





Abstract
In natural populations, genetic variation affects resistance to disease. Knowing how much variation exists, and understanding the genetic architecture of this variation, is important for medicine, for agriculture, and for understanding evolutionary processes. To investigate the extent and nature of genetic variation affecting resistance to pathogens, we are studying a tractable model system: Drosophila melanogaster and its natural pathogen the vertically transmitted sigma virus. We show that considerable genetic variation affects transmission of the virus from parent to offspring. However, maternal and paternal transmission of the virus is affected by different genes. Maternal transmission is a simple Mendelian trait: most of the genetic variation is explained by a polymorphism in ref(2)P, a gene already well known to affect resistance to sigma. In contrast, there is considerable genetic variation in paternal transmission that cannot be explained by ref(2)P and is caused by other loci on chromosome 2. Furthermore, we found no genetic correlation between paternal transmission of the virus and resistance to infection by the sigma virus following injection. This suggests that different loci affect viral replication and paternal transmission.
GENETIC variation affects disease resistance in a wide range of taxa, from humans (reviewed in Hirschhorn and Daly 2005) to mosquitoes (Riehleet al. 2006), from plants (reviewed in Holub 2001) to fruit flies (Henter 1995a,b; Lazzaroet al. 2004; Tinsleyet al. 2006; Dubuffetet al. 2007). Rather few studies have formally estimated how much genetic variation exists in natural populations for resistance to pathogens (Frank 1994; Bergelsonet al. 2001; Niareet al. 2002). However, the existence of this genetic variation in disease resistance raises several key questions: How much genetic variation exists?, How many genes underlie this variation?, Which genes are responsible?, and How big are the effects of these genes?
The answers to these questions have important implications for medicine: genetic variation affects whether disease vectors transmit pathogens to humans (Gooding 1996) and how populations respond to disease. Furthermore, the genes underlying this variation will tell us about the molecular mechanisms that make one individual more susceptible to pathogens than another. Genetic variation in disease resistance is also important for agriculture; such variation affects how plants and animals respond to disease, how they respond to selection during breeding programs, and how they respond to chemical and biological disease control strategies. Understanding genetic variation is also crucial for understanding evolution. Genetic variation in pathogen resistance underpins theories about the evolution of sex (Bell and Smith 1987) and sexual selection (Hamilton and Zuk 1982). The extent and type of genetic variation in disease resistance also affects models of coevolution between parasites and their hosts. Natural selection is expected to remove variation that affects fitness, so finding out why variation in resistance to pathogens is maintained will give insights into the mechanisms of coevolution.
Carefully chosen model systems could provide answers to these questions. We are studying a particularly tractable genetic model: Drosophila melanogaster and the sigma virus. The sigma virus is a natural pathogen of D. melanogaster that is transmitted vertically, from parent to offspring. Unusually for an exclusively vertically transmitted parasite, both males and females pass the virus on to their offspring. Sigma is found in natural populations of D. melanogaster all over the world, at frequencies of 0–15% (Carpenteret al. 2007). A useful feature of this model system is that infected flies are paralyzed or killed when exposed to high concentrations of carbon dioxide, which provides a simple assay of sigma virus infection.
Variation in one gene, ref(2)P, is already known to affect the replication of the sigma virus. ref(2)P encodes a protein that is thought to function both in sperm development and in the Toll pathway (an important component of the innate immune system) (Dezeleeet al. 1989; Avilaet al. 2002). A complex mutation in ref(2)P reduces the replication rate of the sigma virus (Brun and Plus 1980; Carre-Mloukaet al. 2007). This polymorphism has a worldwide distribution and the resistant allele occurs on an average of 20% of chromosomes (C. Schlötterer and P. Orozco, unpublished data on over 2000 alleles from 21 populations). Other loci affecting host responses to sigma have also been approximately mapped. One locus (on chromosome 3) affects vertical transmission from an infected male to its progeny, and several other loci (located on chromosomes 2 and 3) are known to affect replication of the sigma virus after it has been injected into flies but are not thought to affect transmission through males (Gay 1978). However, little more is known about these loci, or whether these are naturally occurring polymorphisms (reviewed in Brun and Plus 1980). Several researchers have studied genetic variation in resistance to sigma, but these experiments have almost all been designed in ways that detect only the effects of ref(2)P (although see Fleuriet 1991).
