





Abstract
Origination of new genes is an important mechanism generating genetic novelties during the evolution of an organism. Processes of creating new genes using preexisting genes as the raw materials are well characterized, such as exon shuffling, gene duplication, retroposition, gene fusion, and fission. However, the process of how a new gene is de novo created from noncoding sequence is largely unknown. On the basis of genome comparison among yeast species, we have identified a new de novo protein-coding gene, BSC4 in Saccharomyces cerevisiae. The BSC4 gene has an open reading frame (ORF) encoding a 132-amino-acid-long peptide, while there is no homologous ORF in all the sequenced genomes of other fungal species, including its closely related species such as S. paradoxus and S. mikatae. The functional protein-coding feature of the BSC4 gene in S. cerevisiae is supported by population genetics, expression, proteomics, and synthetic lethal data. The evidence suggests that BSC4 may be involved in the DNA repair pathway during the stationary phase of S. cerevisiae and contribute to the robustness of S. cerevisiae, when shifted to a nutrient-poor environment. Because the corresponding noncoding sequences in S. paradoxus, S. mikatae, and S. bayanus also transcribe, we propose that a new de novo protein-coding gene may have evolved from a previously expressed noncoding sequence.
THE total number of different proteins in all organisms on earth is estimated to be 1010–1012 (Choi and Kim 2006). How the protein repertoire evolved to this giant diversity that underlies the evolution of the complexity of life is the basis of attracting many evolutionary biologists to the field. Discussions began 40 years ago (Perutz et al. 1965); however, with the accomplishment of complete genome sequences, we have begun to get a more comprehensive view of this complex issue. Comparative genomic study supports the notion that novel protein genes derive from preexisting genes or parts of them. For example, exon shuffling, gene duplication, retroposition, and gene fusion and fission all contribute to the origin of new genes (Long et al. 2003). But the de novo gene origination process that a whole protein-coding gene evolves from a fragment of noncoding sequence is considered seldom and receives little attention. A computational analysis of several archeal and proteobacterial species' genomes suggests that at least 240 and 320 genes, respectively, originated de novo along the branches leading to the Archea and Proteobacteria. Furthermore, there are also many de novo origination events among the species within each of the lineages (Snel et al. 2002). On the basis of the analysis, the author ranked the de novo gene origination process quantitatively the second most important process after gene loss among gene loss, de novo origination, gene duplication, gene fusion/fission, and horizontal gene transfer. This study suggests that de novo evolution not only plays an important role in generating the initial common ancestral protein repertoire but also contributes to the subsequent evolution of an organism. However, it is nearly impossible to identify the noncoding origin of the initial ancestral proteins because of long-term accumulation of mutations. Recently evolved novel protein-coding genes provide us the opportunity to investigate the de novo evolution mechanism of protein-coding genes. This methodology on gene origination has been developed in Drosophila by Long et al. (Long and Langley 1993), which has led to many advances in understanding the mechanism of new gene origination, including gene duplication, retroposition, exon shuffling, and gene fission and fusion (Nurminsky et al. 1998; Wang et al. 2002, 2004; Arguello et al. 2006; Yang et al. 2008). However, only recently did Begun et al. (2006, 2007), Levine et al. (2006), and S. T. Chen et al. (2007) find cases of whole-gene de novo origination in Drosophila melanogaster, D. yakuba, and D. erecta. The de novo genes may be functional on the basis of the RNA expression analysis, although the protein-coding potential of those de novo ORFs still needs to be proven.
Saccharomyces sensu stricto is a complex of Saccharomyces species relevant in the fermentation industry. Novel traits of those lineages, especially Saccharomyces cerevisiae, are of great interest. Studies have shown that the ancestors of Saccharomyces sensu stricto experienced a whole-genome duplication after their divergence from Kluyveromyces waltii some 100–150 million years ago (Wolfe and Shields 1997; Kellis et al. 2003). The subsequent divergence between duplicated genes and massive gene losses played an important role in the evolution of these yeast species (Dujon 2006; Wapinski et al. 2007). It would be of interest to know if de novo gene origination also occurred in yeast, in addition to Drosophila. Partial de novo gene origination has been found to contribute to the genome complexity of Saccharomyces sensu stricto (Giacomelli et al. 2007). Giacomelli et al. (2007) found several cases of partial de novo protein gene evolution through stop codon extension in four species of Saccharomyces sensu stricto (Giacomelli et al. 2007). But whether it is possible for a whole gene to evolve by the de novo way in yeast is unknown.
In this study, we identified a novel protein-coding gene BSC4 that completely evolved from a noncoding sequence in S. cerevisiae. This gene first caught our attention as a species-specific protein-coding gene in our genome comparison analysis among Saccharomyces species (H.-F. Jiang and W. Wang, unpublished data). Previously the BSC4 gene was found as one of the stop codon readthrough genes in baker's yeast by Namy et al. (2003). They found that BSC4 has a typical readthrough nucleotide context around its stop codon and its readthrough frequency is 9% when cloned into a plasmid with reporter genes (Namy et al. 2003). Although the BSC4 gene has been included in many large-scale studies, no specific study has been done with an aim to characterize it. The Saccharomyces Genome Database (SGD) (http://www.yeastgenome.org/) curates dozens of data sets, most of which were carried out using the gene chips of S. cerevisiae. In all the gene chips there are probes designed against the BSC4 gene along with other genes in S. cerevisiae. These data sets provide much expression information for BSC4 under different culture conditions. This gene was also included in the systematic gene deletion project in which ORFs of yeast genes were deleted and subsequent phenotypic analyses were carried out on those derived gene deletion strains (Saccharomyces Genome Deletion Project, http://www-sequence.stanford.edu/group/yeast_deletion_project/deletions3.html). On the basis of the panel of those gene deletion strains, whole-genome synthetic lethal analyses were carried out by Pan et al. (2006) that deleted two genes to see if that would be lethal to S. cerevisiae. Their result shows that deletion of gene DUN1 or RPN4 is lethal to S. cerevisiae if BSC4 is also deleted (Pan et al. 2006). In addition, there are multiple tandem mass-spectrometry analysis results of yeast protein samples deposited into the “Peptide Atlas” (http://www.peptideatlas.org/repository). Our analysis of these proteomics data supports the existence of the BSC4-coded peptides and our population genetic analysis suggests that the ORF of this novel protein-coding gene is under strong negative selection at the nonsynonymous sites. Our expression data show that its orthologous noncoding sequences have detectable expression at the RNA level, across the closely related species of baker's yeast. On the basis of these data, we suggest that a novel protein gene can wholly evolve from a noncoding sequence.
MATERIALS AND METHODS
Yeast strains and culture condition:
Yeast species used in this study are listed in Table 1 and were provided by Jin-Qiu Zhou at Shanghai Institutes for Biological Sciences. The strains of S. cerevisiae used in this study are listed in Table 2. YP medium (1% weight-to-volume yeast extract and 2% weight-to-volume peptone) (Sherman 1991) supplemented with 2% weight-to-volume glucose (YPD media) was used to grow these yeasts. Cultures were grown at 30° and shaken at 250–300 rpm overnight. The culture volume did not exceed 25% of the flask capacity.
