Lif1 C-terminal mutants
YKU80 | yku80Δ605–629 | |||
|---|---|---|---|---|
| LIF1 | Total | HO(+2) | Total | HO(+2) |
| wt | 1a | 1 | 2 | 8 |
| Δ∷kanMX4 | 16 | >1000 | 20 | ∞ |
| Δ410-421 | 1.6 | 1.5 | 16 | 68 |
| T387A | 1 | 1.2 | 4 | 14 |
| S383A, S385A, T387A | 1 | 1.4 | 4 | 13 |
| T413A | 1.6 | 5 | 1 | 10 |
| S415A | 1 | 1 | 4 | 16 |
| T417A | 1.4 | 1.7 | 14 | 79 |
| T417E | NDb | 1.4 | 10 | 74 |
| T417D | 1 | 1 | 8 | 58 |
| T421A | 1 | 1 | 3 | 8 |
| T413A, S415A, T417A, T421 | 1.5 | 1.6 | 19 | 100 |
| T387A, T417A | 2 | 1 | 20 | >1000 |
YKU80 | yku80Δ605–629 | |||
|---|---|---|---|---|
| LIF1 | Total | HO(+2) | Total | HO(+2) |
| wt | 1a | 1 | 2 | 8 |
| Δ∷kanMX4 | 16 | >1000 | 20 | ∞ |
| Δ410-421 | 1.6 | 1.5 | 16 | 68 |
| T387A | 1 | 1.2 | 4 | 14 |
| S383A, S385A, T387A | 1 | 1.4 | 4 | 13 |
| T413A | 1.6 | 5 | 1 | 10 |
| S415A | 1 | 1 | 4 | 16 |
| T417A | 1.4 | 1.7 | 14 | 79 |
| T417E | NDb | 1.4 | 10 | 74 |
| T417D | 1 | 1 | 8 | 58 |
| T421A | 1 | 1 | 3 | 8 |
| T413A, S415A, T417A, T421 | 1.5 | 1.6 | 19 | 100 |
| T387A, T417A | 2 | 1 | 20 | >1000 |
wt, wild type.
Numbers indicate the average fold decrease as compared to YKU80 XRS2 yeast for total and HO(+2) NHEJ in the suicide deletion assay.
Not determined.
NHEJ phenotype of Lif1 C-terminal mutants
YKU80 | yku80Δ605–629 | |||
|---|---|---|---|---|
| LIF1 | Total | HO(+2) | Total | HO(+2) |
| wt | 1a | 1 | 2 | 8 |
| Δ∷kanMX4 | 16 | >1000 | 20 | ∞ |
| Δ410-421 | 1.6 | 1.5 | 16 | 68 |
| T387A | 1 | 1.2 | 4 | 14 |
| S383A, S385A, T387A | 1 | 1.4 | 4 | 13 |
| T413A | 1.6 | 5 | 1 | 10 |
| S415A | 1 | 1 | 4 | 16 |
| T417A | 1.4 | 1.7 | 14 | 79 |
| T417E | NDb | 1.4 | 10 | 74 |
| T417D | 1 | 1 | 8 | 58 |
| T421A | 1 | 1 | 3 | 8 |
| T413A, S415A, T417A, T421 | 1.5 | 1.6 | 19 | 100 |
| T387A, T417A | 2 | 1 | 20 | >1000 |
YKU80 | yku80Δ605–629 | |||
|---|---|---|---|---|
| LIF1 | Total | HO(+2) | Total | HO(+2) |
| wt | 1a | 1 | 2 | 8 |
| Δ∷kanMX4 | 16 | >1000 | 20 | ∞ |
| Δ410-421 | 1.6 | 1.5 | 16 | 68 |
| T387A | 1 | 1.2 | 4 | 14 |
| S383A, S385A, T387A | 1 | 1.4 | 4 | 13 |
| T413A | 1.6 | 5 | 1 | 10 |
| S415A | 1 | 1 | 4 | 16 |
| T417A | 1.4 | 1.7 | 14 | 79 |
| T417E | NDb | 1.4 | 10 | 74 |
| T417D | 1 | 1 | 8 | 58 |
| T421A | 1 | 1 | 3 | 8 |
| T413A, S415A, T417A, T421 | 1.5 | 1.6 | 19 | 100 |
| T387A, T417A | 2 | 1 | 20 | >1000 |
wt, wild type.
