





Abstract
The bacterial transposon Tn7 recognizes replicating DNA as a target with a preference for the region where DNA replication terminates in the Escherichia coli chromosome. It was previously shown that DNA double-strand breaks in the chromosome stimulate Tn7 transposition where transposition events occur broadly around the point of the DNA break. We show that individual DNA breaks actually activate a series of small regional hotspots in the chromosome for Tn7 insertion. These hotspots are fixed and become active only when a DNA break occurs in the same region of the chromosome. We find that the distribution of insertions around the break is not explained by the exonuclease activity of RecBCD moving the position of the DNA break, and stimulation of Tn7 transposition is not dependent on RecBCD. We show that other forms of DNA damage, like exposure to UV light, mitomycin C, or phleomycin, also stimulate Tn7 transposition. However, inducing the SOS response does not stimulate transposition. Tn7 transposition is not dependent on any known specific pathway of replication fork reactivation as a means of recognizing DNA break repair. Our results are consistent with the idea that Tn7 recognizes DNA replication involved in DNA repair and reveals discrete regions of the chromosome that are differentially activated as transposition targets.
MOBILE genetic elements have a profound effect on genome integrity in bacteria. These elements have also been very important in the laboratory as tools to understand the structure of the genome using genetic experiments that complement microscopic analysis (reviewed in Higgins 2005). We are using the bacterial transposon Tn7 to better understand the nature of chromosome replication, chromosome integrity, and chromosome evolution.
Tn7 uses one of its transposition pathways to target lagging-strand DNA synthesis using the transposon-encoded target site-selecting protein TnsE (Peters and Craig 2001a,b). In addition to the TnsE protein, transposition also requires a core transposition machinery consisting of three proteins: TnsA, TnsB, and TnsC (TnsABC) (Rogers et al. 1986; Waddell and Craig 1988; Flores et al. 1990; Kubo and Craig 1990). Tn7 also controls the orientation of transposition events in the chromosome where they occur in a single left-to-right orientation with the direction of lagging-strand DNA replication (Peters and Craig 2001a). TnsABC+E transposition has a strong regional bias for the area in the chromosome where DNA replication terminates. It was shown that while TnsE actively directs transposition events to the region where DNA replication terminates, transposition events with a mutant core machinery, TnsA, TnsB, TnsCA225V (TnsABC*), that does not require a target site-selecting protein occur randomly across the Escherichia coli chromosome. While active termination in the terminus region focuses TnsABC+E transposition to a smaller region of the chromosome, active termination via the E. coli Tus replication termination protein is not itself required for TnsE-mediated transposition (Peters and Craig 2000).
In previous work it was shown that when DNA double-strand breaks (DSBs) were induced by the excision of Tn10, TnsABC+E transposition was specifically stimulated (Peters and Craig 2000). DSBs were induced through the expression of a mutant form of the Tn10 transposase that causes the Tn10 element to excise, but not reinsert into a new target DNA (Haniford et al. 1989). Tn10 excises through the formation of hairpins on the ends of the element leaving a DSB in the donor DNA (Kennedy et al. 1998), which can be repaired by homologous recombination using a sister chromosome. Tn7 transposition events that were stimulated by the DSB were distributed broadly around the break for many thousands of base pairs, not at the point of the break. The observation that DSBs activated the region proximal to the break as a transposition target suggested that TnsE recognized a facet of DSB repair (Smith 2001). Paradoxically, the distribution of transposition events differed greatly with the two positions where DSBs were induced in this study (Peters and Craig 2000).
We investigated what factors are responsible for stimulating TnsE-mediated transposition as a response to DNA damage. We find that DSBs activate transposition into discrete spans in the chromosome that we call “hotspots.” These hotspots are at fixed positions and are activated only by DSBs in the same general region of the chromosome. The stimulatory effect of DSBs does not require the RecBCD enzyme that normally processes DSBs. We find that multiple types of DNA damage stimulate TnsABC+E transposition. However, TnsE-mediated transposition does not require the replication restart proteins and is not stimulated by SOS induction. Our data are consistent with a model where TnsE is capable of recognizing DNA replication involved in the repair of DNA DSBs.
MATERIALS AND METHODS
Bacterial strains and plasmids:
Strains were constructed using standard P1 transduction methods (Miller 1992; Peters 2007) (Table 1). All Tn10 insertion alleles were confirmed in the final strain by DNA sequencing a template made using arbitrary PCR (see below). Drug markers were removed from frt-containing constructs with the FLP recombinase produced from pCP20, which was subsequently lost at 42° (Datsenko and Wanner 2000). Transduction of the dnaC809 allele was by linkage to thrA-34∷Tn10 and confirmed by PCR amplification followed by restriction digestion with HinFI (the dnaC809 mutation abolishes a HinFI site within the dnaC gene) (Sandler et al. 1996). Strain AP457 was constructed by the method described by Datsenko and Wanner (2000). Briefly, primers JEP191 (5′-AGA AAA ACT CAT CGA GCA TCA AAT GAA ATG AAA CTG CAA TTT ATT CAT A GT GTA GGC TGG AGC TGC TTC-3′) and JEP192 (5′-ATG AGC CAT ATT CAA CGG GAA ACG TCT TGC TCG AGG CCG CGA TTA AAT TCA TAT GAA TAT CCT CCT TAG-3′) were used to amplify the CamR gene (and flanking frt sites) from plasmid pKD3 with 40 bp homology to KanR gene cassettes. The PCR product was crossed into AP429 carrying pKD46, which was induced with 0.2% arabinose and the plasmid subsequently lost by growth at 42° giving AP453. Strain AP699 (BW25113 attTn7∷frt-CamR-frt) was made in a similar fashion with primers JEP295 (5′-ATG TTT TTA ATC AAA CAT CCT GCC AAC TCC ATG TGA CAA AGT GTA GGC TGG AGC TGC TTC-3′) and JEP196 (5′-TCA GTC TGA TTT AAA TAA GCG TTG ATA TTC AGT CAA TTA CCA TAT GAA TAT CCT CCT TAG-3′). This amplicon was used to cross the CamR marker into a site 59 bp downstream of attTn7 without loss of host sequence. The ΔsbcDC∷dhfr allele was also constructed by the method described by Datsenko and Wanner (2000). Primers JEP126 (5′-CCA TGC TTT TTC GCC AGG GAA CCG TTA TGC GCA TCC TTC ACA CCT CAG ACC TGG AAA GTA CGT TTG CAG TGA AAT AAC TAT TCA GCA GGA TAA TGA ATA CGC TAT TTA GGT GAC ACT ATA G-3′) and JEP127 (5′-GTA TTC ATT ATC CTG CTG AAT AGT TAT TTC ACT GCA AAC GTA CTT TCC AGT AAT ACG ACT CAC TAT AGG G-3′) were used to amplify a dhfr gene that imparts resistance to trimethoprim. The resulting PCR fragment was crossed into the chromosome as described above. PCR analysis with flanking primers and DNA sequencing confirmed that all, but the most 5′ five amino acids of coding information of sbcD through the most 3′ five amino acids of coding information of sbcC were replaced with the dhfr cassette. Plasmid vectors were constructed with standard techniques as described (Sambrook et al. 1989) (Table 2).
Strains used in this study
Strain | Genotype | Reference |
|---|---|---|
| MC4100 | araD139 Δ(argF-lac)169 rpsL150 relA1 flhD5301 deoC1 ptsF25 rbsR22 e14− Δ(fimB-fimE)632∷IS1 Δ(fruK-yeiR)725 | Casadaban (1976); Peters et al. (2003) |
| NLC28 | MC4100 ValR | McKown et al. (1987) |
| JP617 | NLC28 attTn7∷miniTn7(R90-lacZYA′-KanR-L166) | McKown et al. (1988); Peters and Craig (2000) |
| JP1588 | JP617 priA∷CamR | Nancy Craig |
| AP44 | JP617 dnaC809 | P1(JC19008) × AP40 |
| AP48 | JP617 dnaC809 priA∷CamR | P1(JP1588) × AP44 |
| JP1386 | NLC28 Δara714 | Peters and Craig (2001a) |
| AP429 | JP1386 attTn7∷miniTn7(R90-lacZYA′-KanR-L166) | P1(JP617) × JP1386 |
| AP453 | JP1386 attTn7∷miniTn7(R90-lacZYA′-frt-CamR-frt-L166) | This work |
| AP457 | JP1386 attTn7∷miniTn7(R90-lacZYA′-frt-L166) | This work |
| AP523 | AP457 priC303∷KanR | P1(JC19008) × AP457 |
| AP767 | AP457 ΔsulA6209∷TetR | P1(GM7542) × AP457 |
| AP964 | AP457 lexA71∷Tn5 | P1(KL788) × AP457 |
| AP965 | AP457 lexA71∷Tn5 ΔsulA6209∷TetR | P1(KL788) × AP767 |
| AP40 | JP617 thrA-34∷Tn10 | P1(CAG18442) × JP617 |
| JP949 | NLC28 attTn7∷Tn7 | Nancy Craig |
| AP699 | BW25113 attTn7∷CamR | This work |
| AP724 | NLC28 attTn7∷CamR | This work |
| JP2178 | NLC28 attTn7∷miniTn7(R90-lacZYA′-frt-L166) srlD∷Tn10 ΔsbcDC∷dhfr | This work |
| BW25113 | MG1655 Δ(araD-araB)567 lacZ-4787Δ(∷rrnB-3) Δ(rhaD-rhaB)568 hsdR514 | Datsenko and Wanner (2000) |
| CAG12135 | MG1655 recD-1901∷Tn10 | Singer et al. (1989); Nichols et al. (1998) |
| CAG12153 | MG1655 b3219-6∷Tn10 | Singer et al. (1989); Nichols et al. (1998) |
| CAG12071 | MG1655 smg-3082∷Tn10 | Singer et al. (1989); Nichols et al. (1998) |
| CAG18433 | MG1655 zbf-3057∷Tn10 | Singer et al. (1989); Nichols et al. (1998) |
| CAG18442 | MG1655 thrA-34∷Tn10 | Singer et al. (1989); Nichols et al. (1998) |
| CAG18456 | MG1655 yhfT-3084∷Tn10 | Singer et al. (1989); Nichols et al. (1998) |
| CAG18493 | MG1655 zbi-29∷Tn10 | Singer et al. (1989); Nichols et al. (1998) |
| CAG18642 | MG1655 srlD-3131∷Tn10 | Singer et al. (1989); Nichols et al. (1998) |
| GM7542 | MG1655 ΔsulA6209∷TetR | Martin Marinus via Mark Sutton |
| JC19008 | JC18983 dnaC809 thrA+ priC303∷KanR | Sandler et al. (1996) |
| KL788 | Δ(gpt-lac)5 tsx-35 sulA3 e14- rfbC1 recA441(ts) relA1 rpsL31 kdgK51 mtl-1 spoT1 thi-1 lexA71∷Tn5 creC510 | Miller and Low (1984) |
| STL7206 | MG1655 sbcB∷frt-KanR-frt rph-1 | Susan Lovett |
| STL7886 | MG1655 recC∷frt-CamR-frt rph-1 | Susan Lovett |
Strain | Genotype | Reference |
|---|---|---|