Species used for Southern hybridization
Species | Strain name |
|---|---|
| Saccharomyces cerevisiae | YCM53 |
| S. paradoxus | YCM55 |
| S. mikatae | YCM61 |
| S. kudriavzevii | YCM59 |
| S. bayanus | YCM57 |
Species | Strain name |
|---|---|
| Saccharomyces cerevisiae | YCM53 |
| S. paradoxus | YCM55 |
| S. mikatae | YCM61 |
| S. kudriavzevii | YCM59 |
| S. bayanus | YCM57 |
Species used for Southern hybridization
Species | Strain name |
|---|---|
| Saccharomyces cerevisiae | YCM53 |
| S. paradoxus | YCM55 |
| S. mikatae | YCM61 |
| S. kudriavzevii | YCM59 |
| S. bayanus | YCM57 |
Species | Strain name |
|---|---|
| Saccharomyces cerevisiae | YCM53 |
| S. paradoxus | YCM55 |
| S. mikatae | YCM61 |
| S. kudriavzevii | YCM59 |
| S. bayanus | YCM57 |
Saccharomyces cerevisiae strains for population study
Strains | Description of strain source | Sequence accession no. in GenBank |
|---|---|---|
| AS2.101a | Distilled spirit yeast | EU375917 |
| AS2.1406a | Sake yeast, Japan | EU375913 |
| AS2.148a | Champagne yeast | EU375922 |
| AS2.179a | Soy sauce, Japan | EU375918 |
| AS2.2a | Beer yeast, England | EU375914 |
| AS2.2079a | Grape, China | EU375924 |
| AS2.2080a | Grape, China | EU375920 |
| AS2.3a | Beer yeast, England | EU375921 |
| AS2.7a | Whiskey yeast, United States | EU375919 |
| AS2.724a | Medicinal liquor, China | EU375923 |
| AS2.771a | Leaven, China | EU375925 |
| AS2.820a | Medicinal liquor, China | EU375912 |
| AS2.93a | Distilled spirit yeast | EU375915 |
| XH1549a | Sputum, China | EU375916 |
| BC187b | Barrel fermentation, Napa Valley, California | |
| DBVPG1373b | Soil, Netherlands | |
| DBVPG1788b | Soil, Finland | |
| DBVPG1853b | White Tecc, Ethiopia | |
| DBVPG6044b | Fermenting fruit juice, The Netherlands | |
| DBVPG6765b | Unknown | |
| L_1374b | Wine, Chile | |
| L_1528b | Wine, Chile | |
| YPS128b | Oak, Pennsylvania | |
| SK1b | Soil, United States | |
| Y55b | Wine, France | |
| YGPMb | Rotting fig, California | |
| RM11-1ac | Vineyard, California | CH408055 AAEG01000000 |
| YJM789d | Lung of an AIDS patient | AAFW02000067 |
| S288Ce | Laboratory strain | NC_001146 |
Strains | Description of strain source | Sequence accession no. in GenBank |
|---|---|---|
| AS2.101a | Distilled spirit yeast | EU375917 |
| AS2.1406a | Sake yeast, Japan | EU375913 |
| AS2.148a | Champagne yeast | EU375922 |
| AS2.179a | Soy sauce, Japan | EU375918 |
| AS2.2a | Beer yeast, England | EU375914 |
| AS2.2079a | Grape, China | EU375924 |
| AS2.2080a | Grape, China | EU375920 |
| AS2.3a | Beer yeast, England | EU375921 |
| AS2.7a | Whiskey yeast, United States | EU375919 |
| AS2.724a | Medicinal liquor, China | EU375923 |
| AS2.771a | Leaven, China | EU375925 |
| AS2.820a | Medicinal liquor, China | EU375912 |
| AS2.93a | Distilled spirit yeast | EU375915 |
| XH1549a | Sputum, China | EU375916 |
| BC187b | Barrel fermentation, Napa Valley, California | |
| DBVPG1373b | Soil, Netherlands | |
| DBVPG1788b | Soil, Finland | |
| DBVPG1853b | White Tecc, Ethiopia | |
| DBVPG6044b | Fermenting fruit juice, The Netherlands | |
| DBVPG6765b | Unknown | |
| L_1374b | Wine, Chile | |
| L_1528b | Wine, Chile | |
| YPS128b | Oak, Pennsylvania | |
| SK1b | Soil, United States | |
| Y55b | Wine, France | |
| YGPMb | Rotting fig, California | |
| RM11-1ac | Vineyard, California | CH408055 AAEG01000000 |
| YJM789d | Lung of an AIDS patient | AAFW02000067 |
| S288Ce | Laboratory strain | NC_001146 |
The genes BSC4 of these strains were sequenced by us. These strains were provided by Feng-Yan Bai (Systematic Mycology and Lichenology Laboratory, Institute of Microbiology, Chinese Academy of Sciences).
These strains were sequenced by the Saccharomyces Genome Resequencing Project at the Sanger Institute in collaboration with Ed Louis's group at the Institute of Genetics, University of Nottingham. All the sequences are downloaded from ftp://ftp.sanger.ac.uk/pub/dmc/yeast.
This strain was sequenced by the Broad Institute [Saccharomyces cerevisiae RM11-1a Sequencing Project, Broad Institute of Harvard and MIT (http://www.broad.mit.edu)].
This strain was sequenced by the Stanford Genome Technology Center (Wei et al. 2007).
The reference genome sequence (Saccharomyces cerevisiae systematic sequencing project).