We report considerable genetic variation affecting transmission of the sigma virus among flies that carry a “wild-type” second chromosome derived from natural populations. Most of the genetic variation on chromosome 2 affecting virus transmission by females is explained by a polymorphism in ref(2)P. However, much of the genetic variation in male transmission cannot be explained by ref(2)P. Male transmission is also affected by other genes on chromosome 2. We also look at the susceptibility of individuals to infection following injection of the sigma virus and show that, again, genes other than ref(2)P affect susceptibility to infection.
MATERIALS AND METHODS
D.melanogaster stocks, viral isolates, and general methods:
The effect of the D. melanogaster second chromosome on sigma virus transmission was measured using 88 chromosome-substitution lines (created by Lazzaroet al. 2004). Each of these lines has a different homozygous second chromosome that had been sampled from a population in Pennsylvania in 1998 and 1999 and that had been substituted into a common isogenic genetic background. These lines carry the visible marker spapol on chromosome 4, which helps ensure that there has been no contamination of these lines from flies of other genotypes. A mutation in a gene called ref(2)P is already well-known to affect the susceptibility of D. melanogaster to the sigma virus (Druet al. 1993; Wayneet al. 1996; Banghamet al. 2007). This is a complex mutation, and to take it into account in our experiments, we sequenced the 88 chromosome-substitution lines for this mutation (Banghamet al. 2007). The lines that we refer to as carrying the “resistant” ref(2)P allele all carry the d and p mutations described by Carre-Mloukaet al. (2007), while the lines described as carrying the “susceptible” ref(2)P allele have neither.
Because the sigma virus is transmitted only vertically, and because the chromosome-substitution lines were not infected with sigma at the outset, we created a fly stock that was infected with the sigma virus and used this to cross the virus into our fly lines. We used the viral isolate AP30, which was collected by Jennifer Carpenter in 2005 in Apshawa, Florida (Carpenteret al. 2007). Flies from a wild-caught stock infected with the virus were homogenized in Ringer's solution, and the homogenate was injected into adult female SM5/Pm; spapol flies (henceforth called SM5/Pm), which had the same genetic background as the second-chromosome substitution lines and is homozygous for the susceptible ref(2)P allele. From the progeny of injected SM5/Pm, we selected a line that was infected with the sigma virus and that had a high rate of vertical transmission.
The isogenic P18 strain, used during the assays of transmission from homozygous parents, was generated by A. Fytrou from an isofemale line collected in Pennsylvania and was made isogenic using standard crosses to a balancer stock (SM1/Pm; TM6/Sb; spapol). P18 is homozygous for the susceptible ref(2)P allele.
Throughout the experiment, we used standard procedures for rearing stocks at standard density. To produce standard-density bottle cultures, we collected eggs by putting apple juice–agar plates in cages with live yeast. We washed eggs off these plates and pipetted either 26 μl (for balancer stocks) or 13 μl (for other stocks) into bottles containing standard D. melanogaster maize-sugar-yeast media (Clancy and Kennington 2001). When setting up crosses throughout the experiment, we achieved approximate standard densities of 50 offspring by keeping virgin females for a few days on food that had been sprinkled with live yeast and then setting up crosses with two females in a vial for 2 days, without additional live yeast.
To assay for infection by the sigma virus, adults are exposed to pure CO2 for 15–17 min at 12°. By 2 hr postexposure, uninfected flies are awake from this anesthesia, but flies infected with the sigma virus are dead or paralyzed.
Variation in the rate of sigma virus transmission by females and males:
We carried out two sets of experiments. The sigma virus is vertically transmitted, so in each set of experiments, the panel of second-chromosome-substitution lines were each infected by crossing them to infected SM5/Pm flies. The crosses are summarized in Figure 1.