| MC4100 | araD139 Δ(argF-lac)169 rpsL150 relA1 flhD5301 deoC1 ptsF25 rbsR22 e14− Δ(fimB-fimE)632∷IS1 Δ(fruK-yeiR)725 | Casadaban (1976); Peters et al. (2003) |
| NLC28 | MC4100 ValR | McKown et al. (1987) |
| JP617 | NLC28 attTn7∷miniTn7(R90-lacZYA′-KanR-L166) | McKown et al. (1988); Peters and Craig (2000) |
| JP1588 | JP617 priA∷CamR | Nancy Craig |
| AP44 | JP617 dnaC809 | P1(JC19008) × AP40 |
| AP48 | JP617 dnaC809 priA∷CamR | P1(JP1588) × AP44 |
| JP1386 | NLC28 Δara714 | Peters and Craig (2001a) |
| AP429 | JP1386 attTn7∷miniTn7(R90-lacZYA′-KanR-L166) | P1(JP617) × JP1386 |
| AP453 | JP1386 attTn7∷miniTn7(R90-lacZYA′-frt-CamR-frt-L166) | This work |
| AP457 | JP1386 attTn7∷miniTn7(R90-lacZYA′-frt-L166) | This work |
| AP523 | AP457 priC303∷KanR | P1(JC19008) × AP457 |
| AP767 | AP457 ΔsulA6209∷TetR | P1(GM7542) × AP457 |
| AP964 | AP457 lexA71∷Tn5 | P1(KL788) × AP457 |
| AP965 | AP457 lexA71∷Tn5 ΔsulA6209∷TetR | P1(KL788) × AP767 |
| AP40 | JP617 thrA-34∷Tn10 | P1(CAG18442) × JP617 |
| JP949 | NLC28 attTn7∷Tn7 | Nancy Craig |
| AP699 | BW25113 attTn7∷CamR | This work |
| AP724 | NLC28 attTn7∷CamR | This work |
| JP2178 | NLC28 attTn7∷miniTn7(R90-lacZYA′-frt-L166) srlD∷Tn10 ΔsbcDC∷dhfr | This work |
| BW25113 | MG1655 Δ(araD-araB)567 lacZ-4787Δ(∷rrnB-3) Δ(rhaD-rhaB)568 hsdR514 | Datsenko and Wanner (2000) |
| CAG12135 | MG1655 recD-1901∷Tn10 | Singer et al. (1989); Nichols et al. (1998) |
| CAG12153 | MG1655 b3219-6∷Tn10 | Singer et al. (1989); Nichols et al. (1998) |
| CAG12071 | MG1655 smg-3082∷Tn10 | Singer et al. (1989); Nichols et al. (1998) |
| CAG18433 | MG1655 zbf-3057∷Tn10 | Singer et al. (1989); Nichols et al. (1998) |
| CAG18442 | MG1655 thrA-34∷Tn10 | Singer et al. (1989); Nichols et al. (1998) |
| CAG18456 | MG1655 yhfT-3084∷Tn10 | Singer et al. (1989); Nichols et al. (1998) |
| CAG18493 | MG1655 zbi-29∷Tn10 | Singer et al. (1989); Nichols et al. (1998) |
| CAG18642 | MG1655 srlD-3131∷Tn10 | Singer et al. (1989); Nichols et al. (1998) |
| GM7542 | MG1655 ΔsulA6209∷TetR | Martin Marinus via Mark Sutton |
| JC19008 | JC18983 dnaC809 thrA+ priC303∷KanR | Sandler et al. (1996) |
| KL788 | Δ(gpt-lac)5 tsx-35 sulA3 e14- rfbC1 recA441(ts) relA1 rpsL31 kdgK51 mtl-1 spoT1 thi-1 lexA71∷Tn5 creC510 | Miller and Low (1984) |
| STL7206 | MG1655 sbcB∷frt-KanR-frt rph-1 | Susan Lovett |
| STL7886 | MG1655 recC∷frt-CamR-frt rph-1 | Susan Lovett |
Strains used in this study
Strain | Genotype | Reference |
|---|---|---|
| MC4100 | araD139 Δ(argF-lac)169 rpsL150 relA1 flhD5301 deoC1 ptsF25 rbsR22 e14− Δ(fimB-fimE)632∷IS1 Δ(fruK-yeiR)725 | Casadaban (1976); Peters et al. (2003) |
| NLC28 | MC4100 ValR | McKown et al. (1987) |
| JP617 | NLC28 attTn7∷miniTn7(R90-lacZYA′-KanR-L166) | McKown et al. (1988); Peters and Craig (2000) |
| JP1588 | JP617 priA∷CamR | Nancy Craig |
| AP44 | JP617 dnaC809 | P1(JC19008) × AP40 |
| AP48 | JP617 dnaC809 priA∷CamR | P1(JP1588) × AP44 |
| JP1386 | NLC28 Δara714 | Peters and Craig (2001a) |
| AP429 | JP1386 attTn7∷miniTn7(R90-lacZYA′-KanR-L166) | P1(JP617) × JP1386 |
| AP453 | JP1386 attTn7∷miniTn7(R90-lacZYA′-frt-CamR-frt-L166) | This work |
| AP457 | JP1386 attTn7∷miniTn7(R90-lacZYA′-frt-L166) | This work |
| AP523 | AP457 priC303∷KanR | P1(JC19008) × AP457 |
| AP767 | AP457 ΔsulA6209∷TetR | P1(GM7542) × AP457 |
| AP964 | AP457 lexA71∷Tn5 | P1(KL788) × AP457 |
| AP965 | AP457 lexA71∷Tn5 ΔsulA6209∷TetR | P1(KL788) × AP767 |
| AP40 | JP617 thrA-34∷Tn10 | P1(CAG18442) × JP617 |
| JP949 | NLC28 attTn7∷Tn7 | Nancy Craig |
| AP699 | BW25113 attTn7∷CamR | This work |
| AP724 | NLC28 attTn7∷CamR | This work |
| JP2178 | NLC28 attTn7∷miniTn7(R90-lacZYA′-frt-L166) srlD∷Tn10 ΔsbcDC∷dhfr | This work |
| BW25113 | MG1655 Δ(araD-araB)567 lacZ-4787Δ(∷rrnB-3) Δ(rhaD-rhaB)568 hsdR514 | Datsenko and Wanner (2000) |
| CAG12135 | MG1655 recD-1901∷Tn10 | Singer et al. (1989); Nichols et al. (1998) |
| CAG12153 | MG1655 b3219-6∷Tn10 | Singer et al. (1989); Nichols et al. (1998) |
| CAG12071 | MG1655 smg-3082∷Tn10 | Singer et al. (1989); Nichols et al. (1998) |
| CAG18433 | MG1655 zbf-3057∷Tn10 | Singer et al. (1989); Nichols et al. (1998) |
| CAG18442 | MG1655 thrA-34∷Tn10 | Singer et al. (1989); Nichols et al. (1998) |
| CAG18456 | MG1655 yhfT-3084∷Tn10 | Singer et al. (1989); Nichols et al. (1998) |
| CAG18493 | MG1655 zbi-29∷Tn10 | Singer et al. (1989); Nichols et al. (1998) |
| CAG18642 | MG1655 srlD-3131∷Tn10 | Singer et al. (1989); Nichols et al. (1998) |
| GM7542 | MG1655 ΔsulA6209∷TetR | Martin Marinus via Mark Sutton |
| JC19008 | JC18983 dnaC809 thrA+ priC303∷KanR | Sandler et al. (1996) |
| KL788 | Δ(gpt-lac)5 tsx-35 sulA3 e14- rfbC1 recA441(ts) relA1 rpsL31 kdgK51 mtl-1 spoT1 thi-1 lexA71∷Tn5 creC510 | Miller and Low (1984) |
| STL7206 | MG1655 sbcB∷frt-KanR-frt rph-1 | Susan Lovett |
| STL7886 | MG1655 recC∷frt-CamR-frt rph-1 | Susan Lovett |
Strain | Genotype | Reference |
|---|---|---|
| MC4100 | araD139 Δ(argF-lac)169 rpsL150 relA1 flhD5301 deoC1 ptsF25 rbsR22 e14− Δ(fimB-fimE)632∷IS1 Δ(fruK-yeiR)725 | Casadaban (1976); Peters et al. (2003) |
| NLC28 | MC4100 ValR | McKown et al. (1987) |
| JP617 | NLC28 attTn7∷miniTn7(R90-lacZYA′-KanR-L166) | McKown et al. (1988); Peters and Craig (2000) |
| JP1588 | JP617 priA∷CamR | Nancy Craig |
| AP44 | JP617 dnaC809 | P1(JC19008) × AP40 |
| AP48 | JP617 dnaC809 priA∷CamR | P1(JP1588) × AP44 |
| JP1386 | NLC28 Δara714 | Peters and Craig (2001a) |
| AP429 | JP1386 attTn7∷miniTn7(R90-lacZYA′-KanR-L166) | P1(JP617) × JP1386 |
| AP453 | JP1386 attTn7∷miniTn7(R90-lacZYA′-frt-CamR-frt-L166) | This work |
| AP457 | JP1386 attTn7∷miniTn7(R90-lacZYA′-frt-L166) | This work |
| AP523 | AP457 priC303∷KanR | P1(JC19008) × AP457 |
| AP767 | AP457 ΔsulA6209∷TetR | P1(GM7542) × AP457 |
| AP964 | AP457 lexA71∷Tn5 | P1(KL788) × AP457 |
| AP965 | AP457 lexA71∷Tn5 ΔsulA6209∷TetR | P1(KL788) × AP767 |
| AP40 | JP617 thrA-34∷Tn10 | P1(CAG18442) × JP617 |
| JP949 | NLC28 attTn7∷Tn7 | Nancy Craig |
| AP699 | BW25113 attTn7∷CamR | This work |
| AP724 | NLC28 attTn7∷CamR | This work |
| JP2178 | NLC28 attTn7∷miniTn7(R90-lacZYA′-frt-L166) srlD∷Tn10 ΔsbcDC∷dhfr | This work |
| BW25113 | MG1655 Δ(araD-araB)567 lacZ-4787Δ(∷rrnB-3) Δ(rhaD-rhaB)568 hsdR514 | Datsenko and Wanner (2000) |
| CAG12135 | MG1655 recD-1901∷Tn10 | Singer et al. (1989); Nichols et al. (1998) |
| CAG12153 | MG1655 b3219-6∷Tn10 | Singer et al. (1989); Nichols et al. (1998) |
| CAG12071 | MG1655 smg-3082∷Tn10 | Singer et al. (1989); Nichols et al. (1998) |
| CAG18433 | MG1655 zbf-3057∷Tn10 | Singer et al. (1989); Nichols et al. (1998) |
| CAG18442 | MG1655 thrA-34∷Tn10 | Singer et al. (1989); Nichols et al. (1998) |
| CAG18456 | MG1655 yhfT-3084∷Tn10 | Singer et al. (1989); Nichols et al. (1998) |
| CAG18493 | MG1655 zbi-29∷Tn10 | Singer et al. (1989); Nichols et al. (1998) |
| CAG18642 | MG1655 srlD-3131∷Tn10 | Singer et al. (1989); Nichols et al. (1998) |
| GM7542 | MG1655 ΔsulA6209∷TetR | Martin Marinus via Mark Sutton |
| JC19008 | JC18983 dnaC809 thrA+ priC303∷KanR | Sandler et al. (1996) |
| KL788 | Δ(gpt-lac)5 tsx-35 sulA3 e14- rfbC1 recA441(ts) relA1 rpsL31 kdgK51 mtl-1 spoT1 thi-1 lexA71∷Tn5 creC510 | Miller and Low (1984) |
| STL7206 | MG1655 sbcB∷frt-KanR-frt rph-1 | Susan Lovett |
| STL7886 | MG1655 recC∷frt-CamR-frt rph-1 | Susan Lovett |
Plasmids used in this study and the source or construction
Name | Relevant information |
|---|---|
| pCW4 | pACYC184 derivative encoding TnsABCDE, tetracycline resistant (Waddell and Craig 1988). |
| pCW15 | pACYC184 derivative encoding TnsABC, chloramphenicol resistant (Waddell and Craig 1988). |
| pCW15* | pACYC184 derivative encoding TnsABCA225V, chloramphenicol resistant (Stellwagen and Craig 1997b). |
| pJP123 | pACYC184 derivative encoding TnsABC+E, chloramphenicol resistant (Peters and Craig 2000). |
| pTA106 | pSC101 replicon, ampicillin-resistant cloning vector (Peters and Craig 2000). |
| pJP104 | pTA106 derivative encoding TnsE, ampicillin resistant (Peters and Craig 2000). |
| pQS100 | pTA106 vector encoding TnsABC constructed by cloning the 4919 bp of PvuII fragment encoding TnsABC from pCW4 into the SmaI site of pTA106 with the tnsC gene proximal to the vector HindIII site. |
| pQS101 | pTA106 vector encoding TnsABC constructed by cloning the 4919 bp of PvuII fragment encoding TnsABC from pCW4 into the SmaI site of pTA106 with the tnsA gene proximal to the vector HindIII site. |
| pQS102 | pTA106 vector encoding TnsABC+E constructed by cloning the 4919 bp of PvuII fragment encoding TnsABC from pCW4 into the SmaI site of pJP104 vector with the tnsC gene proximal to the vector HindIII site. |
| pQS103 | pTA106 vector encoding TnsABC+E constructed by cloning the 4919 bp PvuII fragment encoding TnsABC from pCW4 into the SmaI site of pJP104 vector with the tnsA gene proximal to the vector HindIII site. |
| pQS107 | pTA106 vector encoding TnsABCA225V constructed by cloning a 726 bp KpnI-StuI fragment encoding the Alanine(225) to Valine amino acid change of tnsC from pCW15* into pQS101. |
| pZT383 | pMB1 vector with lacIq regulation expressing Tn10 transposase mutant P167S, ampicillin resistant (Galitski and Roth 1997). |
| pJP135 | pMB1 vector with lacIq regulation expressing Tn10 transposase mutant D97A/D161A, ampicillin resistant (Peters and Craig 2000). |
| pCP20 | Temperature-sensitive plasmid with thermal induction of FLP recombinase (Cherepanov and Wackernagel 1995). |
| pKD46 | Temperature-sensitive plasmid with the λ Red proteins under arabinose control (Datsenko and Wanner 2000). |
| pKD3 | Plasmid encoding ampicillin resistance that allows PCR amplification of a gene cassette encoding chloramphenicol resistance flanked by frt sites recognized by the FLP recombinase (Datsenko and Wanner 2000). |
Name | Relevant information |
|---|---|
| pCW4 | pACYC184 derivative encoding TnsABCDE, tetracycline resistant (Waddell and Craig 1988). |
| pCW15 | pACYC184 derivative encoding TnsABC, chloramphenicol resistant (Waddell and Craig 1988). |
| pCW15* | pACYC184 derivative encoding TnsABCA225V, chloramphenicol resistant (Stellwagen and Craig 1997b). |
| pJP123 | pACYC184 derivative encoding TnsABC+E, chloramphenicol resistant (Peters and Craig 2000). |