Saccharomyces cerevisiae strains for population study
Strains | Description of strain source | Sequence accession no. in GenBank |
|---|---|---|
| AS2.101a | Distilled spirit yeast | EU375917 |
| AS2.1406a | Sake yeast, Japan | EU375913 |
| AS2.148a | Champagne yeast | EU375922 |
| AS2.179a | Soy sauce, Japan | EU375918 |
| AS2.2a | Beer yeast, England | EU375914 |
| AS2.2079a | Grape, China | EU375924 |
| AS2.2080a | Grape, China | EU375920 |
| AS2.3a | Beer yeast, England | EU375921 |
| AS2.7a | Whiskey yeast, United States | EU375919 |
| AS2.724a | Medicinal liquor, China | EU375923 |
| AS2.771a | Leaven, China | EU375925 |
| AS2.820a | Medicinal liquor, China | EU375912 |
| AS2.93a | Distilled spirit yeast | EU375915 |
| XH1549a | Sputum, China | EU375916 |
| BC187b | Barrel fermentation, Napa Valley, California | |
| DBVPG1373b | Soil, Netherlands | |
| DBVPG1788b | Soil, Finland | |
| DBVPG1853b | White Tecc, Ethiopia | |
| DBVPG6044b | Fermenting fruit juice, The Netherlands | |
| DBVPG6765b | Unknown | |
| L_1374b | Wine, Chile | |
| L_1528b | Wine, Chile | |
| YPS128b | Oak, Pennsylvania | |
| SK1b | Soil, United States | |
| Y55b | Wine, France | |
| YGPMb | Rotting fig, California | |
| RM11-1ac | Vineyard, California | CH408055 AAEG01000000 |
| YJM789d | Lung of an AIDS patient | AAFW02000067 |
| S288Ce | Laboratory strain | NC_001146 |
Strains | Description of strain source | Sequence accession no. in GenBank |
|---|---|---|
| AS2.101a | Distilled spirit yeast | EU375917 |
| AS2.1406a | Sake yeast, Japan | EU375913 |
| AS2.148a | Champagne yeast | EU375922 |
| AS2.179a | Soy sauce, Japan | EU375918 |
| AS2.2a | Beer yeast, England | EU375914 |
| AS2.2079a | Grape, China | EU375924 |
| AS2.2080a | Grape, China | EU375920 |
| AS2.3a | Beer yeast, England | EU375921 |
| AS2.7a | Whiskey yeast, United States | EU375919 |
| AS2.724a | Medicinal liquor, China | EU375923 |
| AS2.771a | Leaven, China | EU375925 |
| AS2.820a | Medicinal liquor, China | EU375912 |
| AS2.93a | Distilled spirit yeast | EU375915 |
| XH1549a | Sputum, China | EU375916 |
| BC187b | Barrel fermentation, Napa Valley, California | |
| DBVPG1373b | Soil, Netherlands | |
| DBVPG1788b | Soil, Finland | |
| DBVPG1853b | White Tecc, Ethiopia | |
| DBVPG6044b | Fermenting fruit juice, The Netherlands | |
| DBVPG6765b | Unknown | |
| L_1374b | Wine, Chile | |
| L_1528b | Wine, Chile | |
| YPS128b | Oak, Pennsylvania | |
| SK1b | Soil, United States | |
| Y55b | Wine, France | |
| YGPMb | Rotting fig, California | |
| RM11-1ac | Vineyard, California | CH408055 AAEG01000000 |
| YJM789d | Lung of an AIDS patient | AAFW02000067 |
| S288Ce | Laboratory strain | NC_001146 |
The genes BSC4 of these strains were sequenced by us. These strains were provided by Feng-Yan Bai (Systematic Mycology and Lichenology Laboratory, Institute of Microbiology, Chinese Academy of Sciences).
These strains were sequenced by the Saccharomyces Genome Resequencing Project at the Sanger Institute in collaboration with Ed Louis's group at the Institute of Genetics, University of Nottingham. All the sequences are downloaded from ftp://ftp.sanger.ac.uk/pub/dmc/yeast.
This strain was sequenced by the Broad Institute [Saccharomyces cerevisiae RM11-1a Sequencing Project, Broad Institute of Harvard and MIT (http://www.broad.mit.edu)].
This strain was sequenced by the Stanford Genome Technology Center (Wei et al. 2007).
The reference genome sequence (Saccharomyces cerevisiae systematic sequencing project).
Database homology search:
We carried out a tBLASTN search with the protein sequence of BSC4 as the query against the genome sequences of 81 fungal species. The fungal genome database is available at the SGD (http://www.yeastgenome.org/). A tBLASTN search was performed online using the BLAST service provided by the SGD with default parameters. We retained only those hits whose aligned length was >80% of the query length (105 amino acids) and whose identity of aligned fragment was >30%.
Southern blot:
We extracted genomic DNAs of S. bayanus, S. kudriavzevii, S. mikatae, S. paradoxus, and S. cerevisiae using the Puregene DNA isolation kit (Gentra Systems, Research Triangle Park, NC). We digested DNAs with EcoRI (New England BioLabs, Beverly, MA), separated them on an agarose gel, and transferred them to a nylon membrane (Roche Molecular Biochemicals, Indianapolis) by Southern blotting. We prepared the probe for the new gene BSC4 by labeling its PCR product with digoxigenin. We first amplified the gene from genomic DNA using primers AACAAGCAAGTTTTATACAATAC and CTGGGTTGCATGGGTAATTT and then used the PCR product as template to run the second round of PCR with a dNTP mixture containing digoxigenin-labeled dUTP. We hybridized the BSC4 probe to the membrane to evaluate copy number and level of sequence conservation in different species.
DNA sequencing and population analyses:
The BSC4 gene was amplified from genomic DNAs of S. cerevisiae strains listed in Table 2 using primer A (AAATAAATACGATATCAAGGCACCA) and primer D (CCGTCCTTGTTAAATAGTCACCTAA), which are located upstream and downstream of the BSC4 ORF as indicated by the Saccharomyces Genome Deletion Project Consortium (http://www-sequence.stanford.edu/group/yeast_deletion_project/Deletion_primers_PCR_sizes.txt). The PCR products were purified using a Tiangen (Beijing) DNA purification kit and checked by 2% agarose gels before sequence analysis. Bidirectional sequencing was performed for all samples with primers A and D separately by using a BigDye Terminators v 3.0 cycle sequencing kit (Applied Biosystems, Foster City, CA), according to the manufacturer's instructions. Sequences were read by an ABI3100 sequencer (Applied Biosystems). Sequence trace data were trimmed, assembled, and aligned with Sequencher 4.0.5 and by manual verification.
To detect if the BSC4 is a functional sequence and thus subject to selection, Tajima's D test and Fu and Li's tests were carried out with DnaSP 4.00.6 (Tajima 1989; Fu and Li 1993; Rozas et al. 2003) on the basis of population data. For a functional protein-coding gene, a more direct test for ORF functionality is to compare substitution rates at nonsynonymous sites (dN) and synonymous sites (dS). dN should be significantly smaller than dS for a protein-coding gene under functional constraint. The Nei–Gojobori codon model with Jukes–Cantor correction in MEGA 3.1 was used to calculate the overall average dN and dS and their standard errors were computed with the bootstrap method (1000 replicates; seed = 25,000), and the Z-test embedded in MEGA 3.1 was applied to test the difference between dN and dS (Kumar et al. 2004).
Reverse transcriptase–PCR and rapid amplification of cDNA ends:
Yeast cells were harvested from 20 ml of culture at OD600 = 1.0 and then resuspended in RNAlater solution (Ambion, Austin, TX). Total RNA was isolated using the RNeasy kit (QIAGEN, Valencia, CA) and then subjected to DNase I digestion (Invitrogen, San Diego). Retrotranscription was carried out using a Takara (Berkeley, CA) one-step reverse-transcriptase (RT)–PCR kit. A series of forward and reverse primers were designed on the basis of BSC4 and its ortholog sequences: S. cerevisiae forward (F), CCATTGCCATTGGAGAAAGCC, and reverse (R), AAAAGTTGCACAAAAATGTAGTTG; S. paradoxus F, AGAAAGACTTTCGCTCTGATG, and R, TGGTCATATGGACTGTTGTTG; S. mikatae F, GATATACATCGGAGTAAAGTTATT, and R, ATACTTCGTTTCCCACAGTTCT; and S. bayanus F, CATGCACAGCAAGAGGATTAT, and R, GCGGTTGTTGCCCAAATGAG. 3′-Rapid amplification of cDNA ends (RACE) was carried out using the FirstChoice RLM–RACE kit (Ambion).