Numbers indicate the average fold decrease as compared to YKU80 XRS2 yeast for total and HO(+2) NHEJ in the suicide deletion assay.
Not determined.
Importantly, there are many other conserved Lif1 residues in the regions containing T387A and T417A. Specifically, S383 has been implicated in the interaction with Xrs2, although the effect of its single mutation on NHEJ was not assessed (Matsuzaki et al. 2008). To confirm that we were not being misled by assumptions about the Xrs2 FHA domain and analogy to XRCC4, we also created single and multiple alanine substitutions for other nearby conserved Lif1 serine and threonine residues, including S383A. Although some had modest effects, none resulted in incremental NHEJ defects as severe as combined T387A and T417A mutation (Table 2) suggesting that the latter have uniquely important functions. We also replaced T417 with aspartate or glutamate in an attempt to mimic threonine phosphorylation. The resulting strains displayed NHEJ defects similar to alanine substitution (Table 2). Because phosphorylation effects can be mediated by properties other than charge, this finding neither supports nor refutes the possibility that the Xrs2–Lif1 interaction depends on T417 phosphorylation.
If the Xrs2 FHA domain does indeed bind the identified Lif1 threonine residues, then the Xrs2 and Lif1 point mutations described above would be expected to be epistatic. Consistent with this, xrs2-R32A or S47A mutation combined with lif1-T387A/T417A mutation conferred total and HO(+2) suicide deletion NHEJ defects that were no more severe than each mutant alone, in both YKU80 and yku80Δ605–629 backgrounds (Figure 6, A and B and data not shown). Importantly, the YKU80 background would have been quantitatively able to reveal a further decrease in NHEJ efficiency if it had occurred. To confirm these findings by another method, we also transformed linear plasmids into these various strains, where circularization by NHEJ is required for plasmid maintenance (Figure 6C). As with suicide deletion, yku80Δ605–629 mutation impaired NHEJ in the plasmid assay more substantially than either xrs2-R32A or lif1-T387A/T417A mutations. Also, combining yku80Δ605–629 with either the XRS2 or LIF1 point mutations caused an NHEJ defect as severe as yku70Δ mutation. Finally, the xrs2-R32A and lif1-T387A/T417A mutations again behaved epistatically in the YKU80 background, consistent with these mutations each impairing the same NHEJ function.
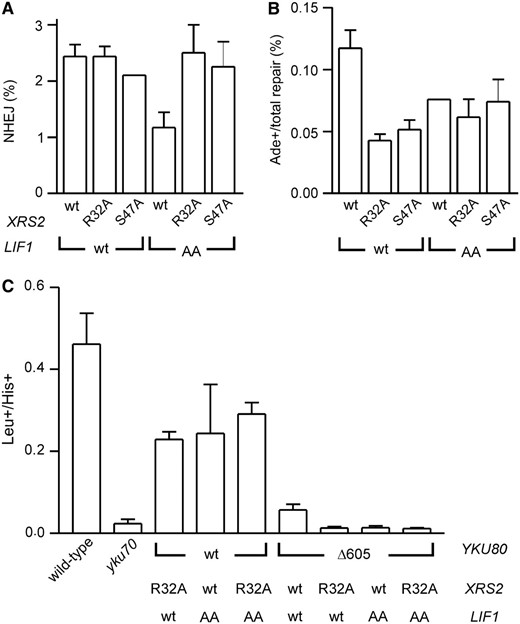
XRS2 and LIF1 mutations that impair the Xrs2–Lif1 interaction are epistatic. (A and B) Suicide deletion analysis similar to Figure 3, B and C, except now mutating both XRS2 and LIF1 in YKU80 wild-type yeast, a background which could reveal additive defects of XRS2 and LIF1 alterations if they existed. (C) An AgeI-cut LEU2-marked plasmid was transformed into yeast strains with the indicated YKU80, XRS2, and LIF1 alleles. Repair of this broken plasmid is expressed as the Leu+ transformation frequency normalized to the His+ transformation frequency from a cotransformed circular HIS3-marked control plasmid. In each assay, lif1-T387A/T417A (“AA”) and xrs2-R32A or -S47A mutations were epistatic with respect to NHEJ.