| pTA106 | pSC101 replicon, ampicillin-resistant cloning vector (Peters and Craig 2000). |
| pJP104 | pTA106 derivative encoding TnsE, ampicillin resistant (Peters and Craig 2000). |
| pQS100 | pTA106 vector encoding TnsABC constructed by cloning the 4919 bp of PvuII fragment encoding TnsABC from pCW4 into the SmaI site of pTA106 with the tnsC gene proximal to the vector HindIII site. |
| pQS101 | pTA106 vector encoding TnsABC constructed by cloning the 4919 bp of PvuII fragment encoding TnsABC from pCW4 into the SmaI site of pTA106 with the tnsA gene proximal to the vector HindIII site. |
| pQS102 | pTA106 vector encoding TnsABC+E constructed by cloning the 4919 bp of PvuII fragment encoding TnsABC from pCW4 into the SmaI site of pJP104 vector with the tnsC gene proximal to the vector HindIII site. |
| pQS103 | pTA106 vector encoding TnsABC+E constructed by cloning the 4919 bp PvuII fragment encoding TnsABC from pCW4 into the SmaI site of pJP104 vector with the tnsA gene proximal to the vector HindIII site. |
| pQS107 | pTA106 vector encoding TnsABCA225V constructed by cloning a 726 bp KpnI-StuI fragment encoding the Alanine(225) to Valine amino acid change of tnsC from pCW15* into pQS101. |
| pZT383 | pMB1 vector with lacIq regulation expressing Tn10 transposase mutant P167S, ampicillin resistant (Galitski and Roth 1997). |
| pJP135 | pMB1 vector with lacIq regulation expressing Tn10 transposase mutant D97A/D161A, ampicillin resistant (Peters and Craig 2000). |
| pCP20 | Temperature-sensitive plasmid with thermal induction of FLP recombinase (Cherepanov and Wackernagel 1995). |
| pKD46 | Temperature-sensitive plasmid with the λ Red proteins under arabinose control (Datsenko and Wanner 2000). |
| pKD3 | Plasmid encoding ampicillin resistance that allows PCR amplification of a gene cassette encoding chloramphenicol resistance flanked by frt sites recognized by the FLP recombinase (Datsenko and Wanner 2000). |
Plasmids used in this study and the source or construction
Name | Relevant information |
|---|---|
| pCW4 | pACYC184 derivative encoding TnsABCDE, tetracycline resistant (Waddell and Craig 1988). |
| pCW15 | pACYC184 derivative encoding TnsABC, chloramphenicol resistant (Waddell and Craig 1988). |
| pCW15* | pACYC184 derivative encoding TnsABCA225V, chloramphenicol resistant (Stellwagen and Craig 1997b). |
| pJP123 | pACYC184 derivative encoding TnsABC+E, chloramphenicol resistant (Peters and Craig 2000). |
| pTA106 | pSC101 replicon, ampicillin-resistant cloning vector (Peters and Craig 2000). |
| pJP104 | pTA106 derivative encoding TnsE, ampicillin resistant (Peters and Craig 2000). |
| pQS100 | pTA106 vector encoding TnsABC constructed by cloning the 4919 bp of PvuII fragment encoding TnsABC from pCW4 into the SmaI site of pTA106 with the tnsC gene proximal to the vector HindIII site. |
| pQS101 | pTA106 vector encoding TnsABC constructed by cloning the 4919 bp of PvuII fragment encoding TnsABC from pCW4 into the SmaI site of pTA106 with the tnsA gene proximal to the vector HindIII site. |
| pQS102 | pTA106 vector encoding TnsABC+E constructed by cloning the 4919 bp of PvuII fragment encoding TnsABC from pCW4 into the SmaI site of pJP104 vector with the tnsC gene proximal to the vector HindIII site. |
| pQS103 | pTA106 vector encoding TnsABC+E constructed by cloning the 4919 bp PvuII fragment encoding TnsABC from pCW4 into the SmaI site of pJP104 vector with the tnsA gene proximal to the vector HindIII site. |
| pQS107 | pTA106 vector encoding TnsABCA225V constructed by cloning a 726 bp KpnI-StuI fragment encoding the Alanine(225) to Valine amino acid change of tnsC from pCW15* into pQS101. |
| pZT383 | pMB1 vector with lacIq regulation expressing Tn10 transposase mutant P167S, ampicillin resistant (Galitski and Roth 1997). |
| pJP135 | pMB1 vector with lacIq regulation expressing Tn10 transposase mutant D97A/D161A, ampicillin resistant (Peters and Craig 2000). |
| pCP20 | Temperature-sensitive plasmid with thermal induction of FLP recombinase (Cherepanov and Wackernagel 1995). |
| pKD46 | Temperature-sensitive plasmid with the λ Red proteins under arabinose control (Datsenko and Wanner 2000). |
| pKD3 | Plasmid encoding ampicillin resistance that allows PCR amplification of a gene cassette encoding chloramphenicol resistance flanked by frt sites recognized by the FLP recombinase (Datsenko and Wanner 2000). |
Name | Relevant information |
|---|---|
| pCW4 | pACYC184 derivative encoding TnsABCDE, tetracycline resistant (Waddell and Craig 1988). |
| pCW15 | pACYC184 derivative encoding TnsABC, chloramphenicol resistant (Waddell and Craig 1988). |
| pCW15* | pACYC184 derivative encoding TnsABCA225V, chloramphenicol resistant (Stellwagen and Craig 1997b). |
| pJP123 | pACYC184 derivative encoding TnsABC+E, chloramphenicol resistant (Peters and Craig 2000). |
| pTA106 | pSC101 replicon, ampicillin-resistant cloning vector (Peters and Craig 2000). |
| pJP104 | pTA106 derivative encoding TnsE, ampicillin resistant (Peters and Craig 2000). |
| pQS100 | pTA106 vector encoding TnsABC constructed by cloning the 4919 bp of PvuII fragment encoding TnsABC from pCW4 into the SmaI site of pTA106 with the tnsC gene proximal to the vector HindIII site. |
| pQS101 | pTA106 vector encoding TnsABC constructed by cloning the 4919 bp of PvuII fragment encoding TnsABC from pCW4 into the SmaI site of pTA106 with the tnsA gene proximal to the vector HindIII site. |
| pQS102 | pTA106 vector encoding TnsABC+E constructed by cloning the 4919 bp of PvuII fragment encoding TnsABC from pCW4 into the SmaI site of pJP104 vector with the tnsC gene proximal to the vector HindIII site. |
| pQS103 | pTA106 vector encoding TnsABC+E constructed by cloning the 4919 bp PvuII fragment encoding TnsABC from pCW4 into the SmaI site of pJP104 vector with the tnsA gene proximal to the vector HindIII site. |
| pQS107 | pTA106 vector encoding TnsABCA225V constructed by cloning a 726 bp KpnI-StuI fragment encoding the Alanine(225) to Valine amino acid change of tnsC from pCW15* into pQS101. |
| pZT383 | pMB1 vector with lacIq regulation expressing Tn10 transposase mutant P167S, ampicillin resistant (Galitski and Roth 1997). |
| pJP135 | pMB1 vector with lacIq regulation expressing Tn10 transposase mutant D97A/D161A, ampicillin resistant (Peters and Craig 2000). |
| pCP20 | Temperature-sensitive plasmid with thermal induction of FLP recombinase (Cherepanov and Wackernagel 1995). |
| pKD46 | Temperature-sensitive plasmid with the λ Red proteins under arabinose control (Datsenko and Wanner 2000). |
| pKD3 | Plasmid encoding ampicillin resistance that allows PCR amplification of a gene cassette encoding chloramphenicol resistance flanked by frt sites recognized by the FLP recombinase (Datsenko and Wanner 2000). |
Transposition assay:
A promoter capture assay, called the papillation assay, monitors transposition levels in lawns of bacteria on indicator media (Huisman and Kleckner 1987; Stellwagen and Craig 1997a). A miniTn7 element encoding the lactose utilization genes without the requisite promoter is situated in the tester strain attTn7 site (JP617 or AP457) (Table 1). In this configuration the lac genes are not transcribed and the strain is unable to use lactose (Lac−). If the element transposes to a new position in the correct orientation in an active gene the element will be transcribed resulting in a phenotypically Lac+ bacterium. On MacConkey's lactose indicator media or on LB lactose X-gal media, Lac+ cells form differentially colored microcolonies, or papillae, on the lawn of Lac− cells. The number of Lac+ papillae provides a measure of transposition levels. To rule out any effect of the tested allele or condition on cell viability or papillae formation an internal control is always tested where papillae formation is compared with a background strain that lacks TnsE.
Transposon mapping:
To map Tn7 transposon insertion events, the Lac+ papillae were purified and P1 linkage mapping was used to determine if transposition had occurred or if an unrelated Lac+ mutation occurred, as described previously using strain JP949 or AP724 (Peters and Craig 2000). For each Tn10 DSB position, two Lac+ events were chosen at random from each of the 12 experiments. Two Lac+ events were chosen at random from each of 24 experiments from cells expressing TnsABC+E following UV irradiation. Because the Lac+ events that result from recombination and not Tn7 transposition varies from 30 to 60%, the total number of real Tn7 transposition events that were mapped is not equal. Tn7 transposition events were mapped by sequencing from the left and/or right ends of products made using arbitrary PCR as described previously and the position in the genome determined using the E. coli MG1655 genome sequence version M52 (Blattner et al. 1997; Peters and Craig 2001a). In ∼5% of the cases both the left and right ends were sequenced to ensure that real transposition occurred. Tn10 insertions were mapped using arbitrary PCR as above, but replacing the arbitrary primer (JEP293 5′-GGC CAC GCG TCG ACT AGT CAN NNN NNN NNN GAT CG-3′) and the element specific primers 1 and 2 from (Nichols et al. 1998). Primer 2 was used for sequencing the final Tn10 template.
β-Galactosidase assay:
The β-galactosidase activity was measured as described in triplicate for each sample (Miller 1992).
DNA damage induction:
To determine the effect of DSBs, a mutant Tn10 transposase (pZT383-TnpP167S) was expressed that allows the element to excise, but not to insert (Haniford et al. 1989; Galitski and Roth 1997; Peters and Craig 2000). To ensure that the effect was from the DNA break and not an unrelated effect of the transposase, control experiments utilized the same vector with mutations in the active site of the Tn10 transposase (pJP135-TnpD97A/D161A) (Table 2) (Peters and Craig 2000). Plates were exposed to 10 mJ of UV in a Stratalinker 1800 (Stratagene). Mitomyocin C (Sigma) and phleomycin (Sigma) were diluted in water and added to the media prior to pouring after it cooled.