Proteomics data analysis:
In this equation, i denotes the number of distinct peptides of the investigated protein that are matched with the spectra and j denotes the number of different spectra matched to each peptide. If the spectra assigned to different peptides are considered as independent evidence for their corresponding protein, then the probability P that a protein is present in the sample can be computed as the probability that at least one peptide assignment corresponding to the protein is correct.
Functional analyses using synthetic lethal and microarray data:
Synthetic lethal data are from the genetic interactions of BSC4 on the SGD (found at http://db.yeastgenome.org/cgi-bin/locus.pl?locus=BSC4). The microarray data were downloaded from the website accompanying Gasch et al.'s (2000) article (http://genome-www.stanford.edu/yeast_stress/).
RESULTS
Origin of the de novo gene BSC4:
BSC4 is a S. cerevisiae gene, which has an ORF of 132 amino acids, and with no apparent similarity to any previously characterized protein. BSC4 has no significant homolog when we used tBLASTN to search against genome sequences of S. bayanus, S. kudriavzevii, S. mikatae, and S. paradoxus under the standard parameters. Even if we use the putative translation product of the stop codon bypass event predicted by Namy et al. (2003), which is a peptide of 237 amino acids, there is still no significant homolog in these sibling species. The absence of homolog might be the false negative result due to incompleteness of the genomic databases of those species. However, the multiple-species search makes this possibility less likely, and the genome databases of Saccharomyces species are widely considered as the most reliable compared with genome databases of other species. These results suggest that BSC4 may be a newly evolved gene in S. cerevisiae. To further rule out possible spurious results caused by sequencing gaps in the outgroups, we conducted a genomic Southern blot with the probe designed against BSC4. The southern blot result shows that only the S. cerevisiae genome exhibits obvious hybridization signals (Figure 1). We also carried out a further tBLASTN search against genome sequences of other fungal species to exclude the probability of multiple-gene loss in the four outgroup species. The results showed that this ORF has no homolog in any other fungal species. However, the origination mechanism still remains to be clarified until we find its ancestral sequence because horizontal gene transfer or high divergence of sequences can both explain the above results.
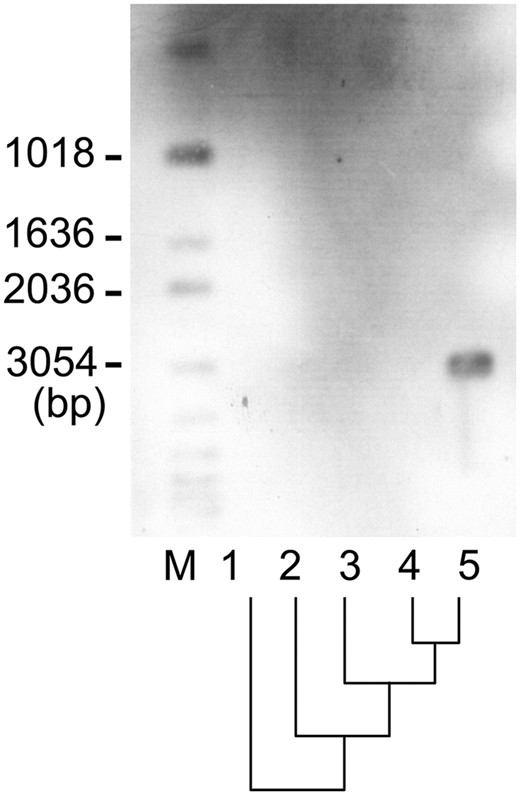
Results of Southern hybridization showing that only S. cerevisiae has a detectable signal of the BSC4 gene. M, marker; 1, S. bayanus; 2, S. kudriavzevii; 3, S. mikatae; 4, S. paradoxus; 5, S. cerevisiae. The phylogenetic relation of the four species is indicated under the lane numbers.
In addition to sequence similarity, the chromosomal context–synteny relationship is another important piece of information for identification of gene relationships. A pair of sequence fragments in two related species can be supposed to be in orthologous relationship if they have weak homology and their flanking genes are in orthologous status, when they do not have BLAST hits of a higher score in other regions of the genome. The Synteny Viewer on the Saccharomyces Genome Database website indicates that the flanking genes of BSC4 have their orthologs in the same synteny blocks of S. bayanus, S. mikatae, and S. paradoxus (Kellis et al. 2003). We cut the intergenic region between the two flanking genes and manually aligned them with BSC4 of S. cerevisiae (Figure 2). Because S. kudriavzevii is not covered in the Synteny Viewer on the Saccharomyces Genome Database website, we did not include it in Figure 2, although we also found by genome comparison that the synteny relationship of the locus in this species is also conserved (data not shown). The alignment shows that there are tracts of homologous sequences and the overall identity across those four Saccharomyces species is 35.71%. Data on the UCSC genome browser also indicate the same orthologous relationship, which is consistent with our analysis. These orthologous regions in the sibling species of S. cerevisiae have very low probability to code for proteins even if we consider stop codon readthrough in those species, because of the existence of a number of premature stop codons (supplemental Figure 1).

Alignment of the orthologous sequences of BSC4 from Saccharomyces bayanus (Sbay), S. mikatae (Smik), S. paradoxus (Spar), and S. cerevisiae (Scer). The conserved nucleotides are shaded.
The flanking genes of BSC4 in the S. cerevisiae genome, ALP1 and LYP1, are a pair of paralogs lined in an inverted direction. This gene order also remains conserved in the more distant yeast genomes of Ashbya gossypii, Kluyveromyces lactis, and S. castelli beyond Saccharomyces sensu stricto complex species (Figure 3). In addition, the length of this intergenic region does not change much across all those species (713 bp in A. gossypii and 889 bp in S. cerevisiae). From these results, we can make an estimate that the origin of the BSC4 ancestral sequence can be dated back at least to the last common ancestor of A. gossypii and S. cerevisiae, i.e., >100 million years ago (Dietrich et al. 2004) when an inverted gene duplication event formed the syntenic orthologs flanking the ancestor of BSC4. However, only after the divergence from S. paradoxus the ancestral noncoding sequence evolved into a protein-coding gene in S. cerevisiae. On the basis of these pieces of evidence, it is very likely that this is a real de novo origination case with clearly defined lineage.
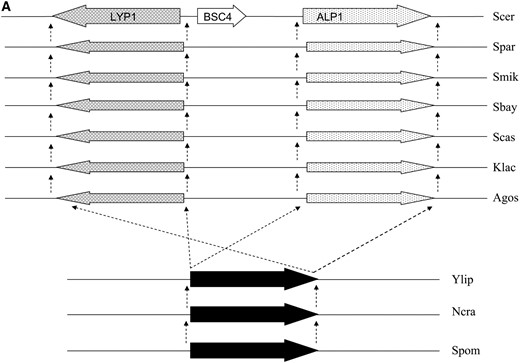
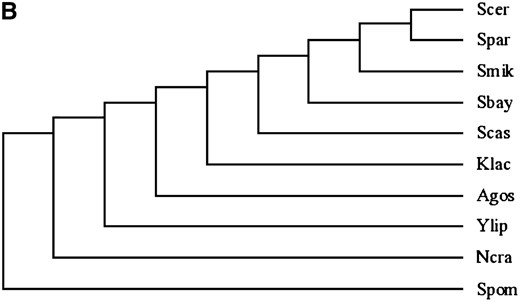
(A) Synteny relationships in the BSC4 region in different fungi species. (B) Phylogenetic tree of the fungi species used in A. Sbay, S. bayanus; Smik, S. mikatae; Spar, S. paradoxus; Scer, S. cerevisiae; Agos, Ashbya gossypii; Spom, Schizosaccharomyces pombe; Ylip, Yarrowia lipolytica; Ncra, Neurospora crassa. The dashed arrows in A indicate an orthologous relationship.
