





Abstract
The genetic basis of hybrid sterility can provide insight into the genetic and evolutionary origins of species barriers. We examine the genetics of hybrid incompatibility between two diploid plant species in the plant clade Solanum sect. Lycopersicon. Using a set of near-isogenic lines (NILs) representing the wild species Solanum pennellii (formerly Lycopersicon pennellii) in the genetic background of the cultivated tomato S. lycopersicum (formerly L. esculentum), we found that hybrid pollen and seed infertility are each based on a modest number of loci, male (pollen) and other (seed) incompatibility factors are roughly comparable in number, and seed-infertility QTL act additively or recessively. These findings are remarkably consistent with our previous analysis in a different species pair, S. lycopersicum × S. habrochaites. Data from both studies contrast strongly with data from Drosophila. Finally, QTL for pollen and seed sterility from the two Solanum studies were chromosomally colocalized, indicating a shared evolutionary history for these QTL, a nonrandom genomic distribution of loci causing sterility, and/or a proclivity of certain genes to be involved in hybrid sterility. We show that comparative mapping data can delimit the probable timing of evolution of detected QTL and discern which sterility loci likely evolved earliest among species.
SPECIATION involves the evolution of reproductive isolating barriers that prevent gene flow between lineages, thereby maintaining their integrity as independently evolving units. Because the tempo and mode of speciation can be strongly influenced by the number and individual effects of changes causing reproductive isolation (Barton and Charlesworth 1984; Gottlieb 1984; Coyne 1992), understanding the genetic basis of these barriers is valuable. One such class of barriers, “intrinsic” hybrid inviability and sterility (that occur in the absence of ploidy differences or apparent ecological factors isolating lineages), is thought to be due to negative epistasis between newly derived alleles that have arisen in isolation but are dysfunctional against the genetic background of the other diverged lineage [also known as “Dobzhansky–Muller” interactions, DMIs, after Dobzhansky 1936 and Muller 1939 (Coyne and Orr 2004)]. Studies have successfully mapped QTL underlying intrinsic hybrid sterility and inviability due to DMIs among species in a diverse set of systems [e.g., Drosophila (e.g., Noor et al. 2001; Barbash et al. 2003; Presgraves 2003; Tao et al. 2003a,b), mosquito (Slotman et al. 2004), and Mimulus (e.g., Sweigart et al. 2006)] to describe the number of loci involved, the size of their individual phenotypic effects, their mode of gene action, and/or their location in the genome. Nonetheless, to assess the generality of these emerging results—and what they might indicate about processes predominantly responsible for the formation of new species—requires studies that examine barriers operating among multiple species pairs within the same group. These studies can provide at least three unique insights: First, they can indicate general mechanisms or dynamics responsible for the evolution of species barriers in specific biological systems; second, they can generate expectations in other systems that share similar biological properties; and third, they can provide the empirical data necessary to inform and evaluate theory on the evolution of hybrid incompatibility.
Despite the potential power of such comparative analyses, there are very few systems in which there are appropriate data available from multiple species pairs. Drosophila, an exceptional group in which multiple studies have examined the genetic basis of intrinsic hybrid incompatibility, provides a clear illustration of the first two benefits of comparative studies. On the basis of genomewide QTL mapping in multiple Drosophila studies, three general patterns have emerged as potentially typical of hybrid incompatibilities among fruitfly species: Incompatibilities that reduce hybrid male sterility appear to evolve most readily and are much more prevalent than incompatibilities causing female sterility or hybrid inviability (e.g., Wu et al. 1996; Tao and Hartl 2003; Tao et al. 2003a,b; and references therein); male sterility is often polygenic and complex, involving interactions between multiple loci to generate sterility phenotypes (e.g., Cabot et al. 1994; Davis and Wu 1996; Tao et al. 2003b; and references therein); and hybrid incompatibilities appear on average to act recessively, such that their phenotypic effects are maximized when in homozygous or hemizygous form (Presgraves 2003; Tao et al. 2003a,b). These patterns suggest that the genetic basis of hybrid incompatibility in Drosophila is likely underpinned by a set of general rules and/or dynamics. In particular, the relative prevalence and genetic complexity of male sterility are thought to be due to the influence both of the Drosophila sex determination system (especially XY heterogamety in males), and of the dynamics of sexual interactions (especially strong male–male competition and/or male–female antagonism) (for details of these inferences, see Coyne and Orr 2004). The identification of these likely underlying causes has also generated a set of expectations for patterns of hybrid incompatibility in other systems that share similar biological properties. These expectations have been evaluated in a number of additional systems [e.g., Anopheles (Slotman et al. 2004) or Aedes (Presgraves and Orr 1998) mosquitoes], especially those that share similar sex determination mechanisms and sexual interaction dynamics with Drosophila (see Coyne and Orr 2004). Conversely, in systems that do not have features such as heterogametic chromosomal sex determination or strong sexual selection, these patterns might be less likely to be observed (Moyle and Graham 2005).
Finally, the Drosophila patterns described above have informed much of the theory currently developed to describe the accumulation and phenotypic expression of Dobzhansky–Muller incompatibilities (Orr 1995; Turelli and Orr 2000; Orr and Turelli 2001; Turelli and Moyle 2007). Nonetheless, appropriate comparative data for assessing some components of this theory have been unavailable to date. For example, comparative data are essential for evaluating the predicted “snowballing” accumulation of hybrid incompatibilities between increasingly divergent lineages (Orr 1995; Coyne and Orr 2004), as well as the assumptions underlying much of the current theoretical work (e.g., independence between different pairs of Dobzhansky–Muller incompatibilities). In addition, data from multiple species crosses within the same phylogenetic group could be used to identify when and where specific incompatibility loci have evolved in the divergence history of lineages (see discussion). These data would be valuable in resolving the relative importance of specific genetic changes in early divergence/speciation processes.
Here we present results from a QTL analysis of a second species cross within the plant clade Solanum sect. Lycopersicon. This group is an attractive model in which to analyze hybrid incompatibility because, in addition to the extensive genetic resources available in this group (Mueller et al. 2005a,b), all members of this clade are closely related hermaphroditic diploids (2n = 2x = 24) (Peralta and Spooner 2001; Nesbitt and Tanksley 2002) that share a high degree of synteny (Chetelat and Ji 2007) and are to some degree intercrossable (Rick 1979). These features both allow formal genetic analysis and indicate that species are largely reproductively isolated by genic loci rather than by changes in large-scale chromosomal organization (Quiros 1991). Our goals in this study are threefold: First, we aim to identify the genomic regions associated with pollen and seed inviability in hybrids between Solanum lycopersicum and S. pennellii and to quantify the number, genomic location, individual effects, and (for seed sterility) mode of gene action of these hybrid incompatibility QTL; second, we aim to directly compare our results—including the location and the frequency of potentially shared hybrid incompatibility QTL—with those from our previous analysis of incompatibilities between S. lycopersicum and S. habrochaites (Moyle and Graham 2005) and to compare results of both studies with Drosophila; and third, we use these comparative data to outline additional empirical insights that could be gained into the evolution of hybrid incompatibility (including pinpointing where specific hybrid incompatibility loci arose in the history of diverging lineages) and to highlight the need for further theoretical work to understand the accumulation of these genetic changes. Finally, note that in this study we do not assess the strength of any potential prezygotic isolating barriers, including pollen–pistil interactions, although these also appear to act between our focal species (Rick 1960; Hogenboom 1972; L. C. Moyle and A. Posto, unpublished results). Accordingly, our analysis cannot address the relative genetic complexity, strength, and/or evolutionary importance during speciation of barriers acting at prezygotic vs. postzygotic stages (see, however, Moyle 2007). Rather, as with many genetic studies of hybrid incompatibility, our analysis focuses on the quantitative description of the genetic basis of postzygotic isolation and what might be inferred about evolution at this stage of reproductive isolation.
MATERIALS AND METHODS
Study system:
Solanum section Lycopersicon is a relatively small plant group within the large and diverse Solanaceae family; the group consists of 14 closely related diploid species or subspecies, including the domesticated tomato, S. lycopersicum (Mill.) (D'Arcy 1979; Peralta et al. 2005; Spooner et al. 2005). Although formerly classified as a separate genus (Lycopersicon), recent taxonomic revision indicates that this group is a monophyletic clade nested within the genus Solanum and renames Lycopersicon species accordingly (Peralta and Spooner 2001). Here we use the revised nomenclature. [Note that the classical nomenclature was used in the previous analysis of hybrid incompatibility QTL (Moyle and Graham 2005).] The two parental species analyzed here differ in several biologically significant features. S. pennellii (formerly Lycopersicon pennellii) is a wild, short-lived, herbaceous, perennial species that occurs predominantly from low to mid-elevations in northwestern South America. Most populations of S. pennellii are obligately outcrossing due to gametophytic self-incompatibility and exhibit high nucleotide diversity (Miller and Tanksley 1990; Stephan and Langley 1998). In contrast, S. lycopersicum (formerly L. esculentum)—the cultivated tomato—is a domesticated, self-pollinating species with comparatively low genetic variation. The putative wild progenitor of S. lycopersicum is also predominantly selfing (Miller and Tanksley 1990; Kondo et al. 2002), and self-compatibility is thought to have preceded domestication (Rick 1995). Phylogenetic resolution and chronological dating of speciation events in Solanum sect. Lycopersicon have proved difficult. However, recent estimates suggest the group began its initial radiation ∼7 million years ago (Nesbitt and Tanksley 2002). Nucleotide divergence between S. lycopersicum and S. pennellii estimated from six independent noncoding regions averages 0.042 substitutions/bp, indicating these species are closely related (Nesbitt and Tanksley 2002). F1 hybrids between the two parental species are successful in one direction of the cross (with S. lycopersicum as maternal parent); F1's show partial hybrid sterility, with both reduced seed yield and reduced pollen fertility (∼30 and 75% of parental levels, respectively; Rick 1960).
Plant material:
Each near-isogenic introgression line (NIL) analyzed here contained one small, individual, overlapping chromosomal segment of the wild species S. pennellii (SP) in an S. lycopersicum (SL) background. All lines were previously generated by Eshed and Zamir (1994, 1995), who provide a detailed description of their construction. Briefly, all lines are the advanced-generation backcross progeny of a single F1 plant produced by crossing S. pennellii accession LA716 as the male parent to S. lycopersicum cv. M82. This F1 individual was backcrossed to the recurrent SL parental line, and 99 BC1 plants were taken through six generations of selfing and then analyzed for genetic markers to assess the locations of S. pennellii introgressions in each, and a subset of lines was taken through three more generations of backcrossing, during which plants were selected for a single targeted introgression and against other additional introgressed regions. Additional BC1S2 families were genotyped and backcrossed to obtain representative introgressions from four regions of the S. pennellii genome that were missing in the first set of BC1S6 lines. Resulting plants were selfed and genotyped for 350 molecular markers, and 50 lines carrying a single homozygous introgression were chosen; together these lines cover the entire wild species genome in the genetic background of the cultivated tomato (Eshed and Zamir 1994). The high density of screened molecular markers and multiple generations of selfing in each homozygous line make residual heterozygosity at regions adjacent to marker-delimited sites unlikely.