RESULTS
Our objective is to understand processes that contribute to the distribution of Tn7 transposition events around induced DSBs at specific locations in the chromosome. It was previously shown that a very different distribution of Tn7 insertions was found when DSBs were induced at two different positions in the E. coli chromosome (Peters and Craig 2000). One simple model for why transposition events might occur at some distance from the original position of an induced DSB is that the potent exonuclease activity of the RecBCD enzyme could move the actual genetic position of the DSB. The extent to which the RecBCD enzyme would process DNA is a function of the density of properly oriented chi sites around the point of a DSB. Chi sites are polar, 8-bp sequences that alter the exonuclease activity and RecA-loading activity of the RecBCD complex (Kuzminov 1999; Kowalczykowski 2000). Given that the distribution of chi sites varies in the chromosome, it seemed possible that chi-site density could explain differences in the distribution of transposition events around DSBs (see for example, Smith 2001). This was of special interest because, fortuitously, one of the original DSB positions (srlD-3131∷Tn10, 2826 kb) was in a region that was very sparse for chi sites (J. E. Peters, unpublished data). To address this idea we analyzed the distribution of TnsABC+E transposition events when DSBs were induced at three positions that were not examined previously, zbi-29∷Tn10, recD-1901∷Tn10, and yhfT-3084∷Tn10 (materials and methods). Tn7 transposition was monitored through the use of a promoter capture assay using a Tn7 derivative that contains the lactose utilization genes without a promoter. Transposition of the Tn7 derivative can allow the host to acquire the ability to grow on lactose (Lac+) if the element moves to a transcribed region of the chromosome. On indicator media a Lac+ cell will form a differentially colored microcolony, or papilla, on the lawn of bacteria. The number of papillae provides a measure of the frequency of transposition. DSB induction at any of the sites chosen specifically stimulated TnsABC+E transposition (Figure 1A and Table 3). As found previously, a background level of Lac+ papillae always occurs in the assay that results from genetic events unrelated to Tn7 transposition (Figure 1A and Table 3) (Peters and Craig 2000).
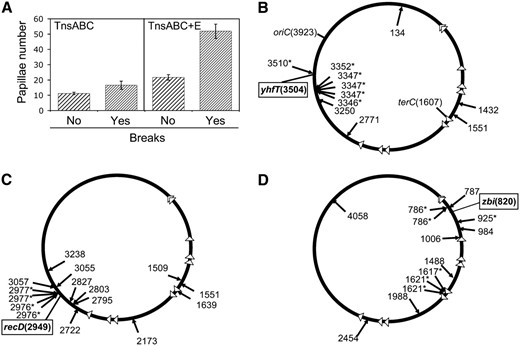
Inducing DNA DSBs specifically stimulates TnsABC+E transposition events that occur proximal to the DSB site with a preference for hotspots. Transposition was monitored with a promoter capture assay using strain JP617 derivatives on MacConkey's media (materials and methods). (A) The graph shows the frequency of transposition indicated by the number of Lac+ papillae after 72 hr at 30° when DSBs were induced with yhfT-3084∷Tn10 on MacConkey's media. Error bars indicate the standard error of the mean (n = 12) (data from other positions are shown in Table 3). Circles represent the E. coli chromosome. Individual insertions from separate experiments using strains expressing the TnsABC+E proteins where DSBs were induced are indicated with arrows. Arrows outside the circle indicate right-to-left insertions, arrows inside the circle indicate left-to-right insertions. The number indicates the position of the insertion on the E. coli genome in kilobases. Insertions that occurred into one of the hotspots found in this work are indicated with an asterisk (*). The gene where the Tn10-generated DSB was induced is indicated in a box (and its position in kilobase pairs). oriC and its position in kilobases are marked. Triangles indicate the position of the 10 ter sites in the chromosome; terC is indicated with its position in kilobases. The JP617 background strains were used to induce DSBs using a Tn10 insertion at one of three positions: yhfT (yhfT-3084∷Tn10 from CAG18456) (B), recD (recD-1901∷Tn10 from CAG12135) (C), or zbi (zbi-29∷Tn10 from CAG18493) (D). Breaks were induced with pZT383 (Yes breaks) or the strain had a control plasmid pJP135 (No breaks). Tn7 transposition functions were expressed from pJP123 (TnsABC+E) or a vector that does not allow transposition was used, pCW15 (TnsABC).
DSBs stimulate TnsE-mediated transposition
No DSBs | Yes DSBs | Fold increase TnsABC+E transposition above background | |||
|---|---|---|---|---|---|
| Tn10 insertion allele | TnsABC | TnsABC+E | TnsABC | TnsABC+E | |
| zbi-29 | 17a (1.6)b | 11 (1.1) | 22 (1.5) | 35 (7.3) | 3.2 |
| recD-1901 | 12 (1.5) | 28 (1.6) | 22 (1.0) | 71 (4.7) | 2.5 |
| b3219-6 | 7 (0.8) | 12 (1.6) | 16 (2.0) | 44 (4.0) | 3.7 |
| smg-3082 | 1 (0.2) | 1 (0.2) | 2 (0.7) | 6 (1.0) | 6.0 |
| yhfT-3084c | 11 (0.7) | 17 (2.7) | 22 (1.8) | 52 (4.7) | 3.1 |
No DSBs | Yes DSBs | Fold increase TnsABC+E transposition above background | |||
|---|---|---|---|---|---|
| Tn10 insertion allele | TnsABC | TnsABC+E | TnsABC | TnsABC+E | |
| zbi-29 | 17a (1.6)b | 11 (1.1) | 22 (1.5) | 35 (7.3) | 3.2 |
| recD-1901 | 12 (1.5) | 28 (1.6) | 22 (1.0) | 71 (4.7) | 2.5 |
| b3219-6 | 7 (0.8) | 12 (1.6) | 16 (2.0) | 44 (4.0) | 3.7 |
| smg-3082 | 1 (0.2) | 1 (0.2) | 2 (0.7) | 6 (1.0) | 6.0 |
| yhfT-3084c | 11 (0.7) | 17 (2.7) | 22 (1.8) | 52 (4.7) | 3.1 |
Average number of papillae from 12 experiments.
Standard error of the mean.
Data from the experiment shown in Figure 4A.
DSBs stimulate TnsE-mediated transposition
No DSBs | Yes DSBs | Fold increase TnsABC+E transposition above background | |||
|---|---|---|---|---|---|
| Tn10 insertion allele | TnsABC | TnsABC+E | TnsABC | TnsABC+E | |
| zbi-29 | 17a (1.6)b | 11 (1.1) | 22 (1.5) | 35 (7.3) | 3.2 |
| recD-1901 | 12 (1.5) | 28 (1.6) | 22 (1.0) | 71 (4.7) | 2.5 |
| b3219-6 | 7 (0.8) | 12 (1.6) | 16 (2.0) | 44 (4.0) | 3.7 |
| smg-3082 | 1 (0.2) | 1 (0.2) | 2 (0.7) | 6 (1.0) | 6.0 |
| yhfT-3084c | 11 (0.7) | 17 (2.7) | 22 (1.8) | 52 (4.7) | 3.1 |
No DSBs | Yes DSBs | Fold increase TnsABC+E transposition above background | |||
|---|---|---|---|---|---|
| Tn10 insertion allele | TnsABC | TnsABC+E | TnsABC | TnsABC+E | |
| zbi-29 | 17a (1.6)b | 11 (1.1) | 22 (1.5) | 35 (7.3) | 3.2 |
| recD-1901 | 12 (1.5) | 28 (1.6) | 22 (1.0) | 71 (4.7) | 2.5 |
| b3219-6 | 7 (0.8) | 12 (1.6) | 16 (2.0) | 44 (4.0) | 3.7 |
| smg-3082 | 1 (0.2) | 1 (0.2) | 2 (0.7) | 6 (1.0) | 6.0 |
| yhfT-3084c | 11 (0.7) | 17 (2.7) | 22 (1.8) | 52 (4.7) | 3.1 |
Average number of papillae from 12 experiments.
Standard error of the mean.
Data from the experiment shown in Figure 4A.
We examined the DNA sequence between each Tn10 allele used to induce DSBs and the resulting Tn7 insertion events that were stimulated by the DSB for all the data shown in this article. In each case we paid particular attention to whether the Tn7 insertions occurred on the end of the break closest to oriC or closest to terC to judge the contribution of chi sites in the chromosome (Table 4). We also analyzed the data from two Tn10 alleles used in previously published work (see Figure 6 in (Peters and Craig 2000). While noting the number of chi sites and the distance between the position of the DSB and the first Tn7 transposition events in both directions, we arbitrarily ignored events that were >300 kb from the DSB and stopped considering chi sites after 20 correctly oriented sites [chi sites are recognized only 20% of the time in vivo and in vitro (Dixon and Kowalczykowski 1993; Dixon et al. 1994; Kuzminov et al. 1994), therefore, 20 chi sites would stop 99% of the RecBCD enzymes] (Table 4). In examining the distribution of TnsE-mediated transposition events that are stimulated by the DSB, we find no obvious relationship between the frequency of chi sites and the relative distribution of insertions around the DSB (Table 4). While no chi sites were found between the induced DSB and the first insertions in some cases (i.e., srlD-3131∷Tn10), in other cases, many chi sites were found before the first insertions proximal to the DSB (Table 4). Interestingly, TnsE-mediated insertion events still occurred broadly around the DSB in the recD-1901∷Tn10 strain. The RecBCD enzyme complex is responsible for tailoring DNA double-strand breaks and loading RecA for their repair. In a recD strain background the RecBC+(D−) enzyme will not degrade from the initial DSB position (Chaudhury and Smith 1984; Amundsen et al. 1986), but will immediately load RecA for replication-mediated DSB repair (Kuzminov and Stahl 1999) (see discussion).