Large experiments:
The first set of experiments was carried out to assess transmission from heterozygous females to homozygous offspring, and from homozygous males to heterozygous offspring (Figure 1) and was carried out with large sample sizes. Infected SM5/Pm virgin females were collected from standard-density bottle cultures. After 3 days, pairs of females were placed in vials with pairs of males from each of the chromosome-substitution lines and allowed to lay for 2 days. Between one and four replicate crosses were set up for each chromosome-substitution line, depending on the numbers available. The infected SM5/+ female F1 progeny were aged for ∼5 days and then backcrossed to the chromosome-substitution line (+/+). Between two and four replicates were set up from each vial. After 2 days in the vial, the parents were removed from the vials and we checked that the female parent was infected; if either female was uninfected the vial was discarded. We collected the offspring of this cross that were homozygous for the wild-type chromosome 2. These flies were genetically identical to the chromosome-substitution lines, but have been infected with the virus from their mothers. Some of the males were put to one side to be used to measure the rate of sigma virus transmission by infected males (see below). Fourteen days after the cross was set up, the remainder of the flies was tested for infection. This provided an estimate of the effect of the wild-type second chromosomes on transmission from a heterozygous female (SM5/+) to homozygous offspring (+/+). In total we assayed 6647 flies in 367 vials.
To measure the rate at which infected homozygous males transmit the sigma virus to their offspring, 7-day-old infected males derived from the previous cross were mated to 3-day-old females from the isogenic P18 strain. For each vial from the previous generation, between one and four replicates were set up. After 2 days in the vial, the parents were removed from the vials and we checked that the male parent was infected; if either male was uninfected the vial was discarded. Fifteen days after the crosses were set up, the progeny were assayed for sigma infection. In total we assayed 49,063 flies in 1022 vials.
Small experiments:
We then repeated the two experiments described above on a smaller scale, alongside the reciprocal crosses (maternal transmission from a homozygous female and paternal transmission from a heterozygous male) (Figure 1). The experimental design was identical for the two experiments repeated, except for the ages of the flies when the crosses were set up. In the preliminary cross for this second round of experiments, SM5/Pm virgin females were 4–6 days old when they were crossed to +/+ males. For the assay of maternal transmission from a heterozygous female, the SM5/+ females' parents were 6–8 days old when the cross was set up, and offspring were assayed 16 days later. We assayed a total of 8037 flies in 430 vials. To assay paternal transmission from a homozygous male, the male +/+ parents were 4–8 days old when the cross was set up, and offspring were assayed 15 days later. We assayed a total of 27,642 flies in 367 vials.
The remaining two experiments were carried out as follows. To estimate paternal transmission from heterozygous males to homozygous offspring, 6–8-day-old SM5/+ males were crossed to 6–7-day-old +/+ virgin females from their respective chromosome-substitution line. As before, these were left to lay eggs for 2 days, and then we checked that the male parent was infected with the sigma virus. Sixteen days after the cross was originally set up, +/+ offspring were assayed for sigma infection. In total, we assayed 5878 flies in 300 vials.
To estimate maternal transmission from homozygous females to heterozygous offspring, F2 virgin +/+ female offspring were retained from the maternal-transmission cross described above. These were kept without live yeast for 8 days, then given yeast for 24 hr before being placed in vials with pairs of P18 males and allowed to lay eggs for 2 days. Parents were removed and checked that the female parent was infected with the virus, and any vials for which the female parent was not infected were discarded. Fifteen days after the crosses were set up, the offspring were tested for infection. In total, we assayed 9079 flies in 231 vials.
Variation in susceptibility to sigma virus infection following injection:
We used a subset of the second-chromosome-substitution lines to assess how long it takes for flies to become CO2 sensitive after they have been injected with the sigma virus. This trait is correlated with the rate of viral replication in the flies (Brun and Plus 1980). To produce the inoculate, 60 SM5/Pm flies infected with the viral isolate AP30 were crushed on ice with 500 μl of Ringer's solution. The mixture was centrifuged for a few seconds and 10% v/v fetal bovine serum was added to the supernatant. The inoculate was stored at −80°. Thirty-two lines were randomly selected from the 69 second-chromosome-substituted lines that had the susceptible ref(2)P allele. To grow flies from these lines at standard density, adults were kept in tubes with apple juice plates for 9 hours. For each “laying tube,” larvae were picked from the apple juice agar and placed in vials at a density of 50 per vial. We aimed to obtain two vials of larvae from each laying tube each day. This was repeated with the same adult flies over 5 days.
Twelve days after larvae had been placed in the tube (and ∼2 days after they emerged as adults), female flies were injected with the sigma virus. It is possible that diurnal rhythm affects the replication of the virus inside the adult, so we recorded whether individuals were injected in the morning or the afternoon. Flies were injected using a fine glass capillary needle. Injected individuals were kept in vials without yeast for 7 days, and then tested for sensitivity to CO2. Across all 32 genotypes, we injected 587 flies split between 149 replicate vials.