Combined mutation of the Yku80 and Lif1 C termini blocks Dnl4 recruitment to a DSB in vivo:
ChIP experiments previously demonstrated that Lif1 and Ku are each essential for recruitment of DNA ligase IV to a DSB in vivo (Teo and Jackson 2000). To determine whether the Yku80–Dnl4 and Xrs2–Lif1 interactions described above are required for this recruitment, we employed a previously described system in which a single DSB is introduced in chromosome II by transient induction of the HO endonuclease (Wu et al. 2008). PCR-based methods allow determination of the extent of chromosome breakage and subsequent repair, as well as the extent and time course of Dnl4–Myc13 binding to the DSB by ChIP. yku80Δ605–629 and xrs2-R32A or lif1-T387A/T417A mutations, which selectively abolish the Yku80–Dnl4 and Xrs2–Lif1 interactions, respectively, were introduced into this strain background. Approximately 60% of cells of all strains suffered a DSB after 60 min of HO induction, and most of these broken chromosomes were rejoined in the wild-type strain during the 1–2 hr following termination of HO expression (Figure 7, A and B). The combined yku80Δ605–629 lif1-T387A/T417A and yku80Δ605–629 xrs2-R32A mutants showed markedly decreased repair, while each single mutant was not substantially different than wild type. This pattern confirms the mutant phenotypes for simple repair of a single chromosomal DSB, although the extent of the mutant defects is not as pronounced in this assay. As reported previously, Dnl4–Myc13 binding to the DSB in the wild-type strain correlated well with the peak repair period (60–120 min) and then declined (Figure 7, C and D) (Wu et al. 2008). In contrast, the double mutants showed only slight, but detectable, ChIP signal at those same time points. The single mutants were not obviously impaired, but perhaps showed a decreased persistence of Dnl4 at later time points. Thus, efficient recruitment of DNA ligase IV to a DSB depends on the Yku80–Dnl4 and Xrs2–Lif1 interactions.
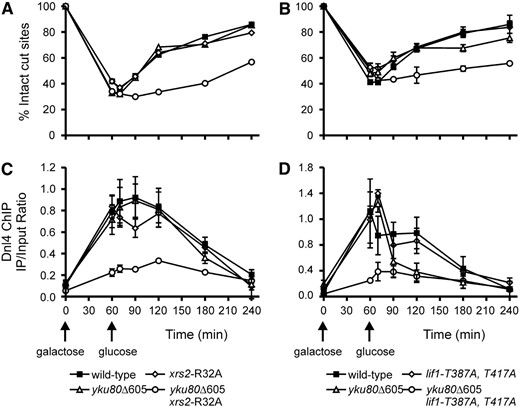
Recruitment of DNA ligase IV to DSBs in vivo depends on combined Yku80–Dnl4 and Xrs2–Lif1 interactions. A DSB was introduced on chromosome II by 60-min exposure to galactose to induce HO endonuclease expression. Glucose was then added to terminate HO expression and allow repair. The fraction of intact HO cut sites was monitored by a competitive PCR assay to determine the extent of initial cutting and subsequent repair. Repair was specifically impaired in yku80Δ605–629 xrs2-R32A (A) and yku80Δ605–629 lif1-T387A/T417A yeast (B). Parallel samples were used for ChIP analysis using α-Myc antibodies against the Dnl4–Myc13 protein present in the strains and PCR primers immediately adjacent to the DSB site. Results are plotted as the ratio of amplification of the DSB site fragment to that of a control fragment amplified with the same primers (see materials and methods). Dnl4–Myc13 localization to the DSB coincided with DSB appearance and repair in the wild-type strain, but was largely absent in the yku80Δ605–629 xrs2-R32A (C) and yku80Δ605–629 lif1-T387A/T417A (D) strains. All results are the mean ± standard deviation of three independent experiments.
DISCUSSION
Yeast NHEJ requires coordination of the Ku, MRX, and DNA ligase IV complexes at a DSB, which is presumably guided by protein–protein interactions between them. We previously identified two such interactions between the C terminus of Yku80 and catalytic domain of Dnl4 and the FHA domain of Xrs2 and the C terminus of Lif1 (Palmbos et al. 2005). Results here extend our molecular understanding of these interactions and validate the hypothesis that they contribute redundantly to recruitment of DNA ligase IV during NHEJ.