The complete set of developed NILs, including an additional 25 sublines with smaller introgressed segments (75 lines total), is publicly available from the Tomato Genetics Resource Center (TGRC) at the University of California at Davis (UC Davis) (http://tgrc.ucdavis.edu). For this experiment, 71 introgression lines were chosen to maximize genomic representation of S. pennellii introgressions in the S. lycopersicum genetic background. These lines provide >98% of the genome of S. pennellii. Four lines were missing from our analysis because insufficient seed were available from the TGRC; as a result, two small genomic regions (on chromosomes 3 and 12) from S. pennellii are not represented in our mapping population [regions 3-E and 12-F on the NIL genome map (http://tgrc.ucdavis.edu); see Figure 2]. Our analysis cannot address whether incompatibility factors are contained within genomic regions that are not represented in our mapping population (see discussion). On the basis of estimates of introgression size (see below), each of these 71 NILs contained on average 25.0 cM of introgressed SP genome (range 2.3–57.2 cM); this corresponds to an average of 1.98% SP genome per NIL (range 0.18–4.54%), assuming a genome size of ∼1260 cM (Tanksley et al. 1992). Finally, note that S. lycopersicum and S. pennellii are separated by one pericentric microinversion on chromosome 7 (Van Der Knaap et al. 2004) contained within IL7-4 (see NIL genome map: http://tgrc.ucdavis.edu); none of our traits of interest map to this genomic location (see results).
Line cultivation and handling:
Each line and both parental accessions were evaluated in a replicated (5 plants/NIL, 15 plants/parental accession), randomized common garden experiment and assayed for male and female fertility and for a set of seven floral, inflorescence, and fruit morphology traits. Results for the morphological traits will be presented elsewhere (L. C. Moyle and T. Nakazato, unpublished results). All plants were propagated under the same conditions, following standard greenhouse cultivation protocols (Moyle and Graham 2005). Seeds were germinated and transplanted as seedlings into flats. Three weeks after transplant, seedlings were transferred to individual 1-gallon pots and grown in a climate-controlled greenhouse at the Indiana University Biology Greenhouse facility. Plants were watered daily, fertilized weekly, and pruned and staked prior to flowering.
Fertility assay:
Pollen and seed fertility was measured in all introgression lines and in the SL and SP parental plants. Pollen fertility was estimated on two unopened flowers on each plant, as previously described (Moyle and Graham 2005). Briefly, all pollen from each flower was collected into lactophenol-aniline blue histochemical stain and a known subsample examined and counted. Pollen inviability was indicated by the absence of a stained cytoplasm; this is a conservative measure of pollen infertility as some grains that stain for cytoplasm may nonetheless be functionally inviable for other reasons (Kearns and Inouye 1993). Pollen fertility was quantified in two ways: total number of pollen grains (PN) and proportion of fertile pollen (PF). Because the pollen counts in each subsample are proportional to the average pollen number per flower, we refer to them here as the number of pollen grains per flower.
Seed fertility (seed set per fruit) was determined by measuring seed production (total seed count) resulting from two different pollination treatments: self-pollination (“self”) and cross-pollination with SL pollen (backcross or “SL cross”). Because all NILs used in the study contain a SP introgression in homozygous form, selfing generates seed that is homozygous for the SP introgression, whereas pollination with S. lycopersicum pollen produces seed that is heterozygous for the SP introgression. For self seed, at least two flowers per plant were allowed to set seed via selfing; where fruits were not set automatically, flowers were hand selfed to ensure that morphological differences were not responsible for lack of seed set. For SL-cross seed, at least two flowers per plant were emasculated preanthesis (in the bud stage) and subsequently hand pollinated with pollen collected and bulked from at least five plants of the SL parental accession. Note that SL cross was not made to SP parent plants because SL pollen on SP stigmas routinely fails due to unilateral incompatibility (Rick 1960; Denettancourt 2001). Upon maturation, fruit was harvested, seeds were extracted by hand from individual fruit, and seed fertility was determined by counting the number of visible seeds from each fruit. Average seed per fruit for each plant was used to generate measures of self-seed set (SSS) and SL-cross seed set (SSC) for each NIL and the SL control parent.
As noted previously (Moyle and Graham 2005), seed set and pollen fertility are not fully independent measures of hybrid incompatibility. Pollen-sterility estimates inviable male gamete production; seed set represents the composite effects of male and female fertility, in addition to early zygotic dysfunction. As such, interpretation of self-seed set requires that individual male fertility within each NIL be taken into account. Several strategies were used to account for the influence of male fertility on self-seed set (see results). In particular, we used a modified partial regression approach to remove the effects of pollen fertility prior to our final QTL analysis of seed fertility. To do this we first analyzed self-seed set data for each NIL with proportion PF as a main effect (arcsine square-root transformed) and then used the residual values of this analysis in our Dunnett's mean-contrast tests to detect QTL (see below). Seed-set QTL that remain significant after this analysis are assumed to confer reduced seed set partially or fully independently of any male infertility effects in the same genomic region.
QTL analysis:
The degree of association between each trait of interest and specific introgression lines was evaluated using Dunnett's mean contrast, which evaluates the mean phenotype of each NIL against the control SL accession with an experimentwise α-level of 0.05, i.e., corrected for multiple comparisons (Dunnett 1955; Zar 1984). For significant lines, results are presented as percentage difference (Δ%) from the isogenic SL control, i.e., the phenotypic effect of the QTL hypothesized to reside within the introgressed segment (Eshed and Zamir 1995). The minimum number and genomic location of QTL underlying differences in each trait were inferred by comparing the positions of introgressed segments having different trait values from the control parent, as previously described (Moyle and Graham 2005). Briefly, we used three rules (following Eshed and Zamir 1995; Tao et al. 2003b): (1) A QTL is counted only if the relevant NIL is significantly different from the corresponding control, (2) each NIL affecting the trait is assumed to carry only a single QTL, and (3) two overlapping introgressions with a significant effect on the trait, in the same direction relative to the control, carry the same QTL. In addition, for adjacent introgressions that contain overlapping regions but whose mean trait values differed significantly, the nonoverlapping portion of the introgression was declared the probable location of the QTL of interest. For seed set, we also estimated the dominance deviation, i.e., the difference between the SL/SP heterozygote (i.e., seeds resulting from NIL × SL crosses) and the midvalue of the homozygote SP (selfed NIL) and SL lines. Significance of this deviation was calculated by contrasting the heterozygous seed set from each NIL with homozygous seed set from the same NIL and seed set from control SL plants, using the LSMeans Contrast test in JMP (see Eshed and Zamir 1995).
Effects of introgression size on hybrid fitness:
If hybrid incompatibility is due to many factors of relatively small effect—i.e., an infinitesimal model of hybrid incompatibility factors—then sterility is expected to be strongly positively associated with the amount of the S. pennellii genome represented in the S. lycopersicum genetic background. We evaluated this expectation by testing the strength of individual regressions of the line means of pollen sterility or of seed sterility against the estimated genetic length (centimorgans) of the introgressed region in each NIL. There is presently no complete physical map of the tomato genome. Therefore the size of individual introgressions within each NIL was estimated in map units (centimorgans), using distances between the relevant genetic markers on the 2002 S. lycopersicum/S. pennellii ultrahigh-density linkage map [see Solanaceae Genome Network website (http://www.sgn.cornell.edu/); distances obtained in March 2007].
Colocalization of sterility loci in different mapping populations:
RESULTS
Pollen fertility:
Multiple introgression lines showed significant reductions in pollen fertility in comparison to both SL and SP parental lines (Figure 1, A and B), with the most substantial reductions observed in proportions of fertile pollen (Figure 1, Table 1). QTL locations inferred from introgression line phenotypes are shown in Figure 2. In total, we detected a minimum of 10 QTL for male fertility—7 QTL for proportion of fertile pollen and 3 QTL for total pollen number (Table 1). At all QTL, the S. pennellii alleles were associated with reduced male fertility in the S. lycopersicum background. Each QTL had a moderate to large effect on the hybrid phenotype, with genomic regions producing ∼25 to >50% hybrid male sterility (Table 1). Of the 10 pollen QTL detected, only 2 appear to be colocated for the two different measures of male fertility (i.e., pn4.1 and pf4.1 on chromosome 4; Figure 2), indicating that these traits are not strongly genetically associated, a result that is supported by the lack of correlation between them (Table 2)
![Distributions of NIL mean phenotypes for pollen and seed traits for 71 NILs. For each trait, the mean phenotypic value of each parental accession (SL, and SP if applicable) and the grand mean phenotypic value of all 71 NILs are indicated with arrows. (A) Total pollen grains per flower (PN). Parental means are SL = 73.5, SP = 100.8 [note that SP (an outcrosser) produces substantially more pollen per flower than SL (a selfer)]. (B) Proportion of fertile pollen grains per flower (PF). Parental means are SL = 0.864, SP = 0.810. (C) Selfed seed set per fruit (SSS). Parental means are SL = 71.54, SP = 101.49. (D) SL-cross seed set per fruit (SSC). SL mean = 50.11.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/genetics/179/3/10.1534_genetics.107.083618/6/m_1437fig1.jpeg?Expires=1717039856&Signature=I3uuay6byoEKdk2PaDixvidG7ZkMtg425LNwf3F3PVDUIOWy6HHcdOOdM1lmc9eTcaboqfHaKGOG5bwveqtU65sPQqJo-FSMzitjnC2EY3wWbiYJKXBX68KmkzjLt7PjExGoDpiFX0tq-n8-JLnKJDYZHJV7KWyNKlZ175chpxbkhU8CM-CKHWOpBeZpmEDSG1A9mDYQJAXc7CoBM8H2Zi3jIN52Nhg1PcFx996JObeb6arH1HcW4dSzw4O~yNvh2DikA~6psfTZLXfLRbpT2aVqfcyx6pM0bMAwvZhFQ1evLp8o-JYc5qCOqMp~tVk0r0I5CZeGggmEhHzc~fNu2A__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Distributions of NIL mean phenotypes for pollen and seed traits for 71 NILs. For each trait, the mean phenotypic value of each parental accession (SL, and SP if applicable) and the grand mean phenotypic value of all 71 NILs are indicated with arrows. (A) Total pollen grains per flower (PN). Parental means are SL = 73.5, SP = 100.8 [note that SP (an outcrosser) produces substantially more pollen per flower than SL (a selfer)]. (B) Proportion of fertile pollen grains per flower (PF). Parental means are SL = 0.864, SP = 0.810. (C) Selfed seed set per fruit (SSS). Parental means are SL = 71.54, SP = 101.49. (D) SL-cross seed set per fruit (SSC). SL mean = 50.11.