Transposition hotspots and chi site frequency around DSBs
Tn10 insertion allelea(position) | Total insertions examineda(% within 300 kb of the DSB) | Tn7 insertions on the oriC or terC side of the break | Chi sites before the insertion/kilobases before the insertion | ||||||
|---|---|---|---|---|---|---|---|---|---|
| First insertion | Second insertion | Third insertion | Fourth insertion | Fifth insertion | Sixth insertion | Seventh insertion | |||
| zbf-3057 (698 kb) | 7 (57) | oriC | 8/74 | >20/>300 | >20/>300 | >20/>300 | >20/>300 | >20/>300 | >20/>300 |
| terC | 6/55 | >20/227b | >20/227b | >20/>300 | >20/>300 | >20/>300 | >20/>300 | ||
| zbi-29 (820 kb) | 13 (46) | oriC | 3/33 | 3/34c | 3/34c | >20/>300 | >20/>300 | >20/>300 | >20/>300 |
| terC | 14/106b | 20/164 | >20/186 | >20/>300 | >20/>300 | >20/>300 | >20/>300 | ||
| recD-1901 (2949 kb) | 15 (73) | oriC | 8/27d | 8/27d | 8/28d | 8/28d | 19/106 | 19/106 | >20/289 |
| terC | 10/122 | 10/146 | 10/154 | 10/227e | >20/>300 | >20/>300 | >20/>300 | ||
| srlD-3131 (2826 kb) | 10 (70) | oriC | 6/37 | >20/150 | >20/>300 | >20/>300 | >20/>300 | >20/>300 | >20/>300 |
| terC | 0/10 | 0/74 | 0/104e | 0/104e | 3/143 | >20/>300 | >20/>300 | ||
| b3219-6 (3364 kb) | 14 (50) | oriC | 8/61 | 11/71f | >20/>300 | >20/>300 | >20/>300 | >20/>300 | >20/>300 |
| terC | 0/19g | 5/126 | 8/150h | 8/150h | 11/>300 | >20/>300 | >20/>300 | ||
| smg-3082 (3430 kb) | 11 (82) | oriC | 0/7f | 11/80i | >20/>300 | >20/>300 | >20/>300 | >20/>300 | >20/>300 |
| terC | 3/78j | 3/83g | 3/83g | 3/83g | 3/83g | 3/83g | 3/84g | ||
| yhfT-3084 (3504 kb) | 28 (54)k | oriC | 2/6i | 7/40 | >20/>300 | >20/>300 | >20/>300 | >20/>300 | >20/>300 |
| terC | 4/152j | 4/157g | 4/157g | 4/157g | 4/157g | 4/157g | 4/157g | ||
Tn10 insertion allelea(position) | Total insertions examineda(% within 300 kb of the DSB) | Tn7 insertions on the oriC or terC side of the break | Chi sites before the insertion/kilobases before the insertion | ||||||
|---|---|---|---|---|---|---|---|---|---|
| First insertion | Second insertion | Third insertion | Fourth insertion | Fifth insertion | Sixth insertion | Seventh insertion | |||
| zbf-3057 (698 kb) | 7 (57) | oriC | 8/74 | >20/>300 | >20/>300 | >20/>300 | >20/>300 | >20/>300 | >20/>300 |
| terC | 6/55 | >20/227b | >20/227b | >20/>300 | >20/>300 | >20/>300 | >20/>300 | ||
| zbi-29 (820 kb) | 13 (46) | oriC | 3/33 | 3/34c | 3/34c | >20/>300 | >20/>300 | >20/>300 | >20/>300 |
| terC | 14/106b | 20/164 | >20/186 | >20/>300 | >20/>300 | >20/>300 | >20/>300 | ||
| recD-1901 (2949 kb) | 15 (73) | oriC | 8/27d | 8/27d | 8/28d | 8/28d | 19/106 | 19/106 | >20/289 |
| terC | 10/122 | 10/146 | 10/154 | 10/227e | >20/>300 | >20/>300 | >20/>300 | ||
| srlD-3131 (2826 kb) | 10 (70) | oriC | 6/37 | >20/150 | >20/>300 | >20/>300 | >20/>300 | >20/>300 | >20/>300 |
| terC | 0/10 | 0/74 | 0/104e | 0/104e | 3/143 | >20/>300 | >20/>300 | ||
| b3219-6 (3364 kb) | 14 (50) | oriC | 8/61 | 11/71f | >20/>300 | >20/>300 | >20/>300 | >20/>300 | >20/>300 |
| terC | 0/19g | 5/126 | 8/150h | 8/150h | 11/>300 | >20/>300 | >20/>300 | ||
| smg-3082 (3430 kb) | 11 (82) | oriC | 0/7f | 11/80i | >20/>300 | >20/>300 | >20/>300 | >20/>300 | >20/>300 |
| terC | 3/78j | 3/83g | 3/83g | 3/83g | 3/83g | 3/83g | 3/84g | ||
| yhfT-3084 (3504 kb) | 28 (54)k | oriC | 2/6i | 7/40 | >20/>300 | >20/>300 | >20/>300 | >20/>300 | >20/>300 |
| terC | 4/152j | 4/157g | 4/157g | 4/157g | 4/157g | 4/157g | 4/157g | ||
Data for the zbf-3057 and srlD-3131 alleles were from the published insertions (Peters and Craig 2000). The remaining data were from the following: zbi-29 (Figure 1D), recD-1901 (Figure 1C), b3219-6 (Figure 4C), smg-3082 (Figure 4B), and yhfT-3084 (Figures 1B and 4A).
Hotspot at 925 kb (serW), 3 insertions 346 bp apart.
Hotspot at 786 kb (aroG), 2 insertions 69 bp apart.
Hotspot at 2976–2977 kb (lysR), 4 insertions 842 bp apart.
Hotspot at 2722–2723 kb (yfiM and kgtP), 3 insertions 333 bp apart.
Hotspot at 3436–3437 (mscL-rplQ), 2 insertions 1074 bp apart.
Hotspot at 3346–3347 kb (mtgA and yrbL), 17 insertion 820 bp apart.
Hotspot at 3214 kb (yqiJ), 3 insertions 282 bp apart (includes insertion induced by the yhfT-3084∷Tn10).
Hotspot at 3510 kb (yhfZ), 2 insertions 14 bp apart.
Hotspot at 3352 kb (yhcC-gltB), 2 insertions 420 bp apart.
An additional 8 insertions were within 300 kb because of the larger number of samples, but the percentage within 300 kb remained the same.
Transposition hotspots and chi site frequency around DSBs
Tn10 insertion allelea(position) | Total insertions examineda(% within 300 kb of the DSB) | Tn7 insertions on the oriC or terC side of the break | Chi sites before the insertion/kilobases before the insertion | ||||||
|---|---|---|---|---|---|---|---|---|---|
| First insertion | Second insertion | Third insertion | Fourth insertion | Fifth insertion | Sixth insertion | Seventh insertion | |||
| zbf-3057 (698 kb) | 7 (57) | oriC | 8/74 | >20/>300 | >20/>300 | >20/>300 | >20/>300 | >20/>300 | >20/>300 |
| terC | 6/55 | >20/227b | >20/227b | >20/>300 | >20/>300 | >20/>300 | >20/>300 | ||
| zbi-29 (820 kb) | 13 (46) | oriC | 3/33 | 3/34c | 3/34c | >20/>300 | >20/>300 | >20/>300 | >20/>300 |
| terC | 14/106b | 20/164 | >20/186 | >20/>300 | >20/>300 | >20/>300 | >20/>300 | ||
| recD-1901 (2949 kb) | 15 (73) | oriC | 8/27d | 8/27d | 8/28d | 8/28d | 19/106 | 19/106 | >20/289 |
| terC | 10/122 | 10/146 | 10/154 | 10/227e | >20/>300 | >20/>300 | >20/>300 | ||
| srlD-3131 (2826 kb) | 10 (70) | oriC | 6/37 | >20/150 | >20/>300 | >20/>300 | >20/>300 | >20/>300 | >20/>300 |
| terC | 0/10 | 0/74 | 0/104e | 0/104e | 3/143 | >20/>300 | >20/>300 | ||
| b3219-6 (3364 kb) | 14 (50) | oriC | 8/61 | 11/71f | >20/>300 | >20/>300 | >20/>300 | >20/>300 | >20/>300 |
| terC | 0/19g | 5/126 | 8/150h | 8/150h | 11/>300 | >20/>300 | >20/>300 | ||
| smg-3082 (3430 kb) | 11 (82) | oriC | 0/7f | 11/80i | >20/>300 | >20/>300 | >20/>300 | >20/>300 | >20/>300 |
| terC | 3/78j | 3/83g | 3/83g | 3/83g | 3/83g | 3/83g | 3/84g | ||
| yhfT-3084 (3504 kb) | 28 (54)k | oriC | 2/6i | 7/40 | >20/>300 | >20/>300 | >20/>300 | >20/>300 | >20/>300 |
| terC | 4/152j | 4/157g | 4/157g | 4/157g | 4/157g | 4/157g | 4/157g | ||
Tn10 insertion allelea(position) | Total insertions examineda(% within 300 kb of the DSB) | Tn7 insertions on the oriC or terC side of the break | Chi sites before the insertion/kilobases before the insertion | ||||||
|---|---|---|---|---|---|---|---|---|---|
| First insertion | Second insertion | Third insertion | Fourth insertion | Fifth insertion | Sixth insertion | Seventh insertion | |||
| zbf-3057 (698 kb) | 7 (57) | oriC | 8/74 | >20/>300 | >20/>300 | >20/>300 | >20/>300 | >20/>300 | >20/>300 |
| terC | 6/55 | >20/227b | >20/227b | >20/>300 | >20/>300 | >20/>300 | >20/>300 | ||
| zbi-29 (820 kb) | 13 (46) | oriC | 3/33 | 3/34c | 3/34c | >20/>300 | >20/>300 | >20/>300 | >20/>300 |
| terC | 14/106b | 20/164 | >20/186 | >20/>300 | >20/>300 | >20/>300 | >20/>300 | ||
| recD-1901 (2949 kb) | 15 (73) | oriC | 8/27d | 8/27d | 8/28d | 8/28d | 19/106 | 19/106 | >20/289 |
| terC | 10/122 | 10/146 | 10/154 | 10/227e | >20/>300 | >20/>300 | >20/>300 | ||
| srlD-3131 (2826 kb) | 10 (70) | oriC | 6/37 | >20/150 | >20/>300 | >20/>300 | >20/>300 | >20/>300 | >20/>300 |
| terC | 0/10 | 0/74 | 0/104e | 0/104e | 3/143 | >20/>300 | >20/>300 | ||
| b3219-6 (3364 kb) | 14 (50) | oriC | 8/61 | 11/71f | >20/>300 | >20/>300 | >20/>300 | >20/>300 | >20/>300 |
| terC | 0/19g | 5/126 | 8/150h | 8/150h | 11/>300 | >20/>300 | >20/>300 | ||
| smg-3082 (3430 kb) | 11 (82) | oriC | 0/7f | 11/80i | >20/>300 | >20/>300 | >20/>300 | >20/>300 | >20/>300 |
| terC | 3/78j | 3/83g | 3/83g | 3/83g | 3/83g | 3/83g | 3/84g | ||
| yhfT-3084 (3504 kb) | 28 (54)k | oriC | 2/6i | 7/40 | >20/>300 | >20/>300 | >20/>300 | >20/>300 | >20/>300 |
| terC | 4/152j | 4/157g | 4/157g | 4/157g | 4/157g | 4/157g | 4/157g | ||
Data for the zbf-3057 and srlD-3131 alleles were from the published insertions (Peters and Craig 2000). The remaining data were from the following: zbi-29 (Figure 1D), recD-1901 (Figure 1C), b3219-6 (Figure 4C), smg-3082 (Figure 4B), and yhfT-3084 (Figures 1B and 4A).
Hotspot at 925 kb (serW), 3 insertions 346 bp apart.
Hotspot at 786 kb (aroG), 2 insertions 69 bp apart.
Hotspot at 2976–2977 kb (lysR), 4 insertions 842 bp apart.
Hotspot at 2722–2723 kb (yfiM and kgtP), 3 insertions 333 bp apart.
Hotspot at 3436–3437 (mscL-rplQ), 2 insertions 1074 bp apart.
Hotspot at 3346–3347 kb (mtgA and yrbL), 17 insertion 820 bp apart.
Hotspot at 3214 kb (yqiJ), 3 insertions 282 bp apart (includes insertion induced by the yhfT-3084∷Tn10).
Hotspot at 3510 kb (yhfZ), 2 insertions 14 bp apart.
Hotspot at 3352 kb (yhcC-gltB), 2 insertions 420 bp apart.
An additional 8 insertions were within 300 kb because of the larger number of samples, but the percentage within 300 kb remained the same.
We tested if a specific interaction between TnsE and the RecBCD complex was required for DSBs to stimulate transposition. We took advantage of the previous finding that the RecFOR proteins can load RecA at DSBs in certain suppressor backgrounds. A RecFOR-dependent DSB repair pathway that is independent of RecBCD can be activated in sbcB sbcC sbcD strains (Clark and Sandler 1994). We determined if inducing DSBs with the srlD3131∷Tn10 would stimulate TnsE-mediated transposition in a ΔrecC background that also contained the ΔsbcB ΔsbcC ΔsbcD alleles (Peters and Craig 2000) (materials and methods). In these experiments we monitored Tn7 transposition on LB lactose X-gal plates given the observation that recC strains are sensitive to bile salts found in MacConkey's media (Prieto et al. 2006). As expected, we found that the ΔrecC allele causes the strain to be sensitive to UV light and mitomycin C, but UV and mitomycin C resistance is reestablished in the ΔrecC ΔsbcB ΔsbcC ΔsbcD strain (data not shown). We found that TnsE-mediated transposition increased significantly (P < 0.05 from a t-test, two tailed, unequal variance) when DSBs were induced in the wild-type or ΔrecC ΔsbcB ΔsbcC ΔsbcD strains, but no significant change was found in the ΔrecC background (Figure 2). Tn7 transposition in the ΔrecC ΔsbcB ΔsbcC ΔsbcD strain was not stimulated to the same extent as was found in the rec+ sbc+ background. This result may be explained by a lower efficiency of DSB repair in the ΔrecC ΔsbcB ΔsbcC ΔsbcD strain as compared to the wild-type strain. This effect is likely compounded in the strains with the TnsABC+E proteins because two DSBs are actually formed, one by the Tn10 and a second DSB at the original chromosomal position of the Tn7 element. These results indicate that the ability of DSBs to stimulate TnsE-mediated transposition is not dependent on an interaction with the RecBCD proteins nor a specific DNA structure made with this enzyme complex.