Statistical analysis of genetic variation:
The statistical analysis was carried out using the R (v.2.2.1) software and language. We determined the factors affecting variation in transmission of sigma using a general linear mixed-effects model implemented using R's LME function (Laird and Ware 1982; Pinheiro and Bates 2000). Viral infection rates were measured as proportions, so these were arcsine-square-root transformed to remove the dependency of the variance of the observation on the observation itself. Because the data were analyzed on a transformed scale, the mean proportion of infected flies reported throughout the results are simply the arithmetic means calculated from the raw data (with equal weights being given to the different genotypes), rather than estimates from the model. For all of the experiments on parental transmission, we excluded any vials that contained fewer than a total of 5 flies.
Maternal transmission:
For both of the female-transmission experiments the infection rates were skewed, so the effect of ref(2)P on the mean transmission was determined using the Wilcoxon's signed-rank test. The subset of lines carrying the resistant ref(2)P allele were more normally distributed, so the genetic variance among these lines was evaluated using linear mixed models. We started with a maximal model and sequentially removed nonsignificant interaction terms and factors. Where we had repeated the experiment on two occasions, we combined the data sets and assigned “experiment” as a fixed effect (to determine the effect of experiment on the mean), and as a random effect, nested within genotype (to determine the effect of experiment on the genotypic variance). Genotype and the levels of replication were assigned as random effects. The residual variance was different in the “large” and “small” data sets, so to address this, we fitted a heteroscedastic model using the VarIdent function of LME, which weights the variances in the two experiments to make them equal (Pinheiro and Bates 2000). We also weighted the residual variance according to the total number of flies in each vial. The significance of each fixed effect was assessed by carrying out an analysis of variance on the appropriate model. The significance of each random effect was assessed by comparing models with and without that effect using a likelihood-ratio test. The likelihood-ratio test statistic, which is twice the difference in the log likelihoods of the models (2Δl), follows the χ2 distribution.
Paternal transmission:
The paternal transmission data sets were also analyzed with general linear mixed-effects models. The analysis was the same as described above (this time using the “combined” data set for paternal transmission to heterozygous offspring, and the small data set for paternal transmission to homozygous offspring), except that this time we were able to include in our analysis lines carrying the resistant allele of ref(2)P and lines carrying the susceptible allele of ref(2)P. Therefore, in both sets of analyses we included the presence/absence of the resistant ref(2)P allele as a fixed effect (see Banghamet al. 2007 for details of genotyping).
We used the between-line variance components (σL2) from these models to estimate the genetic variance (Vg) among the lines. As the data had been arcsine-square-root transformed before the analysis, the variance components and trait means were back-transformed before estimating Vg. The back-transformed overall trait mean (Y′) was obtained as Y′ = sin2Y and the back-transformed variance components (σY2′) were obtained as σY2′ = (sin2Y)2σY2. Assuming that all the genetic variation is additive, Vg = σL′2/2, because all of our lines are homozygous. The coefficient of genetic variation (CVg) was estimated as 100(Vg)1/2/Y′. When estimating CVg from the combined data set on homozygous paternal transmission, the overall trait mean was estimated from the overall trait mean of the large data set alone.
At the beginning of the paternal-transmission assay, we counted the sexes separately in a subset of vials and found that infection rates were the same for male and female offspring. Therefore, for the remainder of the experiment we did not record the infection rates of male and female offspring separately.
Injection experiment:
To analyze the variation in susceptibility to infection following injection with the sigma virus, we used general mixed-effects models. Time of day injected (morning or afternoon) was assigned as a fixed effect, while genotype was assigned as a random effect with any levels of replication nested within this.
We investigated whether the transmission of the virus by homozygous males and the susceptibility to infection following injection are controlled by the same genes. To do this, we analyzed the two experiments in a single model. The paternal-transmission data set had a more complicated structure than the injection data set, with two levels of replication (F2 replicate nested within F1 replicate). So to generate a set of independent estimates of paternal transmission for each genotype, we estimated the value of each F1 replicate from a mixed model. These estimates were then combined with the injection data and analyzed in a model with “experiment type” as a fixed effect and “genotype” as a random effect. The residual variance was slightly higher in the injected data set than in the male-transmission data set. To cope with this, we also fitted a heteroscadistic model using the VarIdent function of LME, which weights the variances in the two experiments to make them equal. This made little difference to the results, and, because it did not significantly improve the fit of the model to the data, we have not presented these results. The approximate confidence limits on the quantitative genetic parameters were estimated using the “intervals” function of LME (Pinheiro and Bates 2000).