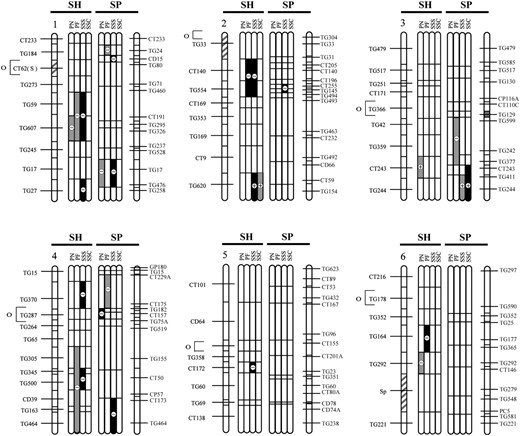
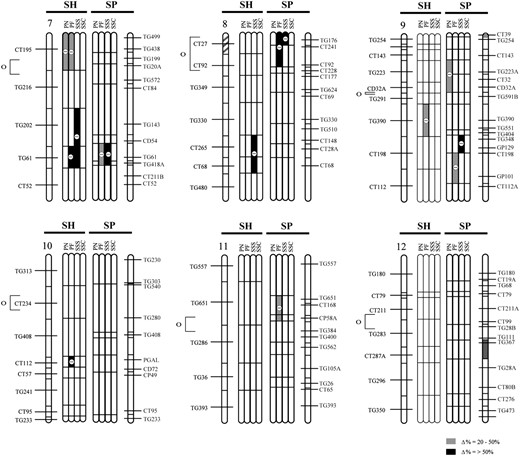
QTL locations for pollen and seed fertility traits for this study (SP) and the previous analysis (SH; Moyle and Graham 2005), with their location on the S. lycopersicum × S. pennellii and S. lycopersicum × S. habrochaites linkage maps, respectively. The shaded bars between the chromosomes show the individual analyses for the following traits: PN, total pollen count per flower; PF, proportion fertile pollen per flower; SSS, self (homozygous) seed set; and SSC, SL-cross (heterozygous) seed set, for each study. Levels of percentage of difference (Δ%) from the SL control parent for 20 > Δ > 50 and Δ > 50 are indicated by the intensity of shading (see key on figure). Chromosomal regions with shaded stripes on the linkage map indicate areas of the genome fixed for SL alleles (i.e., genomic regions not represented in the NIL populations). Centromere location is designated by the open circle and bracket to the left of the SH chromosomal map.
Putative QTL associated with hybrid incompatibility traits in NILs between S. lycopersicum and S. pennellii
Trait | QTL | Direction of effect | Mean phenotype | Additive effect | Δ% | No. NILs observed |
|---|---|---|---|---|---|---|
| Pollen no. (PN) | pn1.1 | — | 47.3 | −13.1 | −35.7 | 1 |
| pn4.1 | — | 34.3 | −19.6 | −53.3 | 1 | |
| pn9.1 | — | 42.6 | −13.7 | −42.0 | 3 | |
| Proportion fertile pollen (PF) | pf1.1 | — | 0.57 | −0.15 | −41.8 | 2 |
| pf3.1 | — | 0.63 | −0.12 | −27.1 | 1 | |
| pf4.1 | — | 0.58 | −0.14 | −33.4 | 1 | |
| pf7.1 | — | 0.54 | −0.16 | −37.7 | 1 | |
| pf8.1 | — | 0.53 | −0.17 | −52.4 | 2 | |
| pf9.1 | — | 0.66 | −0.10 | −31.7 | 2 | |
| pf11.1 | — | 0.67 | −0.10 | −26.9 | 2 | |
| Self-seed set (SSS) | sss1.1 | — | 20.97 | −25.3 | −70.7 | 1 |
| sss1.2 | — | 12.2 | −29.7 | −82.9 | 1 | |
| sss2.1 | — | 26.82 | −22.4 | −62.5 | 2 | |
| sss3.1 | + | 105.00 | 16.7 | 46.8 | 1 | |
| sss4.1 | — | 21.0 | −25.3 | −70.6 | 1 | |
| sss7.1 | — | 27.0 | −22.3 | −62.3 | 1 | |
| sss8.1 | — | 30.1 | −20.7 | −57.9 | 1 | |
| sss9.1 | — | 33.1 | −19.2 | −53.7 | 1 | |
| SL-cross seed set (SSC) | ssc3.1 | + | 80.63 | 15.3 | 60.9 | 2 |
Trait | QTL | Direction of effect | Mean phenotype | Additive effect | Δ% | No. NILs observed |
|---|---|---|---|---|---|---|
| Pollen no. (PN) | pn1.1 | — | 47.3 | −13.1 | −35.7 | 1 |
| pn4.1 | — | 34.3 | −19.6 | −53.3 | 1 | |
| pn9.1 | — | 42.6 | −13.7 | −42.0 | 3 | |
| Proportion fertile pollen (PF) | pf1.1 | — | 0.57 | −0.15 | −41.8 | 2 |
| pf3.1 | — | 0.63 | −0.12 | −27.1 | 1 | |
| pf4.1 | — | 0.58 | −0.14 | −33.4 | 1 | |
| pf7.1 | — | 0.54 | −0.16 | −37.7 | 1 | |
| pf8.1 | — | 0.53 | −0.17 | −52.4 | 2 | |
| pf9.1 | — | 0.66 | −0.10 | −31.7 | 2 | |
| pf11.1 | — | 0.67 | −0.10 | −26.9 | 2 | |
| Self-seed set (SSS) | sss1.1 | — | 20.97 | −25.3 | −70.7 | 1 |
| sss1.2 | — | 12.2 | −29.7 | −82.9 | 1 | |
| sss2.1 | — | 26.82 | −22.4 | −62.5 | 2 | |
| sss3.1 | + | 105.00 | 16.7 | 46.8 | 1 | |
| sss4.1 | — | 21.0 | −25.3 | −70.6 | 1 | |
| sss7.1 | — | 27.0 | −22.3 | −62.3 | 1 | |
| sss8.1 | — | 30.1 | −20.7 | −57.9 | 1 | |
| sss9.1 | — | 33.1 | −19.2 | −53.7 | 1 | |
| SL-cross seed set (SSC) | ssc3.1 | + | 80.63 | 15.3 | 60.9 | 2 |
Direction of effect describes an increase (+) or decrease (−) in the mean phenotype at each locus, compared to the SL parent. Mean phenotype is calculated from all NILs showing the QTL phenotype at each genomic location. Additive effect is calculated as (SP/SP − SL/SL)/2. Δ% describes the percentage of phenotypic change from the SL parent.
Putative QTL associated with hybrid incompatibility traits in NILs between S. lycopersicum and S. pennellii
Trait | QTL | Direction of effect | Mean phenotype | Additive effect | Δ% | No. NILs observed |
|---|---|---|---|---|---|---|
| Pollen no. (PN) | pn1.1 | — | 47.3 | −13.1 | −35.7 | 1 |
| pn4.1 | — | 34.3 | −19.6 | −53.3 | 1 | |
| pn9.1 | — | 42.6 | −13.7 | −42.0 | 3 | |
| Proportion fertile pollen (PF) | pf1.1 | — | 0.57 | −0.15 | −41.8 | 2 |
| pf3.1 | — | 0.63 | −0.12 | −27.1 | 1 | |
| pf4.1 | — | 0.58 | −0.14 | −33.4 | 1 | |
| pf7.1 | — | 0.54 | −0.16 | −37.7 | 1 | |
| pf8.1 | — | 0.53 | −0.17 | −52.4 | 2 | |
| pf9.1 | — | 0.66 | −0.10 | −31.7 | 2 | |
| pf11.1 | — | 0.67 | −0.10 | −26.9 | 2 | |
| Self-seed set (SSS) | sss1.1 | — | 20.97 | −25.3 | −70.7 | 1 |
| sss1.2 | — | 12.2 | −29.7 | −82.9 | 1 | |
| sss2.1 | — | 26.82 | −22.4 | −62.5 | 2 | |
| sss3.1 | + | 105.00 | 16.7 | 46.8 | 1 | |
| sss4.1 | — | 21.0 | −25.3 | −70.6 | 1 | |
| sss7.1 | — | 27.0 | −22.3 | −62.3 | 1 | |
| sss8.1 | — | 30.1 | −20.7 | −57.9 | 1 | |
| sss9.1 | — | 33.1 | −19.2 | −53.7 | 1 | |
| SL-cross seed set (SSC) | ssc3.1 | + | 80.63 | 15.3 | 60.9 | 2 |
Trait | QTL | Direction of effect | Mean phenotype | Additive effect | Δ% | No. NILs observed |
|---|---|---|---|---|---|---|
| Pollen no. (PN) | pn1.1 | — | 47.3 | −13.1 | −35.7 | 1 |
| pn4.1 | — | 34.3 | −19.6 | −53.3 | 1 | |
| pn9.1 | — | 42.6 | −13.7 | −42.0 | 3 | |
| Proportion fertile pollen (PF) | pf1.1 | — | 0.57 | −0.15 | −41.8 | 2 |
| pf3.1 | — | 0.63 | −0.12 | −27.1 | 1 | |
| pf4.1 | — | 0.58 | −0.14 | −33.4 | 1 | |
| pf7.1 | — | 0.54 | −0.16 | −37.7 | 1 | |
| pf8.1 | — | 0.53 | −0.17 | −52.4 | 2 | |
| pf9.1 | — | 0.66 | −0.10 | −31.7 | 2 | |
| pf11.1 | — | 0.67 | −0.10 | −26.9 | 2 | |
| Self-seed set (SSS) | sss1.1 | — | 20.97 | −25.3 | −70.7 | 1 |
| sss1.2 | — | 12.2 | −29.7 | −82.9 | 1 | |
| sss2.1 | — | 26.82 | −22.4 | −62.5 | 2 | |
| sss3.1 | + | 105.00 | 16.7 | 46.8 | 1 | |
| sss4.1 | — | 21.0 | −25.3 | −70.6 | 1 | |
| sss7.1 | — | 27.0 | −22.3 | −62.3 | 1 | |
| sss8.1 | — | 30.1 | −20.7 | −57.9 | 1 | |
| sss9.1 | — | 33.1 | −19.2 | −53.7 | 1 | |
| SL-cross seed set (SSC) | ssc3.1 | + | 80.63 | 15.3 | 60.9 | 2 |
Direction of effect describes an increase (+) or decrease (−) in the mean phenotype at each locus, compared to the SL parent. Mean phenotype is calculated from all NILs showing the QTL phenotype at each genomic location. Additive effect is calculated as (SP/SP − SL/SL)/2. Δ% describes the percentage of phenotypic change from the SL parent.