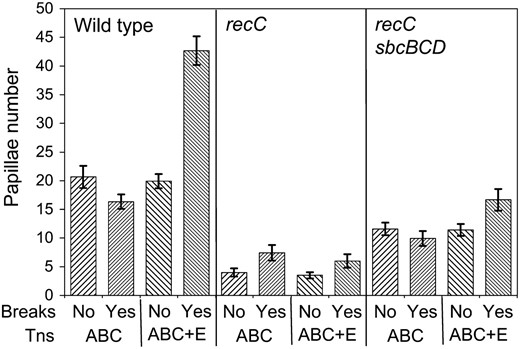
Double-strand DNA breaks stimulate TnsE-mediated transposition in the absence of RecBCD. Transposition was monitored with a promoter capture assay using strain JP617 derivatives on LB media with lactose and X-gal when DSBs were induced at srlD3131∷Tn10 (materials and methods) (Peters and Craig 2000). Breaks were induced with pZT383 (Yes breaks) or the strain had a control plasmid pJP135 (No breaks). Tn7 transposition functions were expressed from pJP123 (TnsABC+E) or a vector that does not allow transposition was used, pCW15 (TnsABC).The graph shows the frequency of transposition indicated by the number of Lac+ papillae after 120 hr at 30° from 12 independent experiments. Error bars indicate the standard error of the mean.
Induced DSBs activate transposition hotspots in the chromosome:
The analysis of multiple DSB positions revealed an interesting finding. TnsE-mediated transposition events seemed to occur at hotspots in the chromosome that appeared to be activated by individual DSBs (Figure 1, B–D). We use the term hotspot to describe when multiple insertions occur within a region of < ∼1 kb of one another in the 4.6 Mbp E. coli chromosome. However, it is important to note that we never found an example where the same insertion site was used (i.e., these are small regional hotspots for insertion). As shown below, these regional hotspots are not explained by a selection bias for highly expressed genes. One of the clearest examples of a hotspot that appeared to be stimulated as a response to a DSB occurred when a break was induced at 3504 kb on the chromosome with yhfT-3084∷Tn10 (Figures 1B and 3A). Thirty-six percent (4/11) of the TnsE-mediated transposition events that were stimulated by the break occurred within a small ∼500-bp patch of sequence that was almost 160 kb away from the yhfT-3084∷Tn10 (Figures 1B and 3A). In another example, 27% (4/15) of the transposition events that were stimulated by inducing breaks at 2949 kb on the chromosome with recD-1901∷Tn10 occurred in a ∼800-bp region that was >27 kb away from the DSB (Figure 1C). It is of additional interest to consider the incidence and distribution of transposition hotspots in the recD-1901∷Tn10 strain (see discussion). All positions where DSBs were induced seemed to activate one or more transposition hotspots (Figure 1). This was also found to be true when we reexamined the TnsE-mediated transposition events collected in an earlier work (Peters and Craig 2000) (Table 4). By comparing the Tn7 insertions that resulted from inducing DSBs at 698 kb (zbf-3057∷Tn10) (Peters and Craig 2000) and 820 kb (zbi-29∷Tn10) (Figure 1D) we found something especially intriguing; DSBs at both of these positions caused a transposition hotspot around the gene serW (925 kb), even though this gene was 227 kb and 106 kb from the position of the DSBs, respectively (Table 4). This observation suggested that the hotspots were at fixed positions in the chromosome and could be activated by DSBs at many positions.
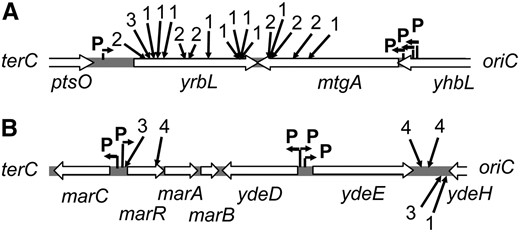
DSBs stimulate TnsABC+E transposition at multiple different positions in the yrbL-mtgA and marR/ydeE-ydeH hotspots. Solid arrows indicate the relative position of TnsE-mediated insertion events, filled arrows above the line indicate left-to-right insertions, filled arrows below the line indicate right-to-left insertions. (A) The yrbL-mtgA hotspot (3346–3347 kb). Insertions were from Figures 1B and 4, A–C. (B) The marR/ydeE-ydeH hotspots (1617/1620–1621). Insertions were from Figure 1D and 4, A –C. The number indicates the Tn10 allele that was used to induce DSBs in the strain; yhfT-3084∷Tn10 (1), smg-3082∷Tn10 (2), and b3219-6∷Tn10 (3), and zbi-29∷Tn10 (4). Open arrows indicate the orientation of the open reading frames. Predicted sigma 70 promoters are shown with bent arrows and indicated (P) from PEC (http://www.shigen.nig.ac.jp/ecoli/pec/). The oriC and terC proximal sides of the region are shown.
Hotspots are fixed in the chromosome and are activated only by DSBs in the same general region of the chromosome:
To confirm the idea that the hotspots were at fixed positions, we examined the effect of inducing DSBs at different distances (157, 83, and 19 kb) from the most active transposition hotspot, yrbL-mtgA (3346-3347) (Figures 3A and 4). We reconstructed the strain where DSBs were induced using the yhfT-3084∷Tn10 at 3504 kb in the chromosome and retested this strain to rule out any unknown factors related to the original construction (Figure 4A). We found that 35% (6/17) of the insertions again occurred in the yrbL-mtgA hotspot which was 157 kb away (Figures 3A and 4A). When we induced DSBs in smg (smg-3082∷Tn10), a position that was 83 kb from the yrbL-mtgA hotspot, we found that 55% (6/11) of the insertions occurred in the yrbL-mtgA hotspot (Figures 3A and 4B). When we induced DSBs in b3219 (b3219-6∷Tn10), a position 19 kb from the yrbL-mtgA hotspot, there seemed to be a drop in the usage of the yrbL-mtgA hotspot; one in 13 insertions occurred in the hotspot (Figures 3A and 4C). However, insertions could now also be found at a more terC proximal hotspot region at 3214 kb (yqjI gene) on the chromosome which was 150 kb away from the DSB (Figure 4C). This yqjI hotspot was also utilized when DSBs were induced at yhtT-3084∷Tn10, which is 290 kb away (Figure 4A). Closer examination of the results indicates that DSBs at multiple positions also induce an oriC proximal hotspot at 3510 kb on the chromosome (the yhfZ gene) (Figures 1B and 4B; Table 4). Taken together, these results demonstrate that there are fixed potential transposition hotspots in the chromosome that can be activated by DSBs over a region of the chromosome that can be hundreds of kilobases from the hotspot (Figures 1 and 4).
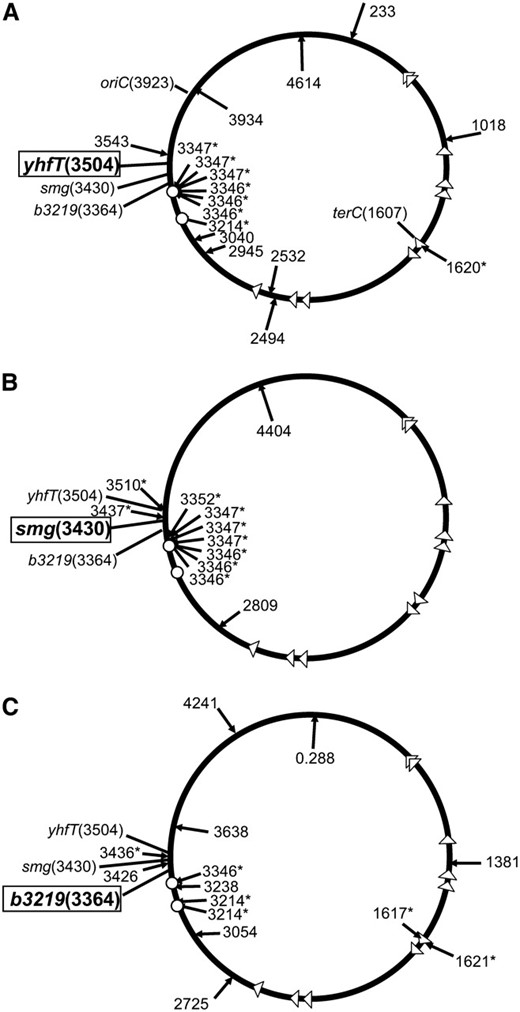
Inducing DSBs specifically stimulates TnsABC+E transposition events that occur proximal to the DSB site with a preference for fixed regional hotspots. Transposition was monitored with a promoter capture assay using strain JP617 derivatives on MacConkey's media (materials and methods). The position of transposition events from strains with the TnsABC+E proteins where DSBs were induced are shown. Designations are as in Figure 1. The three relevant loci, yhfT, smg, and b3219, are indicated (and the position shown in kilobases). The position where the DSB was induced is indicated in boldface and boxed. The JP617 background strains were used to induce DSBs using a Tn10 insertion at one of three positions: yhfT (yhfT-3084∷Tn10 from CAG18456) (A), smg (smg-3082∷Tn10 from CAG12071) (B), or b3219 (b3219-6∷Tn10 from CAG12153) (C). Breaks were induced with pZT383 (Yes breaks) or the strain had a control plasmid pJP135 (No breaks). Tn7 transposition functions were expressed from pJP123 (TnsABC+E) or a vector that does not allow transposition was used, pCW15 (TnsABC).The locations of two relevant hotspots are indicated with small circles, yqiJ (3214 kb) and yrbL-mtgA (3346–3347 kb).
It appears that closer hotspots are generally used preferentially; for example, when DSBs were induced with yhfT-3084∷Tn10, 6 of 16 insertions occurred at the hotspot that was 157 kb away, but only 1 of the 16 insertions occurred at the hotspot 290 kb away (Figure 4A). However, examining the results found when DSBs were induced at b3219-6∷Tn10 suggests that inducing the DSB at a position too close might diminish the attraction to the hotspot; only one of the 14 insertions occurred into a hotspot 19 kb away, but insertions (2/14) could be detected in a hotspot 150 kb away (Figure 4C).
Multiple forms of DNA damage induce TnsABC+E transposition:
Mitomycin C produces covalent interstrand crosslinks in DNA and can lead to the formation of DSBs. We find that mitomycin C stimulates TnsABC+E transposition, resulting in an average of an additional 103 Lac+ papillae per lawn of cells when compared to growth without mitomycin C (Figure 5A). There was only a small (21 Lac+ papillae per experiment) increase in Lac+ events in the control strain that was likely due to repair-related mutagenesis. We also tested the effect of phleomycin, which causes a high frequency of DSBs (Huang et al. 1981; Moore 1989), on Tn7 transposition. TnsE-mediated transposition was stimulated with very low levels of phleomycin that resulted in essentially no increase in the level of background Lac+ events (Figure 5B). We found a significant increase in TnsABC+E transposition at lower concentrations of phleomycin (2.5 ng/ml). Higher concentrations of phleomycin also stimulated transposition, but cell viability started to be affected (50–100 ng/ml) (data not shown).
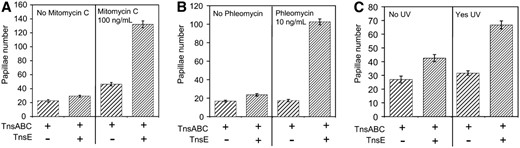
Mitomycin C, phleomycin, and UV exposure stimulate TnsABC+E transposition. Tn7 functions were expressed from pQS101 (TnsABC) or pQS103 (TnsABC+ TnsE). Transposition was monitored with a promoter capture assay using strain JP617 on MacConkey's media (materials and methods). (A) The frequency of transposition is indicated on the y-axis as the number of Lac+ papillae after 72 hr at 30° on MacConkey's plates with and without mitomycin C. (B) The frequency of transposition is indicated on the y-axis as the number of Lac+ papillae after 72 hr at 30° on MacConkey's plates with and without phleomycin. (C) After 18 hr of incubation at 30° one set of plates was exposed to 10 mJ of UV light (Yes UV) and the other was left unexposed as a control (No UV). The frequency of transposition is indicated on the y-axis as the number of Lac+ papillae after 72 hr at 30° post-UV light on MacConkey's plates. Error bars indicate the standard error of the mean (n = 12).