The similarity between BSC4 and its orthologous sequences is indeed much lower compared with other protein-coding orthologs (≥80%) (Kellis et al. 2003). The overall identity of the BSC4 orthologous sequences in the four Saccharomyces species (35.71%) is comparable to that of the essential non-protein-coding RNA (ncRNA) gene TLC1, which is 47.98%. Interestingly, RT–PCR results show that all the BSC4 orthologous sequences in S. bayanus, S. mikatae, and S. paradoxus can transcribe (Figure 4). Pang et al. (2006) reported that the ncRNA except snoRNA and miRNA are on the whole poorly conserved: most display <70% identity between human and mouse (Pang et al. 2006). Therefore, those ancestral orthologs of BSC4 are probably non-protein-coding RNA genes.
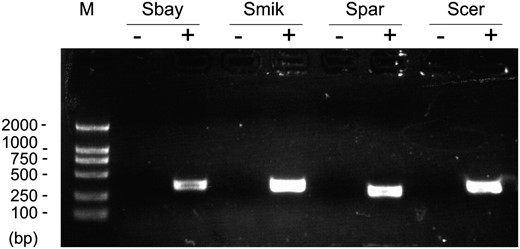
RT–PCR results. “−” indicates the RT–PCR-negative controls in which everything is the same as the positive (+) except omitting reverse transcriptase. Sbay, S. bayanus; Smik, S. mikatae; Spar, S. paradoxus; Scer, S. cerevisiae.
Evidence from DNA, RNA, protein, and phenotype levels supports BSC4 as a functional protein-coding gene:
Population evidence supports the protein-coding potential of BSC4:
To investigate if BSC4 is a protein-coding gene, we first conducted population analysis (Table 2). Although this gene evolved recently only in the S. cerevisiae lineage, sequences from 29 strain samples that are from different localities or origins showed that it is fixed and the ORFs are conserved in all S. cerevisiae populations.
Tajima's D test and Fu and Li's test give population statistics showing whether the observed polymorphism pattern deviates from neutrality caused by selection, indirectly reflecting if the sequence is functional or not (Tajima 1989; Fu and Li 1993). Both tests show a consistent deviation from neutrality (for Tajima's D test, D = −1.70065, 0.10 > P > 0.05; for Fu and Li's test, D = −3.33262, P < 0.02), suggesting the probable existence of purifying selection at this locus. Another population genetics test, for protein-coding ability, is to see if purifying selection has led to lower polymorphism at nonsynonymous sites than at synonymous sites. For BSC4, the substitution rates at synonymous sites (dS) and nonsynonymous sites (dN) are 0.03546 and 0.00951, respectively. dN is significantly smaller than dS (Z-test, P = 0.02354 < 0.05), strongly indicating purifying selection on the ORF of this gene. Thus, these population genetics results strongly suggest that BSC4 is undergoing purifying selection and functional constraint against amino acid change in baker's yeast.
Proteomics data support the protein-coding potential of BSC4:
We found 29 peptides assigned to the gene BSC4 in the tandem mass spectrometry (MS/MS) database (http://www.peptideatlas.org/repository) (Table 3). For each hit, the machine-learning program SEQUEST assigned a statistical validation value. We computed the combined probability of the existence of BSC4 protein to be 0.6078 according to the method of Nesvizhskii et al. (2003). If we take many other hits with scores <0.0001 into account, the probability could be higher. As more MS/MS experiments are being carried out in yeast and the yeast proteome data coverage that is now only 63% (Desiere et al. 2006) becomes deeper, we will be able to find more pieces of peptide evidence for BSC4 in the proteomics data.
Proteomic database search results
Experiment name | Spectrum name | Peptide probability | Peptide sequence |
|---|---|---|---|
| PAe000155 | 005b.4248.4248.2 | 0.0518 | APIAIGESPYVEWSCL |
| PAe000155 | 020b.1059.1059.2 | 0.0002 | KSHINNKLPMQP |
| PAe000120 | 060.0078.0078.3 | 0.3386 | KIVIIYVVR |
| PAe000155 | 900a.1593.1593.2 | 0 | CQALKRRDSKTYILCR |
| PAe000155 | 900a.3944.3944.2 | 0.0032 | EWSCLQVVFR |
| PAe000095 | Mark_T50_18_00.1142.1142.2 | 0 | MTPFSPRKSHINNKLPMQPR |
| PAe000095 | Mark_T50_26_01.1754.1754.3 | 0 | QPRKKKIVIIYVVRFH |
| PAe000095 | Mark_T50_27_00.3167.3167.3 | 0.0001 | NCITSKFYTIHIIKISTPVFRAP |
| PAe000095 | Mark_T50_28_00.2231.2231.2 | 0 | KDMVTKKTTFAQLITRLNH |
| PAe000095 | Mark_T50_28_03.1883.1883.2 | 0.0001 | DMVTKKTTFAQLITRL |
| PAe000095 | Mark_T50_39_00.1105.1105.3 | 0.0001 | PRKSHINNKLPMQPR |
| PAe000095 | Mark_T50_39_00.2179.2179.2 | 0.0872 | NKNCITSKFYTIHIIK |
| PAe000146 | ystcicat_1722_1.2153.2153.3 | 0.2893 | TYILCRTAVFGAMTPFSPR |
| PAe000146 | ystcicat_1722_1.3654.3654.3 | 0 | SHINNKLPMQPRKKKI |
| PAe000146 | ystcicat_1722_2.2386.2386.1 | 0 | TAVFGAMT |
| PAe000146 | ystcicat_23_2.2133.2133.1 | 0 | TAVFGAMT |
| PAe000146 | ystcicat_24_1.0184.0184.2 | 0 | FYTIHIIKISTPVFRAPIAIGESPYVEW |
| PAe000146 | ystcicat_25_1.3790.3790.3 | 0 | SHINNKLPMQPRKKKI |
| PAe000146 | ystcicat_27_2.3763.3763.3 | 0 | SHINNKLPMQPRKKKI |