Correlations between trait scores from 71 NILs between S. lycopersicum and S. pennellii
PN | PF | SSS | |
|---|---|---|---|
| PF | 0.042 | ||
| SSS | 0.180 | 0.238** | |
| SSC | 0.134 | 0.207* | 0.447*** |
PN | PF | SSS | |
|---|---|---|---|
| PF | 0.042 | ||
| SSS | 0.180 | 0.238** | |
| SSC | 0.134 | 0.207* | 0.447*** |
PN, total pollen count per flower; PF, proportion fertile pollen per flower; SSS, self- (homozygous) seed set; SSC, SL-cross (heterozygous) seed set. *0.10 < P < 0.05, **P < 0.05, ***P < 0.0001.
Correlations between trait scores from 71 NILs between S. lycopersicum and S. pennellii
PN | PF | SSS | |
|---|---|---|---|
| PF | 0.042 | ||
| SSS | 0.180 | 0.238** | |
| SSC | 0.134 | 0.207* | 0.447*** |
PN | PF | SSS | |
|---|---|---|---|
| PF | 0.042 | ||
| SSS | 0.180 | 0.238** | |
| SSC | 0.134 | 0.207* | 0.447*** |
PN, total pollen count per flower; PF, proportion fertile pollen per flower; SSS, self- (homozygous) seed set; SSC, SL-cross (heterozygous) seed set. *0.10 < P < 0.05, **P < 0.05, ***P < 0.0001.
.
Seed fertility/viability:
Seed fertility of introgression lines was conspicuously different depending upon whether the resulting seed was heterozygous or homozygous for an SP introgression. Seed set per fruit resulting from self-pollination (SSS), and therefore homozygous for an SP introgression, was substantially reduced in multiple introgression lines in comparison to the parental SL plants (SSS in Table 1, Figure 1C). In total, we detected a minimum of eight QTL for homozygous seed set (Table 1, Figure 2); the only QTL of this group that increased seed set was colocated with a QTL found to increase heterozygous seed set (Figure 2). Each QTL had a relatively large effect on the hybrid phenotype (Table 1); seven genomic regions are associated with >50% reduction in seed fertility, and several individual QTL reduced the number of seeds per fruit by >70% compared to the SL parent (e.g., sss1.1, sss1.2, sss4.1). In contrast, for SL-cross seed (resulting from pollinations with SL pollen), which were heterozygous for SP introgressions, no lines showed a significant reduction in seed fertility in comparison to the pure SL parental line (SSC in Table 1). The only significant line effect observed for heterozygous seed set was a 1.5-fold increase in seed production above the pure parental SL line mean; this resulted in a minimum of one QTL for increased heterozygous seed set (Table 1, Figure 2). The absence of significant seed infertility in crosses between NILs and the SL parent is evidence that observed seed infertility is unlikely to be due entirely to ovule (i.e., strictly female) sterility in the NIL parent; otherwise reduced seed set per fruit would be observed in infertile lines regardless of the genetic source of the pollen. Therefore, it is probable that the majority of seed-infertility QTL identified here are due to zygotic incompatibility expressed as early hybrid seed failure and observed as reduced numbers of seeds per fruit. Direct assessments of ovule fertility in the NILs would be necessary to definitively confirm this inference. We are also currently assessing the influence, if any, of prezygotic pollen–pistil interactions on postzygotic seed set (L. C. Moyle and A. Posto, unpublished data).
Five of the 7 QTL for reduced homozygous seed set are colocated with QTL that confer reduced male fertility (Figure 2). Because homozygous seed set is the product of self-fertilization, QTL for reduced self-seed set may not always be independent of loci for male infertility; that is, lines with low pollen fertility may be relatively ineffective at producing selfed seed. Nonetheless, three lines of evidence support independence between pollen fertility and seed fertility for the majority of the detected QTL. First, 2 QTL detected for reduced self- (homozygous) set seed (sss2.1, sss4.1) are not associated with reduced male fertility (Table 3, Figure 2) and are therefore apparently independent of male function. Second, of the 10 QTL detected for reduced pollen fertility (i.e., both PN and PF QTL), 5 are not colocated with or adjacent to seed-set QTL (Figure 2), indicating that reduced pollen fertility per se does not necessitate low self-seed set, under our experimental conditions. Third, an ANOVA indicates that pollen fertility has a significant but very modest effect on observed variation among NILs in self-seed set (no effect was detected for SL-cross seed set); proportion of fertile pollen explains <6% of self-seed set variation [i.e., R2 = 0.057, p = 0.043; ANOVA with PF (arcsine transformed) as main effect]. Total pollen number had no significant effect on self-seed set (data not shown). This suggests that most of the genetic variation detected for selfed-seed set was due to differences in seed fertility itself, rather a pleiotropic consequence of pollen fertility. Finally, we used the residual seed-set values of this ANOVA in a Dunnett's test of mean differences between each NIL and the control parent. Of eight NILs originally detected with reduced self-seed set, four remained significant after the effects of male fertility were removed. These correspond to 4 QTL: both QTL originally identified as independent of male function and 2 additional QTL (sss1.1, sss1.2) that are colocated with a locus that reduces male function. On the basis of these analyses, our provisional conclusion is that these 4 seed-infertility QTL confer reduced seed set, partially or fully independently of any male infertility effects in the same genomic region. The 3 remaining self-seed QTL (sss7.1, sss8.1, sss9.1; Table 3) might be predominantly the product of male-infertility loci located in these regions.
Pollen fertility at genomic regions containing significant seed-set QTL
Seed QTL | Chromosome | Seed Δ% | PN QTL | PN Δ% | PF QTL | PF Δ% |
|---|---|---|---|---|---|---|
| Associated with pollen QTL | ||||||
| sss1.1a | 1 | −70.69 | — | −29.82 | pf1.1 | −27.05 |
| sss1.2a | 1 | −82.95 | pn1.1 | −35.67 | — | −10.59 |
| sss7.1 | 7 | −62.26 | — | −22.80 | pf7.1 | −37.66 |
| sss8.1 | 8 | −57.93 | — | −18.31 | pf8.1 | −32.17 |
| sss9.1 | 9 | −53.73 | — | −30.29 | pf9.1 | −15.21 |
| Not associated with pollen QTL | ||||||
| sss2.1 | 2 | −62.52 | — | −11.84 | — | −5.40 |
| sss4.1 | 4 | −70.65 | — | −19.40 | — | −12.98 |
| sss3.1 | 3 | 46.77 | — | 11.37 | — | −12.96 |
| ssc3.1 | 3 | 60.90 | — | −0.31 | — | −20.01 |
Seed QTL | Chromosome | Seed Δ% | PN QTL | PN Δ% | PF QTL | PF Δ% |
|---|---|---|---|---|---|---|
| Associated with pollen QTL | ||||||
| sss1.1a | 1 | −70.69 | — | −29.82 | pf1.1 | −27.05 |
| sss1.2a | 1 | −82.95 | pn1.1 | −35.67 | — | −10.59 |
| sss7.1 | 7 | −62.26 | — | −22.80 | pf7.1 | −37.66 |
| sss8.1 | 8 | −57.93 | — | −18.31 | pf8.1 | −32.17 |
| sss9.1 | 9 | −53.73 | — | −30.29 | pf9.1 | −15.21 |
| Not associated with pollen QTL | ||||||
| sss2.1 | 2 | −62.52 | — | −11.84 | — | −5.40 |
| sss4.1 | 4 | −70.65 | — | −19.40 | — | −12.98 |
| sss3.1 | 3 | 46.77 | — | 11.37 | — | −12.96 |
| ssc3.1 | 3 | 60.90 | — | −0.31 | — | −20.01 |
Seed Δ% describes the percentage of phenotypic difference in seeds per fruit from the SL parent. Where applicable, the QTL for total pollen count per flower (PN) or the proportion of fertile pollen (PF) that is colocated with or adjacent to each seed QTL is named. Pollen Δ% describes the percentage of phenotypic change from the SL parent for the corresponding pollen trait at the same chromosomal region.
Pollen-associated seed QTL that remain significant after the effects of the proportion of fertile pollen are removed prior to analysis (see text).
Pollen fertility at genomic regions containing significant seed-set QTL
Seed QTL | Chromosome | Seed Δ% | PN QTL | PN Δ% | PF QTL | PF Δ% |
|---|---|---|---|---|---|---|
| Associated with pollen QTL | ||||||
| sss1.1a | 1 | −70.69 | — | −29.82 | pf1.1 | −27.05 |
| sss1.2a | 1 | −82.95 | pn1.1 | −35.67 | — | −10.59 |
| sss7.1 | 7 | −62.26 | — | −22.80 | pf7.1 | −37.66 |
| sss8.1 | 8 | −57.93 | — | −18.31 | pf8.1 | −32.17 |
| sss9.1 | 9 | −53.73 | — | −30.29 | pf9.1 | −15.21 |
| Not associated with pollen QTL | ||||||
| sss2.1 | 2 | −62.52 | — | −11.84 | — | −5.40 |
| sss4.1 | 4 | −70.65 | — | −19.40 | — | −12.98 |
| sss3.1 | 3 | 46.77 | — | 11.37 | — | −12.96 |
| ssc3.1 | 3 | 60.90 | — | −0.31 | — | −20.01 |
Seed QTL | Chromosome | Seed Δ% | PN QTL | PN Δ% | PF QTL | PF Δ% |
|---|---|---|---|---|---|---|
| Associated with pollen QTL | ||||||
| sss1.1a | 1 | −70.69 | — | −29.82 | pf1.1 | −27.05 |
| sss1.2a | 1 | −82.95 | pn1.1 | −35.67 | — | −10.59 |
| sss7.1 | 7 | −62.26 | — | −22.80 | pf7.1 | −37.66 |
| sss8.1 | 8 | −57.93 | — | −18.31 | pf8.1 | −32.17 |
| sss9.1 | 9 | −53.73 | — | −30.29 | pf9.1 | −15.21 |
| Not associated with pollen QTL | ||||||
| sss2.1 | 2 | −62.52 | — | −11.84 | — | −5.40 |
| sss4.1 | 4 | −70.65 | — | −19.40 | — | −12.98 |
| sss3.1 | 3 | 46.77 | — | 11.37 | — | −12.96 |
| ssc3.1 | 3 | 60.90 | — | −0.31 | — | −20.01 |
Seed Δ% describes the percentage of phenotypic difference in seeds per fruit from the SL parent. Where applicable, the QTL for total pollen count per flower (PN) or the proportion of fertile pollen (PF) that is colocated with or adjacent to each seed QTL is named. Pollen Δ% describes the percentage of phenotypic change from the SL parent for the corresponding pollen trait at the same chromosomal region.
Pollen-associated seed QTL that remain significant after the effects of the proportion of fertile pollen are removed prior to analysis (see text).