RESULTS
Maternal transmission to homozygous offspring:
There is substantial variation in the proportion of offspring infected with the sigma virus from their mother. The first experiment measured transmission from an infected mother heterozygous for the wild-type second chromosome to offspring that were homozygous for that chromosome (Figure 1A). There is a bimodal pattern of transmission: most of the lines were nearly 100% infected and a subset had lower infection rates (Figure 2A). This is explained by the presence or absence of the ref(2)P resistance mutation. For the data set from the large experiment, lines carrying the susceptible ref(2)P allele had a mean rate of transmission of 99%, and for lines carrying the resistant allele the mean rate of transmission was 8% (Wilcoxon's signed-rank test U = 1020, nsusceptible = 60, nresistant = 17, P < 0.0001). When this experiment was repeated on a smaller scale, among lines carrying the susceptible ref(2)P allele, a mean of 96% of the offspring are infected, and for lines carrying the resistant allele, a mean of 23% were infected (Wilcoxon's signed-rank test U = 840, nsusceptible = 60, nresistant = 14, P < 0.0001). Among the genotypes carrying the ref(2)P resistance allele, there was no genetic variation in the transmission rate (log-likelihood ratio test: 2Δl = 0.15; d.f. = 1; P = 0.69).
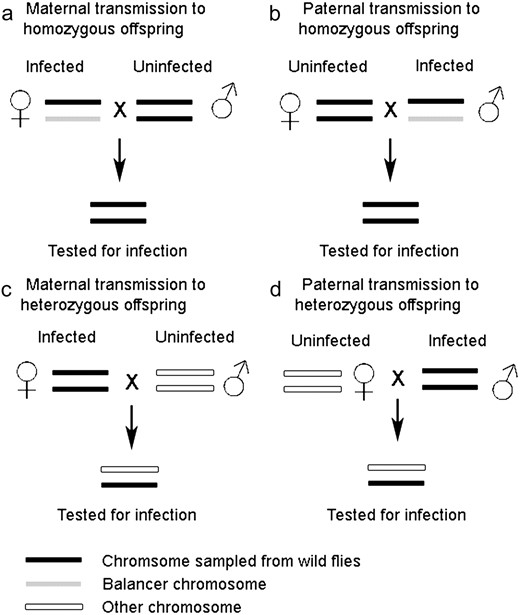
Schematic describing the four types of transmission experiment carried out.
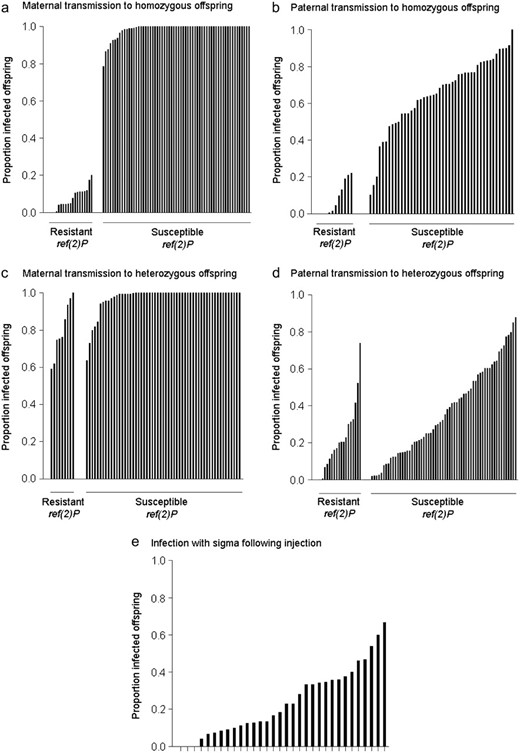
Sigma virus transmission rates across chromosome-extracted lines of D. melanogaster. (a–d) Showing mean proportion of infected offspring in the four transmission experiments in Figure 1. (a and d) Data from the “large” data sets; (b and c) data from the “small” data sets; (e) infection following injection with the virus.