Modeling indicated that the Xrs2 region involved in the Xrs2–Lif1 interaction folds similarly to other FHA domains (Figure 2B). FHA domains consist of β-sheets interspersed with clustered peptide loops (Durocher et al. 2000; Durocher and Jackson 2002). They typically bind to phosphothreonine-containing peptides using these loops, specifically through contacts between two arginines (R32 and R48 in Xrs2) and the phosphorylated threonine and between other residues (S47 and K75) and amino acids flanking the threonine. This provides specificity for both the phosphate and the peptide context. Mutational analysis confirmed that Lif1 binding depends on each of these Xrs2 amino acids (Figure 3A). Interestingly, mutation of R32 and R48 also eliminated background auto-activation by the Xrs2(1–125) two-hybrid bait. Although we do not know the basis of the auto-activation, this result suggests that the arginine mutations severely disturb Xrs2 FHA domain function and/or folding. In contrast, S47 and K75 mutations did not block auto-activation. This is consistent with a specific loss of the Xrs2–Lif1 interaction, although without structural information the precise impact of any mutation cannot be definitely stated.
Concerns about folding of mutant FHA domains notwithstanding, the similarity of the above results to other FHA domains (Durocher et al. 2000; Pike et al. 2001) strongly suggests that Xrs2 binds to Lif1 in a manner characteristic of FHA-peptide interactions. This inference in turn correctly suggested that the Lif1 binding site has critical threonine residues. The C terminus of Lif1 is poorly conserved and does not show any apparent domains. Strikingly, though, two of the most conserved stretches each surround a central threonine (Figure 2C) and bear great similarity to the XRCC4 peptide recently shown to be phosphorylated and bound by the FHA domain of PNKP (Koch et al. 2004). This information allowed us to rapidly validate T417 and T387 as critical Lif1 determinants of Xrs2 binding (Figure 5A). It is unclear why Xrs2 might bind to two different locations on Lif1. XRCC4 also harbors multiple threonine phosphorylation sites in its C terminus, but apparently only one of these is critically required for binding by the PNKP FHA domain (Koch et al. 2004). In yeast, T417 appears to be more important, but T387A mutation alone nonetheless had a measurable effect on binding and NHEJ. Without structural data for the Lif1 C terminus it cannot be determined whether the T387 and T417 regions might form part of a concerted interaction surface, but this is an intriguing possibility.
Our data are generally consistent with recent published work confirming the importance of the Xrs2 FHA domain in mediating interaction with Lif1 (Matsuzaki et al. 2008). However, Matsuzaki et al. (2008) proposed Lif1 S383 as the key FHA target residue. The most plausible explanation for this difference from our work is that S383 and T387 are closely spaced residues in a single well-conserved peptide region (Figure 2C) such that mutation of one residue might easily affect Xrs2 binding to the other. It could be that only one of S383 and T387 is bound in the canonical FHA pocket, or in fact both might be engaged by comparison to the Dun1 FHA domain, which was recently shown to display dual recognition of closely spaced phosphothreonines (Lee et al. 2008). Our NHEJ data cannot resolve the specific contributions of S383 and T387, but are nonetheless consistent with the notion that these residues form parts of a single interaction peptide or surface (Table 2). We also note that Matsuzaki et al. (2008) only measured NHEJ for a combined S383/T387 mutant. Moreover, they did not specifically mutate T417, which we consistently find to have a larger effect than S383/T387 mutations.
An obvious implication of these results is that Lif1 is phosphorylated on S383/T387 and T417. We have attempted to validate this by mass spectroscopy. Preliminary results suggest many possible phosphorylated peptides in the Lif1 protein but conclusive confirmation of specific phosphorylated sites has not been possible due to the high density of serines and threonines. The Xrs2–Lif1 interaction is also difficult to detect in vitro (Chen et al. 2001) and to date has not allowed demonstration of a phosphate-dependent interaction in our hands or others (Matsuzaki et al. 2008). If the interaction is dependent on Lif1 phosphorylation in vivo, the strong two-hybrid signal in the absence of DNA damage suggests that this might be constitutive. Indeed, phosphorylation of XRCC4 T233 recognized by PNKP is constitutively mediated by casein kinase 2 (Koch et al. 2004). However, it is equally possible that Lif1 is phosphorylated in a checkpoint-dependent manner similar to its sister protein Nej1 (Ahnesorg and Jackson 2007), or finally that the Xrs2 interaction might not depend on phosphorylation at all. Regardless, our data demonstrate a strong contribution of the S383/T387 and T417 regions to Xrs2 interaction.