TnsABC+E transposition was also stimulated when the cells received a brief exposure of short-wave UV light (data not shown) (materials and methods). Exposure to UV light leads to pyrimidine dimers resulting in gaps in nascent daughter strands that can lead to DSBs (Rupp and Howard-Flanders 1968; Kuzminov 2001). UV light exposure was still capable of stimulating transposition even if the exposure did not happen until the lawn of cells was fully grown on the plate, 18 hr into the assay (Figure 5C). Similar to the results found with mitomycin C, some Lac+ cells could be attributed to UV-induced mutagenesis, however, this does not account for the larger increase found with TnsABC+E. For example, for the data shown in Figure 5C, an average of 5 additional Lac+ papillae were found in cells without transposition (i.e., compare TnsABC-only strains with and without UV irradiation), as compared to an average of 24 additional Lac+ papillae where TnsE-mediated transposition occurs (i.e., compare TnsABC+E strains with and without UV irradiation). These results suggest that multiple forms of DNA damage are capable of stimulating TnsABC+E transposition and that the process is not specific to DSBs induced by Tn10 excision.
To investigate further the role of DNA damage in stimulating TnsE-mediated transposition we sequenced randomly chosen isolates across 24 experiments from strains expressing TnsABC+E that were exposed to UV irradiation (Figure 6) (materials and methods). TnsE-mediated transposition events occurred at many different regions in the chromosome. Only one of the insertions occurred into a hotspot identified by the direct induction of DSBs (yrbL-mtgA, 3346 kb). Interestingly, we found that insertions did seem to occur at two new hotspots, one in zntA (3605–3606 kb) and another between cspA and an IS150 element (3718 kb) (Figure 6).
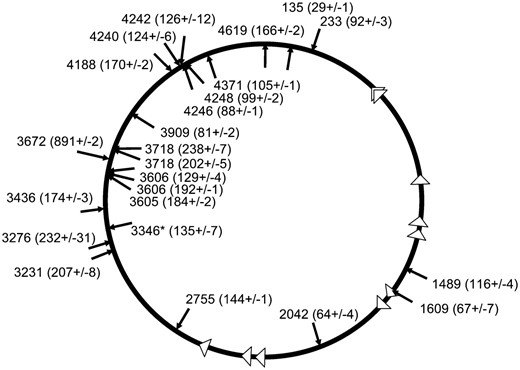
TnsE-mediated transposition events were stimulated to occur at many locations in the chromosome after exposure to UV irradiation. Twenty-four randomly chosen transposition events were mapped and sequences from the Lac+ papillae found in Figure 5B and a replicate of this experiment. Designations are as in Figure 1. Next to each individual insertion the average β-galactosidase activity of the resulting fusion is shown (n = 3) with the standard deviation of the mean (See text for details).
Selection bias does not explain the hotspots in the chromosome:
The pattern of TnsE-mediated transposition cannot be simply explained by the activity level of certain Tn7-mediated Lac+ fusions. This would not explain why the profile of insertions changed when DNA breaks were induced at different positions in the chromosome or with UV light (Figures 1, 4, and 6). However, we were interested in knowing if the activity level of the individual Tn7-mediated Lac+ fusions found with the papillation assay could explain the hotspots. We determined the β-galactosidase activity of 47 isolates and a negative control, each in triplicate (Figures 6 and supplemental S1). These isolates included the samples shown in Figure 6 and other randomly selected isolates from new individual papillation assays. We found that the average β-galactosidase activity level was 261 Miller units, but varied widely from 24 to 1075 with a standard deviation of 262 (supplemental Figure S1). The Tn7-directed Lac+ fusions found in the most prominent hotspot 3346 (yrbL-mtgA) in Figure 6 were below the average β-galactosidase activity level found among the isolates (Figure 6), as were the insertions that seemed to indicate the two new hotspots at zntA and cspA-IS50 found with UV irradiation (Figure 6). These data support the idea that hotspots do not result from a higher or lower transcription activity of certain target genes. To further ensure that hotspots are not explained by the activity of certain genes we determined the position of the insertions that had a β-galactosidase activity level that was greater than one standard deviation above the average (supplemental Figure S1). We found that none of these insertions was located in regional hotspots. Four of these insertions were adjacent to or within genes encoding parts of the ribosome (rrlC, yeaD-rrsH, purH-rrsA, and rpmG). The other four insertions were either between or within unrelated genes (yhjD-yhjE, mscL-yhdM, farR, and thrC). These results indicate that target gene expression level does not explain the hotspots.
TnsE-mediated transposition does not specifically require any one of the known replication restart pathways:
The repair of DNA double-strand breaks and the repair of DNA damage caused by UV irradiation, mitomycin C, and phleomycin require a mechanism to restart DNA replication forks. Given the results described above, it was possible that TnsE required one of the proteins in the replication restart pathway to recognize target DNAs. The attraction of TnsE-mediated transposition to the terminus region could also be explained by an interaction between this pathway and the replication restart apparatus (Bidnenko et al. 2006). Replication fork restart can occur through a number of genetically distinct pathways that likely help address the various ways a DNA replication fork can stall or collapse (Heller and Marians 2006). We determined if any of the multiple pathways for replication fork restart was specifically required for TnsE-mediated transposition during normal growth.
PriA is a 3′–5′ helicase that is required for the major replication fork restart pathways in E. coli (Sandler and Marians 2000). We monitored transposition in a ΔpriA strain and found that this component of the restart apparatus was not required for TnsE-mediated transposition (Figure 7). Surprisingly, we found that TnsE-mediated transposition was actually stimulated in ΔpriA strains. This effect was specific to TnsE-mediated transposition because no change was found in TnsABC* transposition; the TnsABC* core machinery has a mutant TnsCA225V that does not require TnsE for targeting or activation (Stellwagen and Craig 1997b). It is possible that the stimulatory effect found in the ΔpriA background might be caused by TnsE favoring a structure or complex that is processed more slowly in a ΔpriA background. To rule out any requirement for PriA, we utilized a gain-of-function mutation in dnaC, called dnaC809, that allows replication restart in a ΔpriA background. DnaC loads the replicative helicase, DnaB, as part of initiating all DNA replication forks (Kornberg and Baker 1992). The dnaC809 allele bypasses the need for multiple other replication fork restart proteins like PriB and DnaT, in vivo and in vitro (Sandler et al. 1996; Liu et al. 1999). TnsABC+E transposition was unaffected by the dnaC809 allele (Figure 7). The results found with the ΔpriA and dnaC809 strains indicate that the replication fork restart pathway that requires the PriA, PriB, and DnaT proteins is not required for TnsE transposition (i.e., a specific interaction between TnsE and one of these proteins or a specific structure made by these proteins is not required for transposition in vivo).
![The PriA protein is not required for TnsABC+E transposition. Tn7 functions were expressed from pQS100 (TnsABC), pQS107 (TnsABC*), or pQS102 (TnsABC+ TnsE). The TnsABC* mutant core machinery that allows untargeted transposition is included as a positive control to rule out effects that are not associated with TnsE targeted transposition (). Transposition was monitored with a promoter capture assay using strains JP617 [(wild type (WT)], JP1588 (priA), AP44 (dnaC809), and AP48 (dnaC809 priA) (materials and methods). The frequency of transposition is indicated on the y-axis as the number of Lac+ papillae after 72 hr at 30° on MacConkey's media. Error bars indicate the standard error of the mean (n = 12).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/genetics/179/3/10.1534_genetics.108.088161/5/m_1237fig7.jpeg?Expires=1716901052&Signature=rG4WLOayFDl2ln3k8OBQf0JKmvrRmH7qoaVDPYbNghdc76d0a4ywCIfxmos06ixJgwNnz2AW3XaB7aMARePAndtaDYgAUSKpHZWXOIlfNJ-t8brG2jwZkF9KZxVdU0cDRinGdNNZwovLrrzSMXSDg7yJBC1CsUH7U~Itgc66bnMBrzQ~vKbCX6tJhETkSxuzL1gn0W6hN3adrWcUtM~7sMAzZCHtEaEpX5ecoy4yPZnQudS67UX6JufCwASJP0QWXzX4OqhdN7TWFZgwNhu8Wk-uS3CZWdb9zBYoC96BlobrKW16X-mYFaUKG7-Yoh6l2bnivxILc6FafxgpUJli2g__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
The PriA protein is not required for TnsABC+E transposition. Tn7 functions were expressed from pQS100 (TnsABC), pQS107 (TnsABC*), or pQS102 (TnsABC+ TnsE). The TnsABC* mutant core machinery that allows untargeted transposition is included as a positive control to rule out effects that are not associated with TnsE targeted transposition ( ). Transposition was monitored with a promoter capture assay using strains JP617 [(wild type (WT)], JP1588 (priA), AP44 (dnaC809), and AP48 (dnaC809 priA) (materials and methods). The frequency of transposition is indicated on the y-axis as the number of Lac+ papillae after 72 hr at 30° on MacConkey's media. Error bars indicate the standard error of the mean (n = 12).
). Transposition was monitored with a promoter capture assay using strains JP617 [(wild type (WT)], JP1588 (priA), AP44 (dnaC809), and AP48 (dnaC809 priA) (materials and methods). The frequency of transposition is indicated on the y-axis as the number of Lac+ papillae after 72 hr at 30° on MacConkey's media. Error bars indicate the standard error of the mean (n = 12).
PriA is essential for the major replication fork restart pathways involving PriB and a second pathway involving PriC (Sandler 2005). However, PriC can also restart DNA replication forks in a ΔpriA background through the use of a second 3′–5′ DNA helicase, called Rep. We monitored transposition in a ΔpriC strain to determine if the PriC-Rep pathway of replication fork restart was required for TnsE-mediated transposition (Figure 8). We find that TnsABC+E transposition is unaffected by the priC allele when compared with the TnsABC* control strain. Taken together, the observation that PriA and PriC are not required for transposition indicates that no protein that is required for these pathways is an essential host factor for TnsE-mediated transposition. This includes the known pathways that require PriB, DnaT, and Rep. It is unknown why TnsABC+E transposition is stimulated in ΔpriA strains, but this could indicate that slowed DNA repair allows a structure or complex that is preferred by TnsE to linger in the cell. While a priA suppressor mutation is not sufficient to stimulate TnsE-mediated transposition (Figure 7), it remains possible that suppressor mutations in dnaC can facilitate a priA-independent process preferred by TnsE-mediated transposition.
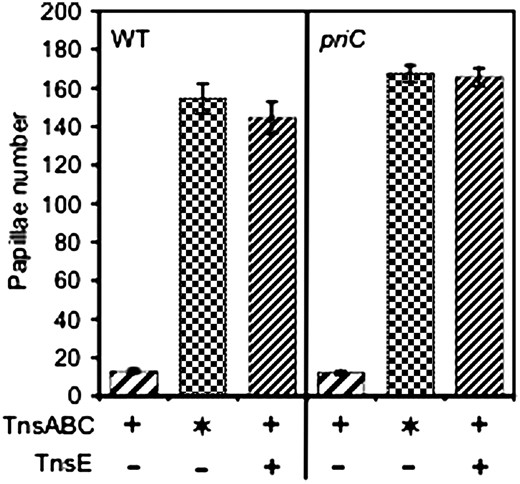
The PriC protein is not required for TnsABC+E transposition. Tn7 functions were expressed from pQS100 (TnsABC), pQS102 (TnsABC+ TnsE), or pQS107 (TnsABC*). The TnsABC* mutant core machinery that allows untargeted transposition is included as a positive control to rule out effects that are not associated with TnsE targeted transposition (). Transposition was monitored with a promoter capture assay using strains AP457 (WT) and AP523 (priC) (materials and methods). The frequency of transposition is indicated on the y-axis as the number of Lac+ papillae after 72 hr at 30° on MacConkey's media. Error bars indicate the standard error of the mean (n = 12).
Induction of the SOS response does not stimulate TnsE-mediated transposition:
The many types of DNA damage that stimulate TnsE-mediated transposition also stimulate the SOS response, thereby leaving the possibility that induction of the SOS response might actually be responsible for the increase in TnsE-mediated transposition. To test this idea we monitored transposition in a strain background that did not have the LexA protein that is responsible for repressing the genes otherwise expressed in the SOS response (materials and methods). To accurately gauge the effect of the SOS response we also included a sulA allele; induction of SulA during the SOS response results in the formation of cell filaments. We find that constitutive SOS induction in the lexA backgrounds does not stimulate TnsE-mediated transposition (Figure 9). Instead we actually find a significant drop in all transposition in the sulA lexA background, which may be explained by one or more of the genes in the SOS regulon effecting cell viability.