In lines with a significant effect for either heterozygous or homozygous seed set, we evaluated the allelic effect of the introgression by determining the size and the significance of the dominance deviation using LSMeans contrasts (see materials and methods). For the one QTL that conferred increased seed set (in both homozygous and heterozygous form) the dominance deviation was marginally significant (Table 4), consistent with the QTL contained within the corresponding genomic region acting dominantly and so increasing both heterozygous seeds per fruit and homozygous seeds per fruit. Alternatively, the data may also be consistent with a QTL that increases female—i.e., ovule—fertility directly, regardless of the pollen source used to sire seeds; if this is the case, then our analysis cannot assess genic action at this QTL. Of the remaining seed QTL, only one showed a significant dominance deviation consistent with recessivity (sss8.1; Table 4); i.e., more heterozygous (SP/SL) seeds were produced than expected on the basis of the midparent value of the homozygous (SP/SP and SL/SL) seed set. Because this seed QTL is likely influenced by pollen sterility at the same genomic location, this pattern of recessivity is probably due solely to differences in fertility of the pollen parent between SP/SP and SP/SL seeds (i.e., lowered male fertility of the SP parent) (Table 4). The remaining seed-fertility QTL show patterns of gene action that do not differ from an additive model (Table 4). However, the absence of significant SSC QTL corresponding to the detected SSS QTL suggests that these loci act at least partially recessively; for several loci the sign of the dominance deviation was in a direction consistent with partial recessivity.
Dominance deviations in self-seed set (SSS) QTL
Self-seed set QTL | SP/SP phenotype (seeds) | SP/SL phenotype (seeds) | SP/SL midparent | Dominance deviation (seeds) | Gene action |
|---|---|---|---|---|---|
| Independent loci | |||||
| sss1.1 | 20.97 | 37.40 | 46.26 | −8.85 | Additive |
| sss1.2 | 12.20 | 32.27 | 41.87 | −9.60 | Additive |
| sss2.1 | 26.82 | 45.26 | 49.12 | −3.92 | Additive |
| sss3.1 | 105.0 | 99.50 | 88.27 | 11.23* | Dominant |
| sss4.1 | 21.00 | 55.56 | 46.27 | 9.27 | Additive |
| Nonindependent loci | |||||
| sss7.1 | 27.00 | 53.72 | 49.27 | 4.45 | Additive |
| sss8.1 | 30.10 | 75.24 | 50.82 | 24.42a | Recessive |
| sss9.1 | 33.10 | 57.25 | 52.32 | 4.93 | Additive |
Self-seed set QTL | SP/SP phenotype (seeds) | SP/SL phenotype (seeds) | SP/SL midparent | Dominance deviation (seeds) | Gene action |
|---|---|---|---|---|---|
| Independent loci | |||||
| sss1.1 | 20.97 | 37.40 | 46.26 | −8.85 | Additive |
| sss1.2 | 12.20 | 32.27 | 41.87 | −9.60 | Additive |
| sss2.1 | 26.82 | 45.26 | 49.12 | −3.92 | Additive |
| sss3.1 | 105.0 | 99.50 | 88.27 | 11.23* | Dominant |
| sss4.1 | 21.00 | 55.56 | 46.27 | 9.27 | Additive |
| Nonindependent loci | |||||
| sss7.1 | 27.00 | 53.72 | 49.27 | 4.45 | Additive |
| sss8.1 | 30.10 | 75.24 | 50.82 | 24.42a | Recessive |
| sss9.1 | 33.10 | 57.25 | 52.32 | 4.93 | Additive |
Phenotypes are calculated as means of all NILs contributing to each QTL. Dominance deviation describes the difference between the observed SP/SL phenotypes (SSC) and the expected phenotype based on the midparent value between the observed SP/SP and SL/SL homozygous seed set. Positive values indicate that SP/SL heterozygotes produce more seed than expected. *P < 0.07.
Significant deviation from the midparent expectation.
Dominance deviations in self-seed set (SSS) QTL
Self-seed set QTL | SP/SP phenotype (seeds) | SP/SL phenotype (seeds) | SP/SL midparent | Dominance deviation (seeds) | Gene action |
|---|---|---|---|---|---|
| Independent loci | |||||
| sss1.1 | 20.97 | 37.40 | 46.26 | −8.85 | Additive |
| sss1.2 | 12.20 | 32.27 | 41.87 | −9.60 | Additive |
| sss2.1 | 26.82 | 45.26 | 49.12 | −3.92 | Additive |
| sss3.1 | 105.0 | 99.50 | 88.27 | 11.23* | Dominant |
| sss4.1 | 21.00 | 55.56 | 46.27 | 9.27 | Additive |
| Nonindependent loci | |||||
| sss7.1 | 27.00 | 53.72 | 49.27 | 4.45 | Additive |
| sss8.1 | 30.10 | 75.24 | 50.82 | 24.42a | Recessive |
| sss9.1 | 33.10 | 57.25 | 52.32 | 4.93 | Additive |
Self-seed set QTL | SP/SP phenotype (seeds) | SP/SL phenotype (seeds) | SP/SL midparent | Dominance deviation (seeds) | Gene action |
|---|---|---|---|---|---|
| Independent loci | |||||
| sss1.1 | 20.97 | 37.40 | 46.26 | −8.85 | Additive |
| sss1.2 | 12.20 | 32.27 | 41.87 | −9.60 | Additive |
| sss2.1 | 26.82 | 45.26 | 49.12 | −3.92 | Additive |
| sss3.1 | 105.0 | 99.50 | 88.27 | 11.23* | Dominant |
| sss4.1 | 21.00 | 55.56 | 46.27 | 9.27 | Additive |
| Nonindependent loci | |||||
| sss7.1 | 27.00 | 53.72 | 49.27 | 4.45 | Additive |
| sss8.1 | 30.10 | 75.24 | 50.82 | 24.42a | Recessive |
| sss9.1 | 33.10 | 57.25 | 52.32 | 4.93 | Additive |
Phenotypes are calculated as means of all NILs contributing to each QTL. Dominance deviation describes the difference between the observed SP/SL phenotypes (SSC) and the expected phenotype based on the midparent value between the observed SP/SP and SL/SL homozygous seed set. Positive values indicate that SP/SL heterozygotes produce more seed than expected. *P < 0.07.
Significant deviation from the midparent expectation.
Correlations among traits:
Moderate to strong correlations were found between two pairs of traits (Table 2): First, the number of self-seed set was strongly correlated with the number of cross-seed set, consistent with a possible pleiotropic mechanism underlying these physiologically and genetically related traits. Second, self-seed set was weakly positively correlated with pollen fertility. While some lines with low pollen fertility could have been relatively ineffective at setting selfed seed (as previously discussed), correlations between pollen and seed sterility could also be due to several other mechanisms, including a common developmental basis that influences formation of pollen, ovules, and seed; chromosomal clustering of loci underlying fertility traits so that correlations are due to the nonrandom distribution of these loci in the genome; and/or the action of loci that confer an overall reduction in plant physiological performance or health. Our analysis cannot differentiate pleiotropic effects of single Dobzhansky–Muller interactions from genomic clustering, although finer-scale mapping and eventual identification of underlying genetic factors could resolve these first two alternatives definitively. There is no evidence supporting the third explanation, as there were no large systematic differences between plants in plant size, biomass, or general health, based on greenhouse observations. Regardless, the correlation between pollen and seed fertility is very modest, indicating that factors other than pollen fertility also contributed to reduced self-seed set—consistent with our detection of QTL that reduce seed set independently of reduced male function, and vice versa.
Introgression size effects on hybrid incompatibility:
We detected statistically significant negative associations between the estimated size of the introgressed genetic region(s) in each NIL and both self-seed set (SSS) and SL-cross seed set (SSC); introgression size was not associated with proportion of fertile pollen or total pollen count per flower (supplemental Figure 1, A and B). Nonetheless, significant introgression-size effects explained at most a minority component of the observed variation between lines in fertility. For example, the strongest relationship observed—between introgression size and self-seed set—explained ∼17% of the among-line variation in this trait (supplemental Figure 1); introgression size explained ∼8, ∼2, and <1% of the among-line variation in SSC, PN, and PF, respectively. In comparison, line effects explained 45, 34, 28, and 27%, respectively, of the experimental variance in SSS, SSC, PN, and PF (main effects of line in individual ANOVAs on plant mean values).
QTL colocalization in the two Solanum mapping populations:
In our previous analysis of hybrid incompatibility that used a NIL mapping population between the wild outcrossing species S. habrochaites and S. lycopersicum (Moyle and Graham 2005), we found that hybrid pollen and seed infertility (PN, PF, SSS, and SSC) were each based on 5–11 QTL (Table 5; Figure 1), which individually reduced hybrid fitness by 36–90%. Seed-infertility QTL acted either additively or recessively, with the exception of a single QTL that increased seeds per fruit by ∼twofold above the control SL in both heterozygous (crossed) and homozygous (selfed) NILs. Five of the 8 SSS QTL that reduced fertility were found to be independent of male fertility effects (Moyle and Graham 2005). In terms of genomically colocated sterility loci detected in the two mapping populations, we found that the two studies shared one and three genomic locations for PF and SSS QTL, respectively; no locations were shared for PN or SSC QTL (Figure 1; Table 6). Tests of genomic colocalization among detected QTL in the two populations found significant or marginally significant associations between PF and SSS QTL (Table 6), although only one association remains significant after statistical correction for multiple testing.
Comparison of features of the two Solanum studies of hybrid incompatibility
Population 1 | Population 2 | |
|---|---|---|
| Wild species features | ||
| Predominant mating system | Outcrossing | Outcrossing |
| Mating system of wild species parental accession | Self-incompatible | Self-compatible |
| Silent-site divergence from SL (%) | 4.4 | 4.2 |
| Mapping population structure/features | ||
| Genome coverage, no. NILs analyzed (%) | 85 | >98 |
| No. NILs analyzed | 71 | 71 |
| No. NILs, single introgression only | 47 | 71 |
| Average introgression length (% of genome) | 48.5 (4) | 25.0 (1.98) |
| Range of introgression lengths (% of genome) | 4.5–135(0.35–10.7) | 2.3–57.2(0.18–4.54) |
| Hybrid incompatibility results: no. QTL detected (average phenotypic effect, Δ%) | ||
| PN | 3a (−39.2) | 3 (−43.7) |
| PF | 8 (−36.8) | 7 (−35.9) |
| SSS | 8a (−78.7) | 7a (−65.8) |
| SSC | 1 (121.7) | 1 (60.9) |
Population 1 | Population 2 | |
|---|---|---|
| Wild species features | ||
| Predominant mating system | Outcrossing | Outcrossing |
| Mating system of wild species parental accession | Self-incompatible | Self-compatible |
| Silent-site divergence from SL (%) | 4.4 | 4.2 |
| Mapping population structure/features | ||
| Genome coverage, no. NILs analyzed (%) | 85 | >98 |
| No. NILs analyzed | 71 | 71 |
| No. NILs, single introgression only | 47 | 71 |
| Average introgression length (% of genome) | 48.5 (4) | 25.0 (1.98) |
| Range of introgression lengths (% of genome) | 4.5–135(0.35–10.7) | 2.3–57.2(0.18–4.54) |
| Hybrid incompatibility results: no. QTL detected (average phenotypic effect, Δ%) | ||
| PN | 3a (−39.2) | 3 (−43.7) |
| PF | 8 (−36.8) | 7 (−35.9) |
| SSS | 8a (−78.7) | 7a (−65.8) |
| SSC | 1 (121.7) | 1 (60.9) |
Population 1 refers to the S. habrochaites × S. lycopersicum study (Moyle and Graham 2005); population 2 refers to the S. pennellii × S. lycopersicum study (this study).