A key finding is that our Xrs2 and Lif1 mutations affect not only their interaction but also NHEJ efficiency. However, like complete deletion of the FHA domain (Palmbos et al. 2005), these point mutations were only significant in yeast that also had a disrupted Yku80–Dnl4 interaction (Tables 1 and 2). This relationship is bidirectional, though, since disruption of the Xrs2–Lif1 interaction also strongly potentiates yku80Δ605 mutation (see especially Figure 7). It is further important that Matsuzaki et al. (2008) consistently see a larger NHEJ phenotype than us when disrupting only the Xrs2–Lif1 interaction. This is likely attributable to differences in strains or specific features of the NHEJ assays employed, but regardless their data provide independent confirmation of a primary importance of the Xr2–Lif1 interaction. Matsuzaki et al. (2008) did not examine combined disruptions of the Xr2–Lif1 and Yku80–Dnl4 interactions to know whether synergy would also be observed in their hands.
The specific role of the Yku80 C terminus in NHEJ assays is presumably mediated by its observed interaction with Dnl4 (Palmbos et al. 2005). An alternative explanation was that it supported Ku–DSB binding but we have now shown that KuΔ605 binds DNA normally (Figure 1), consistent with the fact that the human Ku–DNA cocrystal did not contain the Ku80 C terminus (Walker et al. 2001). This suggests a model in which Ku threads onto a DSB end which, due to the polarity of Ku binding (Walker et al. 2001), orients the Yku80 C terminus toward the DSB to provide a hook for Dnl4 binding. Interestingly, this hook appears to function differently in humans since interaction between human Ku and DNA ligase IV does not require the Ku80 C terminus (Costantini et al. 2007). Instead, this region binds DNA-PKcs (Gell and Jackson 1999), which is absent in yeast.
Considering all results together, we propose that the observed Xr2–Lif1 and Yku80–Dnl4 interactions redundantly stabilize DNA ligase IV at a DSB to promote efficient NHEJ. The failed recruitment of Dnl4 to a DSB in yku80Δ605–629 xrs2-R32A or yku80Δ605–629 lif1-T387A/T417A double mutant yeasts strongly supports this hypothesis (Figure 7). It is also supported by the observation that Dnl4 binding to a DSB is delayed by ∼10 min relative to both Ku and MRX (Wu et al. 2008). We cannot say at present what features might promote the greater dependence on one interaction over another. Indeed, a possibility for experimental artifact is that disruption of one interaction might cause a change in conformation of the overall NHEJ assembly to expose another interaction not ordinarily being used. However, the fact that these interactions can each be demonstrated independently of NHEJ and the combined functional data provided here and by Matsuzaki et al. (2008) make it most likely that both the Xrs2–Lif1 and Yku80–Dnl4 interactions are realized during wild-type NHEJ, but with differing degrees of importance depending on the context.
We finally note that ligase recruitment and NHEJ are not completely diminished even in the absence of both the Yku80–Dnl4 and Xrs2–Lif1 interactions. Moreover, complete Ku loss has a much more severe affect on ligase–DSB association (Wu et al. 2008) than the focused Yku80–Dnl4 interaction mutant studied here. These observations suggest still further important contacts between Ku, MRX, and DNA ligase IV. This is not surprising since, for example, Ku polarity will also position other regions, most notably the Yku70 VWA domain, toward the DSB end (Walker et al. 2001; Daley et al. 2005). The final picture emerges of a complex network of interactions which perform the critical task of recruiting and positioning DNA ligase IV at a DSB.
Footnotes
Present address: Maisonneuve-Rosemont Hospital, Guy Bernier Research Center, University of Montreal, Montreal, Quebec, Canada H1T 2M4.
Footnotes
Communicating editor: S. Keeney
Acknowledgement
We thank the Wilson laboratory for their support and critical readings of the manuscript, and Alan Tomkinson and Melissa Hefferin for helpful discussions of the work. This work was supported by National Institutes of Health grant CA102563 (T.E.W.) and the University of Michigan Loeb Predoctoral Fellowship (P.L.P.).
References
Aparicio, O., J. V. Geisberg, E. Sekinger, A. Yang, Z. Moqtaderi et al.,
Assenmacher, N., and K. P. Hopfner,
Kelley, L. A., R. M. MacCallum and M. J. Sternberg,
Trujillo, K. M., D. H. Roh, L. Chen, S. Van Komen, A. Tomkinson et al.,

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}