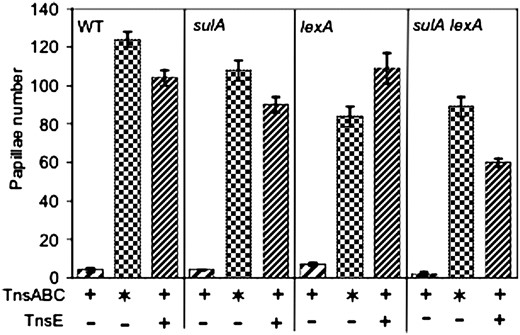
Induction of the SOS response does not stimulate TnsABC+E transposition. Tn7 functions were expressed from pQS100 (TnsABC), pQS102 (TnsABC+ TnsE), or pQS107 (TnsABC*). The TnsABC* mutant core machinery that allows untargeted transposition is included as a positive control to rule out effects that are not associated with TnsE targeted transposition (). Transposition was monitored with a promoter capture assay using strains AP457 (WT), AP767 (sulA), AP964 (lexA), and AP965 (sulA lexA) (materials and methods). The frequency of transposition is indicated on the y-axis as the number of Lac+ papillae after 72 hr at 30° on MacConkey's media. Error bars indicate the standard error of the mean (n = 12).
DISCUSSION
When DSBs are induced at specific positions in the E. coli chromosome, TnsE-mediated transposition is stimulated to occur preferentially at one or more discrete hotspots (Figures 1, 3, and 4). We show that these hotspots are fixed in the chromosome and are activated only when the DSB is induced in the same region of the chromosome (Figure 4; Table 4). We find that multiple forms of DNA damage stimulate TnsABC+E transposition (Figures 5 and 6), however, this is not explained simply by induction of the SOS response (Figure 9). Although replication fork restart is required after DNA damage, no single pathway of replication fork restart nor RecBCD is an essential target for TnsE-meditated transposition (Figures 2, 7, and 8). Our results are consistent with the idea that the replication-mediated repair of DSBs activates regional loci in the chromosome that can be targeted by TnsABC+E transposition.
Replication involved in repair may be recognized by TnsABC+E transposition:
Given that TnsABC+E transposition recognizes an aspect of DNA replication in conjugal plasmids and the chromosome (Peters and Craig 2001a), it seems reasonable to speculate that replication involved in DNA repair could also be a target for TnsABC+E transposition. This idea is supported by the distribution of TnsE-mediated insertions that are stimulated by DSBs in three ways: First, in almost every case, the left-to-right orientation of the ends of the insertions at hotspots are consistent with an orientation predicted if the replication fork started at the DSB (Peters and Craig 2001a). For example, transposition events in hotspots at aroG (786 kb), lysR (2976–2977 kb), mscL-rplQ (3436–3437), and yhfZ (3510 kb) that were all on the oriC proximal side of the induced DSB were in the opposite orientation of the insertions in the terC proximal hotspots at serW (925 kb), yqiJ (3214 kb), yrbL-mtgA (3346–3347 kb), and yhcC-gltB (3352 kb) (Figures 1 and 4) (Peters and Craig 2000). Second, most of the hotspots activated by induced DSBs are proximal to the terminus and not the origin of DNA replication (Figures 1 and 4). The distribution of actively oriented chi sites makes it much more likely that replication forks will be shunted toward the terminus (Kuzminov 1999; Kowalczykowski 2000). The most obvious hotspot that was on the oriC proximal side of the DSB was found in the recD-1901∷Tn10 strain, a strain where chi sites will not be recognized by the RecBC+(D−) enzyme thereby allowing replication forks to progress in either direction from the DSB (Chaudhury and Smith 1984; Amundsen et al. 1986). Third, the one hotspot that appeared to be stimulated by DSBs at the farthest distributed locations (zbi-29∷Tn10, b3219-6∷Tn10, or yhfT-3084∷Tn10) is only 10 kb from terC (Figures 1D, 3B, 4, A and C). Given that replication forks initiated at DSBs throughout the chromosome would be directed to terC, this result is also consistent with the idea that hotspots are activated by DNA replication from DNA repair. DSBs are also associated with UV irradiation, exposure to mitomycin C and phleomycin, and could provide a unifying target across these agents of DNA damage for TnsABC+E transposition (Figure 5). It is especially significant that phleomycin had the greatest stimulatory effect on transposition given that this drug is associated with the direct formation of DSBs (Huang et al. 1981; Moore 1989).
What causes the transposition hotspots?
Even if DNA replication associated with DNA repair is being targeted by TnsE-mediated transposition it would not immediately explain the hotspots found in the chromosome. It is possible that the hotspots are positions where forks initiated for DSB repair stall in the chromosome, much in the same way that normal DNA replication appears to be a preferred target when replication terminates proximal to terC (Figure 10A). There are multiple intriguing examples where hotspots are found where replication forks might be predicted to stall. Replication forks have been found to stall at tRNA genes and other highly expressed genes when transcription complexes may collide “head-on” with DNA replication forks (Deshpande and Newlon 1996; Mirkin and Mirkin 2007). This could indicate that the serW tRNA gene acts like a hotspot because of replication fork stalling. Of some interest, the serW locus may be especially sensitive to recombination given that the serW tRNA gene is also a known position for the occurrence of pathogenicity islands (Taylor et al. 2002). The hotspot at 3436–3437 (mscL-rplQ) is at the end of a large operon that includes many highly transcribed essential genes including ribosomal subunits, the alpha subunit of RNA polymerase, and secY. This hotspot was activated only when DSBs were induced in a region predicted to send DNA replication forks in a head on collision with transcription complexes.
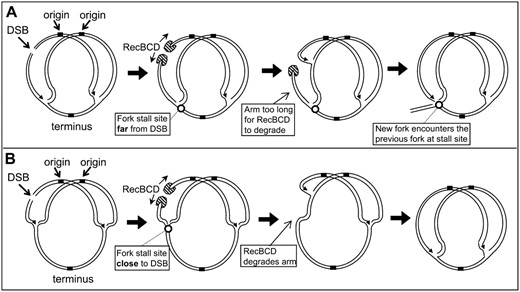
A model explaining how DNA replication forks initiated for DSB repair could lead to hotspots where DNA replication forks stall. Additionally the model explains why sites where replication forks stall that are close to a DSB may not form hotspots, but sites farther from the DSB could form hotspots. A and B show a chromosome that has been partially replicated from the single origin of DNA replication. (A) A hypothetical situation where a fork stall site is far from a DSB; (B) a stall site close to a DSB. In both cases the RecBCD enzyme should degrade at both ends of the DSB. However, the RecBCD enzyme that processes the DSB end toward the origin is quickly converted to a replication fork progressing toward the terminus because of frequent chi sequences in one orientation. The RecBCD enzyme is indicated in crosshatching and the site where replication forks stall is indicated with a small circle. See text for details.
If the hotspots are positions where replication forks can stall, this process could be compounded over time during the assay. This is because DSBs are continually induced during the multiple day assays that would presumably allow replication forks to “catch up” to previous forks at the stall site (Figure 10A). If the DSB was sufficiently close to a replication fork stall site it is possible that the RecBCD enzyme could degrade the noncontiguous arm of the stalled replication fork (i.e., at the DSB, the oriC-proximal broken end is likely used to initiate DNA replication on a sister chromosome, but the terC-proximal broken end must be degraded) (Figure 10B). This could provide an explanation for why hotspots are strongly activated by DSBs 157 and 83 kb from the hotspot, but not when DSBs are induced 19 kb from the hotspot (Figure 4); at larger distances there may be insufficient time for processing of the broken end before arrival of the fork initiated at the DSB (Figure 10B). While the RecBCD enzyme is very processive, it will dissociate after ∼30 kb. This model is also supported by the observation that significant oriC proximal hotspots are found only in the recD strain; the RecBC(D−) enzyme does not require chi sites to load RecA.
Our results with the ΔpriA allele could also support a model where hotspots result from stalled replication forks. Repair pathways involving PriA or PriC must restart DNA replication when forks stall or collapse at DNA damage (Heller and Marians 2005). While neither pathway is specifically required for TnsABC+E transposition, TnsABC+E transposition is sensitive to which pathway is used (Figures 7 and 8). Strains with the ΔpriA allele must use the PriC pathway, which may be less efficient at restarting replication on certain kinds of substrates, possibly resulting in persistently stalled replication forks somewhere in the chromosome (Heller and Marians 2005). Lagging-strand DNA replication may also become especially available in the ΔpriA background when it is uncoupled from leading-strand replication. It has been proposed that the incidence of rearrangements in the ΔpriA background could provide in vivo evidence of this uncoupling (Lovett 2005).
It is important to note that many of the hotspots occur in regions where head on collisions between replication and transcription complexes are not expected. It is possible that replication forks could stall by other mechanisms at these positions. While many of the hotspots are in genes of unknown function, there is no obvious commonality in function between hotspots found in well-studied genes (data not shown) (Table 4). We also find no obvious consensus sequence between the multiple regional hotspots (data not shown).
We find that neither the hotspots nor the differential activation of hotspots results from the expression level of genes in the chromosome. Of 47 randomly selected Tn7-based Lac+ fusions that were examined, none of the most transcriptionally active isolates resulted from insertions into hotspots (Figures 6 and supplemental S1). The insertions into hotspots generally produced below average levels of β-galactosidase activity (Figure 6). Therefore, while there are limitations to the promoter-based assay used in this study, we clearly observe a phenomenon that is specific to the TnsE-mediated pathway of Tn7 transposition following DNA damage.
Hotspots reflect a finer organization feature than any known macrodomain structure of the chromosome:
Various types of barriers have been demonstrated in the E. coli chromosome using a variety of genetic and physical assays. Experiments suggest that the chromosome is organized into six large macrodomains with largely fixed barriers based on the ability of site-specific recombination sequences to interact (Valens et al. 2004). The Tn7 hotspots do not coincide with the barriers of these macrodomains suggesting hotspots do not result from this domain structure. While the transposition hotspots usually seem to be in the same macrodomain as the induced DNA double strand break, this is likely explained by the large size of the macrodomains and that the genetic position of the macrodomain boundaries remains loosely defined. Other experiments have indicated that fluid barriers in the chromosome appear to exist that constrain the chromosome in units of ∼10 kb (Deng et al. 2004; Postow et al. 2004). However, we find transposition hotspots with a wide distribution of the insertions. It is intriguing that another transposon, IS903, has a preference for small regional hotspots (Swingle et al. 2004). However, any mechanistic connection between IS903 and Tn7 is unclear; there is no overlap with the hotspots used with these elements and the IS903 hotspots do not need to be activated by regional DSBs.
Future experiments will focus on determining the precise nature of the transposition hotspots activated by DNA damage. These studies may illuminate biological adaptations for genome stability.
Footnotes
Communicating editor: S. Gottesman
Acknowledgement
We thank Nancy Craig (Johns Hopkins Medical School, Howard Hughes Medical Institute), Susan Lovett (Brandeis University), Steve Sandler (University of Massachusetts at Amherst), Mark Sutton (University at Buffalo), and the E. coli Genetic Stock Center, Yale University for providing strains and plasmids. We thank the anonymous reviewers for suggesting useful experiments, Zaoping Li for comments on the manuscript, and David Miller and Brian Kvitko for preliminary work as rotation students. This work was funded by a grant from the National Science Foundation (MCB-0315316).
References
Amundsen, S. K., A. F. Taylor, A. M. Chaudhury and G. R. Smith,
Chaudhury, A. M., and G. R. Smith,
Datsenko, K. A., and B. L. Wanner,
Deng, S., R. A. Stein and N. P. Higgins,
Dixon, D. A., J. J. Churchill and S. C. Kowalczykowski,
Heller, R. C., and K. J. Marians,
Kuzminov, A.,
Liu, J., L. Xu, S. J. Sandler and K. J. Marians,
McKown, R. L., C. S. Waddell, L. A. Arciszewska and N. L. Craig,
Miller, J. H.,
Moore, C. W.,
Nichols, B. P., O. Shafiq and V. Meiners,
Peters, J. E.,
Peters, J. E., and N. L. Craig,
Sambrook, J., E. F. Fritsch and T. Maniatis,

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}