Maternal transmission to heterozygous offspring:
The next experiment measured transmission from an infected mother that was homozygous for the wild-type second chromosome to offspring that were heterozygous for that chromosome (Figure 1C). Again ref(2)P had a significant effect on maternal transmission (Wilcoxon's signed-rank test U = 516, nsusceptible = 58, nresistant = 12, P = 0.003), but this time the effect was smaller: lines carrying the resistant ref(2)P allele had a mean transmission of 77% and lines carrying the susceptible allele had a mean transmission of 98% (Figure 3).
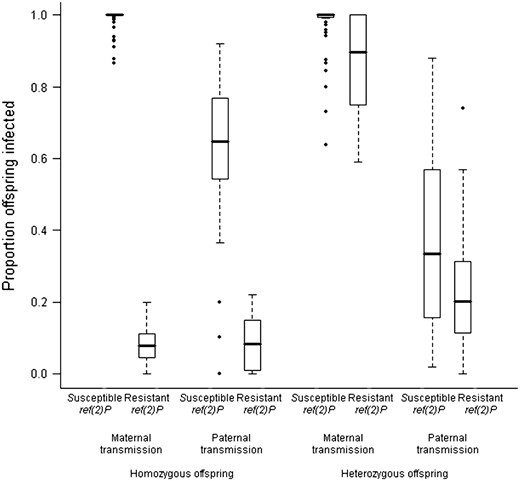
The effect of ref(2)P on viral transmission rates. The plot shows the proportion of infected offspring among lines with the resistant and susceptible ref(2)P alleles in each of the experiments. The horizontal line shows the median infection rate of the different genotypes, boxes show the interquartile range, and whiskers are 1.5 times the interquartile range. Genotypes outside this range are shown as outliers.
We are unable to conclude whether, for this trait, there is additional genetic variation over and above the effect of ref(2)P. An initial analysis of the genotypes carrying the ref(2)P resistance allele suggested there was (log-likelihood ratio test: 2Δl = 6.93; d.f. = 1; P = 0.009). However, there were very few independent replicate measurements on each of these genotypes, and when the analysis was repeated on a model that included the nonsignificant “replicate” term, there was no longer significant genetic variation among the lines (log-likelihood-ratio test: 2Δl = 2.24; d.f. = 1; P = 0.13).
Paternal transmission to homozygous offspring:
In the next two experiments we examined viral transmission through sperm rather than eggs. Again, we examined paternal transmission in two different contexts: transmission from a male heterozygous for the wild-type second chromosome, and transmission from a male homozygous for the wild-type second chromosome.
The first experiment measured transmission from an infected father heterozygous for the wild-type second chromosome to offspring that were homozygous for that chromosome (Figure 1B). The resistant ref(2)P allele was associated with a 59% drop in the transmission (F1,139 = 172.25; P < 0.0001). There was no additional effect of genotype over and above this (log-likelihood-ratio test: 2Δl = 0.31; d.f. = 1; P = 0.58). However, the lack of an effect of genotype may be due to a lack of power, as our sample size is small in this experiment (see methods). The genetic variance of 0.002 and wide 95% confidence intervals (0–0.077), encompasses the genetic variance of the homozygous paternal-transmission data set (Vg = 0.022) (Table 1).
Genetic variation in sigma virus infection and transmission rates
Trait | Parent | Offspring | ref(2)P | Vg (95% confidence interval) | CVg (%) |
|---|---|---|---|---|---|
| Paternal transmission | Heterozygous | Homozygous | In modela | 0.002 (0.000–0.077) | 8 |
| Not in model | 0.053 (0.032–0.088) | 44 | |||
| Paternal transmission | Homozygous | Heterozygous | In modela | 0.022 (0.015–0.032) | 50 |
| Not in model | 0.023 (0.016–0.033) | 50 | |||
| Infection following injection | Homozygous | — | Only susceptible lines | 0.009 (0.004–0.022) | 64 |
Trait | Parent | Offspring | ref(2)P | Vg (95% confidence interval) | CVg (%) |
|---|---|---|---|---|---|
| Paternal transmission | Heterozygous | Homozygous | In modela | 0.002 (0.000–0.077) | 8 |
| Not in model | 0.053 (0.032–0.088) | 44 | |||
| Paternal transmission | Homozygous | Heterozygous | In modela | 0.022 (0.015–0.032) | 50 |
| Not in model | 0.023 (0.016–0.033) | 50 | |||
| Infection following injection | Homozygous | — | Only susceptible lines | 0.009 (0.004–0.022) | 64 |
The presence or absence of the ref(2)P allele that confers resistance to the sigma virus was a fixed effect in the model and is therefore not contributing to the estimate of Vg.