Excludes one detected QTL that increased hybrid fertility.
Comparison of features of the two Solanum studies of hybrid incompatibility
Population 1 | Population 2 | |
|---|---|---|
| Wild species features | ||
| Predominant mating system | Outcrossing | Outcrossing |
| Mating system of wild species parental accession | Self-incompatible | Self-compatible |
| Silent-site divergence from SL (%) | 4.4 | 4.2 |
| Mapping population structure/features | ||
| Genome coverage, no. NILs analyzed (%) | 85 | >98 |
| No. NILs analyzed | 71 | 71 |
| No. NILs, single introgression only | 47 | 71 |
| Average introgression length (% of genome) | 48.5 (4) | 25.0 (1.98) |
| Range of introgression lengths (% of genome) | 4.5–135(0.35–10.7) | 2.3–57.2(0.18–4.54) |
| Hybrid incompatibility results: no. QTL detected (average phenotypic effect, Δ%) | ||
| PN | 3a (−39.2) | 3 (−43.7) |
| PF | 8 (−36.8) | 7 (−35.9) |
| SSS | 8a (−78.7) | 7a (−65.8) |
| SSC | 1 (121.7) | 1 (60.9) |
Population 1 | Population 2 | |
|---|---|---|
| Wild species features | ||
| Predominant mating system | Outcrossing | Outcrossing |
| Mating system of wild species parental accession | Self-incompatible | Self-compatible |
| Silent-site divergence from SL (%) | 4.4 | 4.2 |
| Mapping population structure/features | ||
| Genome coverage, no. NILs analyzed (%) | 85 | >98 |
| No. NILs analyzed | 71 | 71 |
| No. NILs, single introgression only | 47 | 71 |
| Average introgression length (% of genome) | 48.5 (4) | 25.0 (1.98) |
| Range of introgression lengths (% of genome) | 4.5–135(0.35–10.7) | 2.3–57.2(0.18–4.54) |
| Hybrid incompatibility results: no. QTL detected (average phenotypic effect, Δ%) | ||
| PN | 3a (−39.2) | 3 (−43.7) |
| PF | 8 (−36.8) | 7 (−35.9) |
| SSS | 8a (−78.7) | 7a (−65.8) |
| SSC | 1 (121.7) | 1 (60.9) |
Population 1 refers to the S. habrochaites × S. lycopersicum study (Moyle and Graham 2005); population 2 refers to the S. pennellii × S. lycopersicum study (this study).
Excludes one detected QTL that increased hybrid fertility.
Significance of QTL colocalization between the two Solanum studies
Trait 1 | Population 1, no. QTL | Population 2, no. QTL | No. colocalized QTL | Significance (P) |
|---|---|---|---|---|
| PN | 4 | 3 | 0 | NS |
| PF | 8 | 7 | 1 | 0.050b |
| SSS | 9 | 8 | 3 | 0.002 |
| SSSa | 6 | 5 | 1 | 0.069b |
| SSC | 1 | 1 | 0 | NS |
Trait 1 | Population 1, no. QTL | Population 2, no. QTL | No. colocalized QTL | Significance (P) |
|---|---|---|---|---|
| PN | 4 | 3 | 0 | NS |
| PF | 8 | 7 | 1 | 0.050b |
| SSS | 9 | 8 | 3 | 0.002 |
| SSSa | 6 | 5 | 1 | 0.069b |
| SSC | 1 | 1 | 0 | NS |
Population 1 refers to the S. habrochaites × S. lycopersicum study (Moyle and Graham 2005); population 2 refers to the S. pennellii × S. lycopersicum study (this study).
Significance of colocalization when only SSS QTL that are statistically independent of pollen fertility (see text) are considered.
NS after correction for multiple testing.
Significance of QTL colocalization between the two Solanum studies
Trait 1 | Population 1, no. QTL | Population 2, no. QTL | No. colocalized QTL | Significance (P) |
|---|---|---|---|---|
| PN | 4 | 3 | 0 | NS |
| PF | 8 | 7 | 1 | 0.050b |
| SSS | 9 | 8 | 3 | 0.002 |
| SSSa | 6 | 5 | 1 | 0.069b |
| SSC | 1 | 1 | 0 | NS |
Trait 1 | Population 1, no. QTL | Population 2, no. QTL | No. colocalized QTL | Significance (P) |
|---|---|---|---|---|
| PN | 4 | 3 | 0 | NS |
| PF | 8 | 7 | 1 | 0.050b |
| SSS | 9 | 8 | 3 | 0.002 |
| SSSa | 6 | 5 | 1 | 0.069b |
| SSC | 1 | 1 | 0 | NS |
Population 1 refers to the S. habrochaites × S. lycopersicum study (Moyle and Graham 2005); population 2 refers to the S. pennellii × S. lycopersicum study (this study).
Significance of colocalization when only SSS QTL that are statistically independent of pollen fertility (see text) are considered.
NS after correction for multiple testing.
DISCUSSION
The nature of the genes that underlie reproductive incompatibility between species influences both dynamics governing the fixation of these loci and, more broadly, which traits are chiefly responsible for the origin of new taxonomic groups. Here we have presented data from a second Solanum species cross-examining hybrid incompatibility, producing a direct comparison to our previous analysis in Solanum (Moyle and Graham 2005). Multiple biological features of the species and statistical features of the mapping populations (Table 5) make a direct comparison between these studies highly appropriate. We find that both studies support three substantive conclusions about the genetics of hybrid incompatibility in this group. First, hybrid incompatibility—measured as pollen sterility and seed infertility—appears to be based on a relatively modest number of QTL. Second, the numbers of QTL influencing male (pollen) sterility and other forms of hybrid incompatibility are roughly comparable. Third, seed-infertility factors generally appear to act recessively or at most additively in hybrid introgression lines. Together these findings form the basis of an emerging picture of the genetics of hybrid incompatibility in this group. Importantly, the first two findings differ substantially from prior observations in Drosophila, while the third is largely in agreement with previous studies. Below we address each of these findings. Finally, we discuss what additional insights can be gained from these comparative analyses of hybrid incompatibility.
Complexity of hybrid incompatibility:
The number and individual effect sizes of incompatibility loci can suggest how many factors might be required to isolate evolving populations. In this study, interspecific pollen and seed infertility could each be due to the effects of 4–11 QTL. Accounting for colocated QTL within this study, we detect a total of 13 genomic regions associated with one or more components of hybrid incompatibility; 11 regions were detected in the previous analysis (Moyle and Graham 2005). In both studies, most of these regions have relatively large individual effects on hybrid infertility (Table 5), although none individually causes complete sterility. These findings are quite different from general patterns in Drosophila, where interspecific—especially male—sterility is frequently highly polygenic and complex (e.g., Wu and Davis 1993; Davis and Wu 1996). Data from the most comparable analyses in fruit flies (Hollocher and Wu 1996; True et al. 1996; Tao et al. 2003b) indicate that ∼60 QTL contribute to hybrid male sterility when Drosophila mauritiana is introgressed into a D. simulans background (Tao et al. 2003a), a species pair separated for ∼0.3 MY. Time since species divergence does not account for this difference between Drosophila and Solanum pairs in the estimated number of hybrid incompatibility QTL; both our species pairs are likely to have been separated for substantially >1 MY (Nesbitt and Tanksley 2002; Table 5).
At least some of the observed differences between Drosophila and Solanum may be due to differences in the degree of genetic resolution between studies. Our Solanum studies use somewhat larger introgressions compared to the Drosophila studies, although introgression lengths are comparable when expressed as a percentage of the whole genome (Moyle and Graham 2005; Table 5). Greater genetic resolution within our mapped regions might, however, reveal a more complex genetic basis underlying each of the QTL identified here. Nonetheless, as we have argued previously (Moyle and Graham 2005), three lines of evidence suggest that the number of loci we have detected are a reasonable estimate of the relative complexity of hybrid incompatibility in Solanum. First, our estimates are roughly in line with previous analyses that suggest a modest number of Dobzhansky–Muller interactions can control hybrid incompatibility among plant species (e.g., Liu et al. 2001; Matsubara et al. 2003; Sweigart et al. 2006; see Moyle and Graham 2005 for additional references). Second, our own analysis suggests that incompatibility is not due to many genes of relatively small individual effect. Both Solanum studies detected relatively weak introgression size effects, for only some measures of hybrid incompatibility (i.e., this association was never significant for measures of male sterility); in comparison, genetic differences between introgression lines explained up to five times the variation observed for these traits. These results suggest that an infinitesimal model of hybrid incompatibility is inappropriate for both Solanum species pairs, a conclusion that is consistent with the limited number of incompatibility QTL we detected and the many genomic regions apparently unassociated with hybrid incompatibility in both studies. Finally, while we do not yet know the molecular genetic basis of QTL identified here, single QTL have regularly been cloned to one or a few individual loci of large effect in several plant systems (Paran and Zamir 2003; Alonso-Blanco et al. 2005), including Solanum [e.g., fw2.2 (Frary et al. 2000) and Brix9-2-5 (Fridman et al. 2000)]. If the fine-scale genetic basis of hybrid incompatibility is similar to these traits, then each of our QTL may be limited to one or few molecular loci of moderate to large effect. Clearly, however, further genetic dissection using shorter recombinants of the current introgressions will be helpful in resolving the number of individual molecular loci contributing to each identified QTL.