| PAe000146 | ystcicat_29_2.0345.0345.2 | 0 | PMQPRKKKIVIIYVVRFH |
| PAe000146 | ystcicat_30_1.4670.4670.2 | 0 | AIGESPYVEWSCLQVVFRK |
| PAe000146 | ystcicat_30_2.4774.4774.2 | 0.0006 | AIGESPYVEWSCLQVVFRK |
| PAe000146 | ystcicat_34_1.3160.3160.2 | 0 | KDMVTKKTTFAQLITRLNHFLC |
| PAe000146 | ystcicat_34_2.5617.5617.2 | 0.0318 | DSKTYILCRTAVFGAMTPFSPR |
| PAe000146 | ystcicat_35_1.0036.0036.2 | 0 | KTYILCRTAVFGAMTPFSPRK |
| PAe000146 | ystcicat_36_2.1526.1526.2 | 0 | PRKSHINNKLPMQPRK |
| PAe000146 | ystcicat_4142_1.4611.4611.3 | 0 | NCITSKFYTIHIIKISTPVFRAPIAIGESPY |
| PAe000146 | ystcicat_4142_1.4815.4815.2 | 0 | KNKNCITSKFYTIHIIKISTP |
| PAe000146 | ystcicat_4344_2.0838.0838.2 | 0 | CQALKRRDSKTYILCR |
| Total probability | 0.6078 |
Experiment name | Spectrum name | Peptide probability | Peptide sequence |
|---|---|---|---|
| PAe000155 | 005b.4248.4248.2 | 0.0518 | APIAIGESPYVEWSCL |
| PAe000155 | 020b.1059.1059.2 | 0.0002 | KSHINNKLPMQP |
| PAe000120 | 060.0078.0078.3 | 0.3386 | KIVIIYVVR |
| PAe000155 | 900a.1593.1593.2 | 0 | CQALKRRDSKTYILCR |
| PAe000155 | 900a.3944.3944.2 | 0.0032 | EWSCLQVVFR |
| PAe000095 | Mark_T50_18_00.1142.1142.2 | 0 | MTPFSPRKSHINNKLPMQPR |
| PAe000095 | Mark_T50_26_01.1754.1754.3 | 0 | QPRKKKIVIIYVVRFH |
| PAe000095 | Mark_T50_27_00.3167.3167.3 | 0.0001 | NCITSKFYTIHIIKISTPVFRAP |
| PAe000095 | Mark_T50_28_00.2231.2231.2 | 0 | KDMVTKKTTFAQLITRLNH |
| PAe000095 | Mark_T50_28_03.1883.1883.2 | 0.0001 | DMVTKKTTFAQLITRL |
| PAe000095 | Mark_T50_39_00.1105.1105.3 | 0.0001 | PRKSHINNKLPMQPR |
| PAe000095 | Mark_T50_39_00.2179.2179.2 | 0.0872 | NKNCITSKFYTIHIIK |
| PAe000146 | ystcicat_1722_1.2153.2153.3 | 0.2893 | TYILCRTAVFGAMTPFSPR |
| PAe000146 | ystcicat_1722_1.3654.3654.3 | 0 | SHINNKLPMQPRKKKI |
| PAe000146 | ystcicat_1722_2.2386.2386.1 | 0 | TAVFGAMT |
| PAe000146 | ystcicat_23_2.2133.2133.1 | 0 | TAVFGAMT |
| PAe000146 | ystcicat_24_1.0184.0184.2 | 0 | FYTIHIIKISTPVFRAPIAIGESPYVEW |
| PAe000146 | ystcicat_25_1.3790.3790.3 | 0 | SHINNKLPMQPRKKKI |
| PAe000146 | ystcicat_27_2.3763.3763.3 | 0 | SHINNKLPMQPRKKKI |
| PAe000146 | ystcicat_29_2.0345.0345.2 | 0 | PMQPRKKKIVIIYVVRFH |
| PAe000146 | ystcicat_30_1.4670.4670.2 | 0 | AIGESPYVEWSCLQVVFRK |
| PAe000146 | ystcicat_30_2.4774.4774.2 | 0.0006 | AIGESPYVEWSCLQVVFRK |
| PAe000146 | ystcicat_34_1.3160.3160.2 | 0 | KDMVTKKTTFAQLITRLNHFLC |
| PAe000146 | ystcicat_34_2.5617.5617.2 | 0.0318 | DSKTYILCRTAVFGAMTPFSPR |
| PAe000146 | ystcicat_35_1.0036.0036.2 | 0 | KTYILCRTAVFGAMTPFSPRK |
| PAe000146 | ystcicat_36_2.1526.1526.2 | 0 | PRKSHINNKLPMQPRK |
| PAe000146 | ystcicat_4142_1.4611.4611.3 | 0 | NCITSKFYTIHIIKISTPVFRAPIAIGESPY |
| PAe000146 | ystcicat_4142_1.4815.4815.2 | 0 | KNKNCITSKFYTIHIIKISTP |
| PAe000146 | ystcicat_4344_2.0838.0838.2 | 0 | CQALKRRDSKTYILCR |
| Total probability | 0.6078 |
The total probability is calculated according to Equation 1 in materials and methods. The peptide probability <0.0001 is denoted as 0.
Proteomic database search results
Experiment name | Spectrum name | Peptide probability | Peptide sequence |
|---|---|---|---|
| PAe000155 | 005b.4248.4248.2 | 0.0518 | APIAIGESPYVEWSCL |
| PAe000155 | 020b.1059.1059.2 | 0.0002 | KSHINNKLPMQP |
| PAe000120 | 060.0078.0078.3 | 0.3386 | KIVIIYVVR |
| PAe000155 | 900a.1593.1593.2 | 0 | CQALKRRDSKTYILCR |
| PAe000155 | 900a.3944.3944.2 | 0.0032 | EWSCLQVVFR |
| PAe000095 | Mark_T50_18_00.1142.1142.2 | 0 | MTPFSPRKSHINNKLPMQPR |
| PAe000095 | Mark_T50_26_01.1754.1754.3 | 0 | QPRKKKIVIIYVVRFH |
| PAe000095 | Mark_T50_27_00.3167.3167.3 | 0.0001 | NCITSKFYTIHIIKISTPVFRAP |
| PAe000095 | Mark_T50_28_00.2231.2231.2 | 0 | KDMVTKKTTFAQLITRLNH |
| PAe000095 | Mark_T50_28_03.1883.1883.2 | 0.0001 | DMVTKKTTFAQLITRL |
| PAe000095 | Mark_T50_39_00.1105.1105.3 | 0.0001 | PRKSHINNKLPMQPR |
| PAe000095 | Mark_T50_39_00.2179.2179.2 | 0.0872 | NKNCITSKFYTIHIIK |
| PAe000146 | ystcicat_1722_1.2153.2153.3 | 0.2893 | TYILCRTAVFGAMTPFSPR |
| PAe000146 | ystcicat_1722_1.3654.3654.3 | 0 | SHINNKLPMQPRKKKI |
| PAe000146 | ystcicat_1722_2.2386.2386.1 | 0 | TAVFGAMT |
| PAe000146 | ystcicat_23_2.2133.2133.1 | 0 | TAVFGAMT |
| PAe000146 | ystcicat_24_1.0184.0184.2 | 0 | FYTIHIIKISTPVFRAPIAIGESPYVEW |
| PAe000146 | ystcicat_25_1.3790.3790.3 | 0 | SHINNKLPMQPRKKKI |
| PAe000146 | ystcicat_27_2.3763.3763.3 | 0 | SHINNKLPMQPRKKKI |
| PAe000146 | ystcicat_29_2.0345.0345.2 | 0 | PMQPRKKKIVIIYVVRFH |
| PAe000146 | ystcicat_30_1.4670.4670.2 | 0 | AIGESPYVEWSCLQVVFRK |
| PAe000146 | ystcicat_30_2.4774.4774.2 | 0.0006 | AIGESPYVEWSCLQVVFRK |
| PAe000146 | ystcicat_34_1.3160.3160.2 | 0 | KDMVTKKTTFAQLITRLNHFLC |
| PAe000146 | ystcicat_34_2.5617.5617.2 | 0.0318 | DSKTYILCRTAVFGAMTPFSPR |
| PAe000146 | ystcicat_35_1.0036.0036.2 | 0 | KTYILCRTAVFGAMTPFSPRK |