Three final factors need to be considered when interpreting the number of hybrid incompatibility QTL we have detected: the possible influence of complex epistasis or of other genetic interactions not tested in these populations (both of which might contribute to undercounting QTL) and the influence of unconditionally deleterious loci (which might contribute to overcounting QTL). First, introgression lines containing single chromosomal regions (i.e., NILs) are individually unable to detect complex epistasis, i.e., epistasis involving multiple loci from one species in the genetic background of another. This is a notable limitation of using NILs for QTL analyses; if complex epistatic interactions commonly underlie a specific trait, then introgression line analysis will give an underestimate of the loci contributing to this trait. Studies in Drosophila have indicated that hybrid incompatibility could be exacerbated by interactions among multiple introgressions from a single species in a foreign genetic background (e.g., Wu and Hollocher 1998; Tao et al. 2003b; Chang and Noor 2007). Nonetheless, there is still somewhat mixed evidence that complex epistasis is generally important in hybrid incompatibility (e.g., Kim and Rieseberg 2001; Slotman et al. 2004). In our previous analysis, we identified one genomic region (pf4.1) in which the combined effects of two or more loci were apparently necessary for the expression of significant pollen sterility (Moyle and Graham 2005). This was possible because in this previous S. lycopersicum–S. habrochaites NIL mapping population, approximately one-third of the lines contained two or three introgressed regions (Table 5). Nonetheless, on the basis of patterns of sterility in lines with single vs. >1 introgressed regions in this other population, we found no evidence that interactions between introgressions systematically act to exacerbate the expression of sterility; in some cases, co-introgression of >1 chromosomal region may actually alleviate sterility phenotypes (Moyle and Graham 2006). In the current study we have no power to detect complex epistasis, as all S. lycopersicum–S. pennellii NILs contain single introgressed regions only. We are currently serially combining NILs containing putative hybrid incompatibility QTL to directly assess the strength of pairwise interactions among conspecific introgressions on a foreign genetic background (T. Nakazato and L. C. Moyle, unpublished data). This ongoing analysis will indicate the extent to which the current study might have underestimated the number of hybrid incompatibility loci in our analyses.
Second, the structure and history of the mapping populations make the detection of some potential additional genetic interactions impossible. In particular, two small genomic regions are missing from the population analyzed here because low seed yield in the corresponding NILs prevented their distribution by the TGRC (see materials and methods). Similarly, in our previous analysis two missing genomic regions might potentially have contained sterility loci (Moyle and Graham 2005). These small missing regions could explain up to two additional QTL in each population. In addition, we did not assess the possible incompatibility effects of nuclear–mitochondrial or nuclear–chloroplast interactions because both populations are uniform for a single cytoplasmic type (S. lycopersicum); both populations were also evaluated under relatively benign greenhouse conditions, precluding detection of loci that might be expressed under different environmental conditions. These latter two factors are unlikely to account for differences between Solanum and comparable Drosophila studies, however, as these also usually do not vary cytoplasmic type and are also performed under relatively benign lab conditions.
Finally, inbreeding depression due to unconditionally deleterious loci could account for one or more putative hybrid incompatibility QTL, if semilethal recessive alleles segregating in the normally outbreeding S. pennellii parent were fixed during the generation of these inbred homozygous lines. However, unlike our previous analysis with the outcrossing species S. habrochaites, the NILs used in this study began with an inbred accession of the (normally outbreeding) S. pennellii (Eshed and Zamir 1994, Table 5). Given its prior history of inbreeding, this accession is unlikely to be segregating many strong unconditionally deleterious alleles; the inbred S. pennellii parental line is highly fertile and shows little evidence of inbreeding depression (Eshed and Zamir 1994; this article). Therefore the bulk of the loci identified here are likely associated with true hybrid incompatibility factors rather than strongly deleterious alleles that were sheltered in the S. pennellii parental population. Given these caveats, on balance our data appear to support an emerging consensus that hybrid incompatibility is substantially less complex in Solanum (and perhaps in other similar biological systems) than in Drosophila.
Expression of male sterility vs. other hybrid incompatibilities:
The second striking result of our analyses in Solanum is the roughly comparable numbers of male-sterility QTL and QTL for other forms of hybrid incompatibility. Analyses in Drosophila indicate that male-sterility factors are one to two orders of magnitude more abundant than loci for female sterility or hybrid inviability (e.g., True et al. 1996; Tao et al. 2003a) and it is generally accepted that male sterility is the first and most rapidly evolving form of hybrid incompatibility in Drosophila (Wu and Hollocher 1998). In our study the relative numbers of loci for male sterility and other forms of hybrid incompatibility are difficult to assess precisely. First, pollen and seed fertility are not strictly analogous measures of hybrid fertility; the former assesses male gamete inviability whereas it is likely that most of the seed sterility observed here is due to zygote incompatibility (lethality) causing early seed failure after fertilization, rather than ovule sterility per se. Second, some measures of hybrid incompatibility may be nonindependent, including pollen and seed sterility (as discussed above). Nonetheless, assuming that our estimated numbers are largely correct (10 pollen QTL vs. 4–7 QTL for seed set), these relative counts for different components of hybrid incompatibility are a substantial departure from the orders of magnitude difference detected in Drosophila studies. In addition, the seed-infertility QTL we detected have, on average, much stronger negative effects on hybrid fitness than the pollen-sterility QTL (Tables 1 and 5).
As hypothesized previously (Moyle and Graham 2005), the difference between our results and those typical of Drosophila might be due to different modes of sex determination, different propensities for sexual interactions, different reproductive biology, and/or different patterns of covariation between traits underlying sex function, between the two groups. For example, in Drosophila, the faster evolution of male sterility is thought to be due to overall faster evolution of male traits, driven by sexual selection or sex-ratio selection, or to sex chromosome–autosome interactions that differ between hybrid males and females (Wu and Davis 1993; Wu et al. 1996; Turelli and Orr 2000; Tao and Hartl 2003). In contrast, Solanum species are hermaphroditic and do not have heteromorphic sex chromosomes, and the relative importance of sexual interactions such as male–male competition is currently unknown in this (and many) plant system(s). Accordingly, differences in sex determination and/or sexual interactions might result in a comparatively slow accumulation of male-sterility factors in Solanum, thereby resulting in the rough parity between pollen sterility and other forms of hybrid incompatibilities. Alternatively, large differences in reproductive biology between Drosophila and Solanum might plausibly explain the differences detected here (Moyle and Graham 2005). For example, hybrid seed inviability (abortion) is a common early interspecific barrier among many plant groups (e.g., Brink and Cooper 1947; Johnston et al. 1980; Katsiotis et al. 1995) and has frequently been attributed to species-specific changes in the endosperm (the triploid tissue that provisions embryonic development within the seed) among Solanaceous species (e.g., Hogenboom 1972; Ehlenfeldt and Ortiz 1995; Lester and Kang 1998). Coordination of unique tissues—such as the endosperm—during reproductive development is one way in which the reproductive biology of different groups might facilitate very different mechanisms of early evolution of reproductive isolation (see also Turelli and Moyle 2007). In our study, this apparent propensity for hybrid seed incompatibility might contribute to equalizing the relative frequency of zygotic-inviability and male-sterility loci. More data on the relative complexity of male vs. female gametogenesis, on the number of genes contributing directly to male and female function (and therefore, for example, their relative sensitivity to mutational change), and on the relative rates of change in these traits will be necessary to assess which forms of hybrid incompatibility typically evolve faster, which traits are differentially involved, and which biological factors are most important in affecting consistent differences among groups.
The mode of gene action for hybrid seed set:
The third major finding of our two Solanum studies is that loci influencing hybrid seed set are generally recessive or at most additive in their gene action. In particular, even at genomic locations where homozygous introgressions produce reduced seed fertility (i.e., our SSS QTL), seed that was heterozygous for these introgressions was not significantly more sterile than the control SL parent. There are very few direct studies of the genomewide dominance relationships of sterility factors in plants (see Sano 1986, Ikehashi and Araki 1988, and Matsubara et al. 2007, for examples of data from specific loci). Several studies in Drosophila (e.g., Hollocher and Wu 1996; True et al. 1996; Presgraves 2003) and other insects (e.g., Slotman et al. 2004) have indicated that factors underlying hybrid male and female sterility, and hybrid inviability, generally act partly or fully recessively. Our findings therefore appear to be consistent with patterns in these other groups. This conclusion relies on the assumption that dominant seed incompatibility loci were not differentially lost during the development of the introgression lines we used. Only two small genomic regions are missing from the NIL population analyzed here, indicating that loss of many dominant factors is unlikely. We have little reason to expect that these missing regions differentially harbor dominant as opposed to recessive incompatibility factors. In our previous analysis, there was evidence that only one of four missing genomic regions might potentially have contained dominant sterility loci (Moyle and Graham 2005). Given these observations we believe that, for both studies, the failure to detect many dominant factors is an unlikely explanation of our observation that seed sterility generally appears to be recessive.
Comparative hybrid sterility in Solanum:
By directly comparing the results from this study with the previous analysis of pollen and seed sterility between S. lycopersicum and S. habrochaites, we were able to evaluate whether, and how many, QTL appear to be shared in common between the two mapping populations. Our results suggest that seed (SSS) and pollen (PF) fertility loci might be colocated more frequently than expected by chance. Colocation of QTL in the two studies could be due to two alternative mechanisms: First, loci that are involved in reproductive/fertility functions could be nonrandomly distributed in the genome, resulting in apparent “sterility hotspots”; accordingly, changes in these loci that affect between-species fertility will appear to be clustered in the genome. Second, at loci that are colocated, the genetic basis of the QTL could be identical in the two crosses; that is, colocated QTL are due to the evolutionary fixation of the same underlying genetic lesion. These explanations are not necessarily mutually exclusive: Even if there is a propensity for chromosomal clustering of loci affecting particular sterility phenotypes, individual shared QTL could still be due to the same underlying genetic mechanism. Given the current resolution of our data we are unable to differentiate these possibilities. Nonetheless, the second hypothesis can be experimentally tested by evaluating whether chromosomal regions carrying colocated QTL from the different species crosses are able to complement each other genetically, when their respective NILs are combined via hybridization. The inference is that the underlying genetic changes are not identical in cases where hybrid fertility is “rescued.” Conversely, when hybrid fertility remains low, either the underlying genetic lesion is identical or other additional interactions between these loci contribute to maintaining reduced hybrid fertility; these possibilities would need to be resolved with further positional cloning of the underlying genes.
Once resolved, comparative mapping data on colocated and lineage-specific loci can offer unique insights into the evolutionary history of the genes responsible for hybrid incompatibility. First, they could be used to infer the timing of trait changes. Consider the case where colocated genes are due to the same underlying genetic change. For this to be true, this trait must have evolved along an evolutionary branch shared by all species involved in the comparison. For example, in our two mapping populations, the phylogenetic relationships among the three species involved are still not definitively resolved, but a consensus phylogeny based on data from AFLPs, cpDNA restriction sites, and sequences from granule-bound starch synthase and internal transcribed spacers indicates that S. habrochaites (SH) and SP share a more recent common ancestor that either species does with SL (Spooner et al. 2005; Figure 3). In this case, shared hybrid incompatibility loci in the two species crosses must have evolved along either branch a or branch d of this phylogeny (Figure 3). Conversely, QTL that are not shared in common between species must have evolved along nonshared branches of the phylogeny (e.g., branches b and c, Figure 3). Second, narrowing the timing of evolution of specific QTL to specific branches of a phylogeny itself indicates which loci were most likely involved earlier vs. later during evolutionary divergence between species (e.g., those occurring on branch a vs. branch b or branch c for our species pairs; Figure 3). For example, if the colocated loci in our studies are indeed shared, the implication is that these arose closer to the original divergence event between the common ancestors of SL, SH, and SP. Third, consistent differences in the relative timing (e.g., early vs. late) of different kinds of DMIs (e.g., pollen vs. seed sterility) might suggest whether particular forms of incompatibility consistently evolve earlier (closer to the original speciation event) than others during the process of lineage divergence. For example, although we have only two species pairs to examine, if colocated loci identified in our analysis are indeed shared, then our data suggest that loci affecting PF and SSS tend to evolve earlier than loci influencing PN. Adding additional species pairs to these analyses is necessary to confirm whether particular kinds of sterility consistently evolve earlier than others during the process of lineage divergence. If so, these patterns might themselves indicate that consistently different forces and dynamics are governing the early vs. late accumulation of these different types of incompatibility (e.g., stronger selection on particular components of sexual reproduction).
![(A) Schematic of the phylogenetic relationships between the three species involved in the two studies: S. lycopersicum (SL), S. pennellii (SP), and S. habrochaites (SH). Internal and tip branches are labeled for reference. Shared hybrid incompatibility loci in the two species crosses must have evolved along either branch a or branch d. Because hybrid incompatibility is expressed when SH and/or SP alleles are placed in the background of SL, it is most plausible that these loci are due to “derived–derived” interactions (Orr 1995), where the QTL we have detected evolved in the lineage leading to SH and SP, after its split from the MRCA of SL (i.e., along branch a). The alternative is that detected hybrid-sterility QTL are one-half of a pair of interacting DM factors that both arose on branch d, i.e., are due to “derived–ancestral” interactions (Orr 1995). This scenario requires that the detected QTL evolved after its DM partner along this branch, which is mathematically less likely (Orr 1995) and arguably less plausible biologically. (B) A simplified expected distribution of hybrid incompatibility factors along each phylogenetic branch, under the “snowball” prediction (Orr 1995; Orr and Turelli 2001), i.e., where the expected number of hybrid incompatibilities increases as an exponential function of time (E[I] ∝ t2). The graphed expectations assume that DM incompatibilities are based on pairwise interactions only.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/genetics/179/3/10.1534_genetics.107.083618/6/m_1437fig3.jpeg?Expires=1717039856&Signature=On9faAX3d7~sQGmyJuJmTDMuz6MS78PammaR1FuqUTMxChIF0gmishuc0OT6nWdtOySmsY8ZbI90cgeVSu9hZf99XtjIRiiJp7xCLOh~qNdmLJ60yPZHDh8O4uP2nw4xgjDkk53GqD6zadN96c9NuY1OMcpp6-fnQ0mDlJEGcB9s8JqrsuLyCUh5~0CTDh0TQ8Lq-PUGdhbPdY2J9IqXUoYy8vKr-pdhZaawDUKZ21~JZp~hi4duS6IzcMEFwpz43oGjb6ndoSsJHbyg405GtMNKyI9i5FUF5W1SJZG~hsSNudn5WWMZzVEdJKO98hlW3Ddh6G5U0JvadEU~ui0F~Q__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
(A) Schematic of the phylogenetic relationships between the three species involved in the two studies: S. lycopersicum (SL), S. pennellii (SP), and S. habrochaites (SH). Internal and tip branches are labeled for reference. Shared hybrid incompatibility loci in the two species crosses must have evolved along either branch a or branch d. Because hybrid incompatibility is expressed when SH and/or SP alleles are placed in the background of SL, it is most plausible that these loci are due to “derived–derived” interactions (Orr 1995), where the QTL we have detected evolved in the lineage leading to SH and SP, after its split from the MRCA of SL (i.e., along branch a). The alternative is that detected hybrid-sterility QTL are one-half of a pair of interacting DM factors that both arose on branch d, i.e., are due to “derived–ancestral” interactions (Orr 1995). This scenario requires that the detected QTL evolved after its DM partner along this branch, which is mathematically less likely (Orr 1995) and arguably less plausible biologically. (B) A simplified expected distribution of hybrid incompatibility factors along each phylogenetic branch, under the “snowball” prediction (Orr 1995; Orr and Turelli 2001), i.e., where the expected number of hybrid incompatibilities increases as an exponential function of time (E[I] ∝ t2). The graphed expectations assume that DM incompatibilities are based on pairwise interactions only.
These predictions are relatively straightforward. However, in our data we have perhaps the simplest case of relationships among species—two species pairs, in which one species is shared in common. Inferences about when specific DMIs evolved will likely become more challenging when additional species pairs from the same clade, with or without common species, are included in these kinds of data sets. Routinely understanding and predicting where and when DMIs have evolved is also likely to be more complex than for other quantitative traits, because intrinsic hybrid incompatibility is a property of pairs of taxa (i.e., is inherently epistatic), rather than a trait than can evolve within a single lineage (Turelli et al. 2001). An analytical framework designed specifically to assess the evolution of DMIs in a phylogenetic context would clearly be valuable. This framework would also need to accommodate other unique features of DMIs. For example, while there is no inherent “directionality” to standard quantitative traits, this is unlikely to be true for hybrid incompatibility, which only increases in magnitude with increasing time between diverging lineages. Similarly, while quantitative traits are often assumed to change monotonically with time, theoretical predictions suggest that the probability of expressing hybrid incompatibilities should “snowball” (i.e., increase faster than linearly with time) between diverging lineages (Orr 1995). Accordingly, phylogenetic models of DMI accumulation must take into account that the expected number of DMIs between two species increases exponentially as you move from the root to the tips of a phylogenetic tree. This will be important when assessing whether the number of shared hybrid incompatibility loci between different species pairs is greater (or less) than would be expected on the basis of the known phylogenetic relationships between species. Deviations from expected values could occur if a particular evolutionary branch (or branches) had experienced an acceleration or a deceleration of accumulation of DMIs. To provide an approximation of this expectation, Figure 3B illustrates the relative numbers of expected accumulated incompatibilities (E[I]) along each branch of the phylogeny, given the simplified expectation that DMIs are accumulating in direct proportion to the square of time since divergence from a common ancestor (i.e., E[I] ∝ t2, where t is time; Orr 1995), and that each DMI is due to a pairwise interaction (rather than involving three or more genes; Orr 1995). This is undoubtedly a crude oversimplification in comparison to the expectations generated by a formal analytical treatment, but allows a very coarse evaluation of whether our data might be consistent with snowball expectations. Our observed data suggest that the number of DMIs that have accumulated along branches b and c is roughly four to seven times the number that have accumulated on branch a, provided that all shared QTL can be assigned to branch a (see Figure 3A). That is, for PF QTL, one QTL is shared in common whereas six or seven QTL are unique to each species cross; for (independent) SSS QTL, one QTL is shared in common, whereas three or four are unique to each species cross (Figure 2). This distribution of QTL on the phylogeny [i.e., E(I)c = E(I)b = between 4 × E(I)a and 6 × E(I)a] is consistent with crude snowball expectations, provided that the most recent common ancestor (MRCA) of SH and SP is approximately half the age of the MRCA of these two species and SL (i.e., t2/t1 ≈ 2.24–2.65). While this simplified example demonstrates that data on collocated loci could be used to evaluate theoretical predictions of the genetic basis of hybrid incompatibility, establishing precise expectations—and the size of confidence intervals on these expectations—clearly requires more formal analytical modeling.
Conclusions:
Although intensive to generate, comparative analyses of hybrid incompatibility can generate unique insights. Between biological groups, they can identify similarities and differences in the genetic basis of species barriers. Here we have shown that the patterns apparently typical of Solanum differ from those of Drosophila, and identified several biological mechanisms that might underlie these differences. Future research in several complementary areas will be useful in resolving which mechanisms might be most crucial for these differences. Conversely, within species groups, comparative data are essential for understanding the time course and dynamics of accumulation of hybrid incompatibilities. Our exploration of these comparative data indicates that they can reveal information on the origin of individual loci and different classes of isolating barrier, as well as the assessment of key theoretical predictions. To our knowledge, this is the first study to explicitly use comparative hybrid incompatibility mapping to outline these inferences. This exploration highlights the need for an analytical framework to examine these data in an explicitly phylogenetic context. Ultimately, our goal in Solanum is to describe equivalent patterns in multiple species crosses, to more completely assess the comparative evolution of hybrid incompatibility in this group. Two additional, unpublished studies of pollen sterility in other Solanum crosses (L. C. Moyle and E. B. Graham, unpublished data) indicate that our provisional conclusions are robust. Comparable data in other systems would be particularly valuable in assessing the generality of the patterns that emerge.
Footnotes
Communicating editor: D. M. Rand
Acknowledgement
We are grateful to M. W. Hahn, M. Turelli, D. Rand, and two anonymous reviewers, for discussion and/or comments that improved earlier versions of the manuscript. R. Chetelat and the C. M. Rick Tomato Genetics Resource Center (University of California at Davis) kindly provided seed stocks of the introgression lines. This research was supported by a National Science Foundation grant (Division of Environmental Biology, grant 0532097) to L.C.M.
References
Barbash, D. A., D. F. Siino, A. M. Tarone and J. Roote,
Barton, N. H., and B. Charlesworth,
Brink, R. A., and D. C. Cooper,
Chetelat, R. T., and Y. Ji,
D'Arcy, W. G.,
De Nettancourt, D.,
Dunnett, C. W.,
Ehlenfeldt, M. K., and R. Ortiz,
Eshed, Y., and D. Zamir,
Fridman, E., T. Pleban and D. Zamir,
Gottlieb, L. D.,
Hogenboom, N. G.,
Hollocher, H., and C. I. Wu,
Ikehashi, H., and H. Araki,
Katsiotis, A., R. E. Hanneman and R. A. Forsberg,
Kearns, C. A., and D. W. Inouye,
Kim, S. C., and L. H. Rieseberg,
Lester, R. N., and J. H. Kang,
Liu, Y. S., L. H. Zhu, J. S. Sun and Y. Chen,
Moyle, L. C.,
Muller, H. J.,
Noor, M. A. F., K. L. Grams, L. A. Bertucci and J. Reiland,
Orr, H. A., and M. Turelli,
Peralta, I. E., S. K. Knapp and D. M. Spooner,
Quiros, C. F.,
Rick, C. M.,
Rick, C. M.,
Rick, C. M.,
Sano, Y.,
Spooner, D. M., I. E. Peralta and S. Knapp,
Tao, Y., and D. L. Hartl,
Van Der Knaap, E., A. Sanyal, S. A. Jackson and S. D. Tanksley,
Wu, C. I., and H. Hollocher,

{kind=link}
{kind=link}
{kind=link}