| PAe000146 | ystcicat_36_2.1526.1526.2 | 0 | PRKSHINNKLPMQPRK |
| PAe000146 | ystcicat_4142_1.4611.4611.3 | 0 | NCITSKFYTIHIIKISTPVFRAPIAIGESPY |
| PAe000146 | ystcicat_4142_1.4815.4815.2 | 0 | KNKNCITSKFYTIHIIKISTP |
| PAe000146 | ystcicat_4344_2.0838.0838.2 | 0 | CQALKRRDSKTYILCR |
| Total probability | 0.6078 |
Experiment name | Spectrum name | Peptide probability | Peptide sequence |
|---|---|---|---|
| PAe000155 | 005b.4248.4248.2 | 0.0518 | APIAIGESPYVEWSCL |
| PAe000155 | 020b.1059.1059.2 | 0.0002 | KSHINNKLPMQP |
| PAe000120 | 060.0078.0078.3 | 0.3386 | KIVIIYVVR |
| PAe000155 | 900a.1593.1593.2 | 0 | CQALKRRDSKTYILCR |
| PAe000155 | 900a.3944.3944.2 | 0.0032 | EWSCLQVVFR |
| PAe000095 | Mark_T50_18_00.1142.1142.2 | 0 | MTPFSPRKSHINNKLPMQPR |
| PAe000095 | Mark_T50_26_01.1754.1754.3 | 0 | QPRKKKIVIIYVVRFH |
| PAe000095 | Mark_T50_27_00.3167.3167.3 | 0.0001 | NCITSKFYTIHIIKISTPVFRAP |
| PAe000095 | Mark_T50_28_00.2231.2231.2 | 0 | KDMVTKKTTFAQLITRLNH |
| PAe000095 | Mark_T50_28_03.1883.1883.2 | 0.0001 | DMVTKKTTFAQLITRL |
| PAe000095 | Mark_T50_39_00.1105.1105.3 | 0.0001 | PRKSHINNKLPMQPR |
| PAe000095 | Mark_T50_39_00.2179.2179.2 | 0.0872 | NKNCITSKFYTIHIIK |
| PAe000146 | ystcicat_1722_1.2153.2153.3 | 0.2893 | TYILCRTAVFGAMTPFSPR |
| PAe000146 | ystcicat_1722_1.3654.3654.3 | 0 | SHINNKLPMQPRKKKI |
| PAe000146 | ystcicat_1722_2.2386.2386.1 | 0 | TAVFGAMT |
| PAe000146 | ystcicat_23_2.2133.2133.1 | 0 | TAVFGAMT |
| PAe000146 | ystcicat_24_1.0184.0184.2 | 0 | FYTIHIIKISTPVFRAPIAIGESPYVEW |
| PAe000146 | ystcicat_25_1.3790.3790.3 | 0 | SHINNKLPMQPRKKKI |
| PAe000146 | ystcicat_27_2.3763.3763.3 | 0 | SHINNKLPMQPRKKKI |
| PAe000146 | ystcicat_29_2.0345.0345.2 | 0 | PMQPRKKKIVIIYVVRFH |
| PAe000146 | ystcicat_30_1.4670.4670.2 | 0 | AIGESPYVEWSCLQVVFRK |
| PAe000146 | ystcicat_30_2.4774.4774.2 | 0.0006 | AIGESPYVEWSCLQVVFRK |
| PAe000146 | ystcicat_34_1.3160.3160.2 | 0 | KDMVTKKTTFAQLITRLNHFLC |
| PAe000146 | ystcicat_34_2.5617.5617.2 | 0.0318 | DSKTYILCRTAVFGAMTPFSPR |
| PAe000146 | ystcicat_35_1.0036.0036.2 | 0 | KTYILCRTAVFGAMTPFSPRK |
| PAe000146 | ystcicat_36_2.1526.1526.2 | 0 | PRKSHINNKLPMQPRK |
| PAe000146 | ystcicat_4142_1.4611.4611.3 | 0 | NCITSKFYTIHIIKISTPVFRAPIAIGESPY |
| PAe000146 | ystcicat_4142_1.4815.4815.2 | 0 | KNKNCITSKFYTIHIIKISTP |
| PAe000146 | ystcicat_4344_2.0838.0838.2 | 0 | CQALKRRDSKTYILCR |
| Total probability | 0.6078 |
The total probability is calculated according to Equation 1 in materials and methods. The peptide probability <0.0001 is denoted as 0.
3′ RACE data support the translation readthrouth hypothesis of BSC4:
Namy et al. (2003) reported that BSC4 has typical readthrough nucleotide context around its stop codon and this sequence can give a readthrough frequency of 9% when cloned into a plasmid with reporter genes (Namy et al. 2003). When the initial stop codon is bypassed, translation will go on to the second stop codon 321 bp downstream. To confirm their hypothesis we carried out 3′ RACE to characterize the 3′-UTR of the BSC4 transcript. The RACE result shows that this gene has a very long 3′-UTR that is 512 bp from the stop codon, much longer than the average length of ∼200 bp in yeast (Miura et al. 2006). The RACE product matches perfectly with its corresponding genomic sequence, except the additional poly(A) tail, and contains a second stop codon 321 bp away from the first stop codon. This is consistent with the RT–PCR result and the translation readthrough hypothesis of Namy et al. (2003).
Functional implication for BSC4 from evidence of expression and phenotype:
RT–PCR results (Figure 4) show that BSC4 is expressed in normal culture conditions, which suggests that this is a functional gene. The microarray data of Gasch et al. (2000) showed that the expression level of this gene would rise upon entering the stationary stage, which suggests that this gene might function in the cell response of yeast to the stationary phase (Gasch et al. 2000).
On the other hand, Pan et al. (2006) reported that BSC4 had two synthetic lethal partners RPN4 and DUN1, which means that yeast is not viable if both BSC4 and RPN4 or BSC4 and DUN1 are deleted (Pan et al. 2006). We can deduce from the synthetic lethal relationship that BSC4 functions either in a parallel redundant pathway or in the same essential pathway with RPN4 and DUN1. RPN4 is a transcriptional activator of many DNA repair genes and proteasome genes (Xie and Varshavsky 2001; Dohmen et al. 2007). DUN1 is a downstream DNA damage checkpoint kinase (S. H. Chen et al. 2007). Because both DUN1 and RPN4 function in the DNA damage repair pathway, and because RPN4 showed a partial coexpression pattern in stationary phase with BSC4 (Figure 5), BSC4 may also function in the DNA repair pathway.
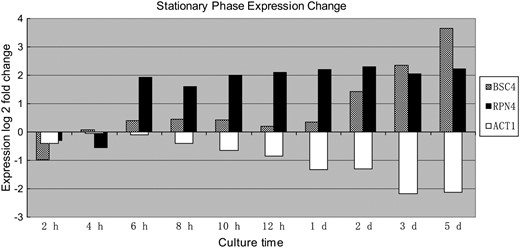
Comparisons of expression changes among BSC4, RPN4, and ACT1 based on the microarray data of Gasch et al. (2000). ACT1 is selected because it is considered as a good internal control in most yeast expression quantification experiments and it also indicates that the upregulation of gene expression is neither a whole-genome pattern nor a system error of the microarray. The y-axis denotes the log2 value of fold change, and the x-axis denotes the culture time after the time point 0. YPD cultures were grown at 30° to OD600 = 0.3, at which point the cell culture was collected to serve as the time = 0 reference. Samples were recovered at 2, 4, 6, 8, 10, and 12 hr and 1, 2, 3, and 5 days of culture incubation. The partial coexpression pattern of BSC4 and RPN4 in stationary phase is shown.
DISCUSSION
Whole-gene de novo origination was first discovered by Levine et al. (2006). They found five de novo genes in D. melanogaster and/or D. simulans. Four of them were found to have noncoding paralogs on the same chromosome X. They also found that at least three of the four noncoding paralogs' sequences have RNA expression in D. melanogaster at low levels and they attributed this phenomenon to the hypertranscription of the male X in Drosophila testis. They also noted that it may be a common phenomenon that de novo gene evolution is more likely to occur in a previously transcribed region (Levine et al. 2006). At the same time, the authors also mentioned that they cannot determine whether the coding sequences or their paralogous noncoding sequences are ancestral. Therefore, if de novo origination occurs before intrachromosomal duplication, which generates another copy of the de novo gene sequence, the paralogous noncoding sequence can also be taken as the degenerated copy of the de novo protein-coding gene. And there are already many reports that many degenerated pseudogenes show RNA expression (Hirotsune et al. 2003; Piehler et al. 2006; Zheng et al. 2007).
Using the same methods, Begun et al. (2006, 2007) also found de novo genes in D. yakuba and/or D. erecta by screening the accessory gland-expressed genes and testis-expressed genes. They found that de novo gene origination tends to be more often X-linked in many lineages of Drosophila. In Begun et al.'s (2007) work, 4 of 11 initial candidate de novo genes in D. yakuba and/or D. erecta are found to have homologous noncoding sequences with RNA expression in the outgroup species D. melanogaster. However, they exclude those 4 genes from further analysis because alignment of the homologous sequences is ambiguous.
In this work, we provide evidence at both the protein and the phenotype levels of a de novo protein-coding gene for the first time and this is also the first fully supported case that a protein-coding gene evolved from a previously transcribed region (or probably an RNA gene). It is plausible that protein-coding genes can originate from previously transcribed regions that contain the necessary transcription elements and provide RNA materials for a protein translation machine. Recent evidence suggests that the majority of the genomes of mammals and other complex organisms are in fact transcribed. These transcripts include microRNAs and snoRNAs and tens of thousands of longer transcripts. These RNAs appear to compose a hidden layer of internal signals that control various levels of gene expression in physiology and development, including chromatin architecture/epigenetic memory, transcription, RNA splicing, editing, translation, and turnover (Mattick and Makunin 2006). In addition, large RNA pools provide enormous potential for de novo protein gene evolution. It is possible that a common path for de novo protein gene evolution involves first a piece of DNA sequence to be transcribed via recruiting the transcription elements and machine, followed by the transcribed sequence giving birth to a novel protein-coding gene through the acquisition of an open reading frame through mutations. The BSC4 case vividly demonstrates this gradual process of de novo gene origination.
In addition to the mechanism of de novo evolution, the driving force behind the evolutionary process is another important issue. Functional data from BSC4 provided some clues as to why the de novo protein-coding gene got fixed in S. cerevisiae during evolution. The functional analysis shows that expression of BSC4 is upregulated when S. cerevisiae enters stationary phase and BSC4 may function in the DNA repair pathway. Like many other microorganisms, S. cerevisiae responds to starvation by stopping growth and entering into a stationary phase. When entering a stationary phase, yeasts stop dividing and experience an aging-like process. Consistent with the free radical theory of aging, Madia et al. (2007) report that yeasts accumulate more DNA mutations in the aging process, similar with the aging process of mammalian dividing cells. Reports also indicate that DNA base excision repair is absolutely essential for cells to survive the stationary phase aging process (MacLean et al. 2003). So the DNA repair pathway may be very critical for yeast to survive a frequent transition from relatively nutrient-rich environments, e.g., ripe grapes and wilted plant leaves, to nutrient-poor environments, e.g., earth and insect body surface (Werner and Braun 1996). Through getting involved in this pathway, the de novo protein gene BSC4 may have contributed to the fitness of S. cerevisiae when S. cerevisiae were shifted to the nutrient-poor environment and finally got fixed in S. cerevisiae during evolution.
Overall, our study identified and characterized a whole-gene de novo evolution case in S. cerevisiae for the first time. This gene originated from a previously noncoding but transcribed sequence. Evidence from population, RNA, proteome, and phenotype levels supports the functionality and coding potential of this gene. Functional data indicate that this de novo gene may be involved in the DNA repair pathway during the stationary phase of S. cerevisiae. Further studies on the function of BSC4 will help us to disclose how the novel protein integrates into the gene network of yeast and how the de novo origination event is related to the evolution of S. cerevisiae.
Regarding de novo gene origination, we propose that there may be two steps of a gradual evolution process for a noncoding DNA sequence that evolves into a protein-coding gene: first, the DNA sequence is transcribed by evolving cis-elements in the DNA sequence to recruit its transcription machine; and second, the transcribed sequence obtains its open reading frame and gets into the translation machine. Our findings support this gradual model of de novo protein-coding gene origination.
Footnotes
These authors contributed equally to this work.
Footnotes
Communicating editor: N. Takahata
Acknowledgement
We thank Xin Li, Qi Zhou, and Dan Li for helpful discussions and Paul Lemetti for English editing of the manuscript. We also thank Jin-Qiu Zhou and Feng-Yan Bai for providing yeast strains. This work was supported by a Chinese Academy of Sciences–Max Planck Society Fellowship, by two National Natural Science Foundation of China key grants (nos. 30430400 and 30623007), and by a 973 Program (no. 2007CB815703-5) to W.W.
References
Choi, I. G., and S. H. Kim,
Giacomelli, M. G., A. S. Hancock and J. Masel,
Levine, M. T., C. D. Jones, A. D. Kern, H. A. Lindfors and D. J. Begun,
Long, M., E. Betran, K. Thornton and W. Wang,
Miura, F., N. Kawaguchi, J. Sese, A. Toyoda, M. Hattori et al.,
Perutz, M. F., J. C. Kendrew and H. C. Watson,
Wang, W., F. G. Brunet, E. Nevo and M. Long,
Wei, W., J. H. McCusker, R. W. Hyman, T. Jones, Y. Ning et al.,
Xie, Y., and A. Varshavsky,

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}