Genetic variation in sigma virus infection and transmission rates
Trait | Parent | Offspring | ref(2)P | Vg (95% confidence interval) | CVg (%) |
|---|---|---|---|---|---|
| Paternal transmission | Heterozygous | Homozygous | In modela | 0.002 (0.000–0.077) | 8 |
| Not in model | 0.053 (0.032–0.088) | 44 | |||
| Paternal transmission | Homozygous | Heterozygous | In modela | 0.022 (0.015–0.032) | 50 |
| Not in model | 0.023 (0.016–0.033) | 50 | |||
| Infection following injection | Homozygous | — | Only susceptible lines | 0.009 (0.004–0.022) | 64 |
Trait | Parent | Offspring | ref(2)P | Vg (95% confidence interval) | CVg (%) |
|---|---|---|---|---|---|
| Paternal transmission | Heterozygous | Homozygous | In modela | 0.002 (0.000–0.077) | 8 |
| Not in model | 0.053 (0.032–0.088) | 44 | |||
| Paternal transmission | Homozygous | Heterozygous | In modela | 0.022 (0.015–0.032) | 50 |
| Not in model | 0.023 (0.016–0.033) | 50 | |||
| Infection following injection | Homozygous | — | Only susceptible lines | 0.009 (0.004–0.022) | 64 |
The presence or absence of the ref(2)P allele that confers resistance to the sigma virus was a fixed effect in the model and is therefore not contributing to the estimate of Vg.
Paternal transmission to heterozygous offspring:
This experiment measured transmission from an infected father homozygous for the wild-type second chromosome to offspring that were heterozygous for that chromosome (Figure 1D). In contrast to the corresponding test on maternal transmission, the resistant ref(2)P allele did not have a significant effect on the transmission of the virus (F1,848 = 3.10; P = 0.08). However, there was substantial variation among the different genotypes (Figure 2D; Table 1) (log-likelihood-ratio test: 2Δl = 48.94; d.f. = 1; P < 0.0001). As would be expected from these results, the ref(2)P genotype explains only 5% of this variation (Table 1), indicating that other genes on chromosome 2 affect this trait.
Because we repeated this experiment twice, we can examine how repeatable our results are. To do this we analyzed the data using a model with the term experiment included both as a fixed effect (to test for differences in the mean between the experiments) and as a random effect nested within genotype (to test for differences in the relative transmission rate of the different genotypes between the two experiments). Although the mean transmission rate is different for the two experiments (log-likelihood-ratio test: 2Δl = 8.86; d.f. = 1; P = 0.003), the relative transmission rate is not (log-likelihood-ratio test: 2Δl = 1.02; d.f. = 1; P = 0.31).
Variation in susceptibility to sigma virus infection following injection:
To examine whether the genes controlling paternal transmission have specific effects on transmission through sperm, or more general effects on viral susceptibility, we injected a subset of the lines with the virus and tested them with our CO2 assay 7 days later. Because the resistance mutation in ref(2)P is already known to affect this trait, we chose lines that all had the susceptible ref(2)P allele. Time of day injected (morning or afternoon), had no significant effect on variation in transmission (F2,115 = 0.300; P = 0.7412). We found that genetic variation significantly affected the proportion of flies infected with the virus 7 days after injection (Figure 2; log-likelihood-ratio test: 2Δl = 105.4; d.f. = 1; P < 0.0001). This trait had a coefficient of genetic variation of 64% (Table 1).
Are the same genes controlling paternal transmission and the onset of CO2 sensitivity after injection? It can be seen in Figure 4 that there is little correlation between the infection rate of each genotype in the two experiments. To see whether different genes are controlling the two traits, we tested whether there were significant differences in the slopes of the lines in Figure 4. First we fitted a fixed slope model, in which the random effect genotype alters the mean transmission in both experiments in the same way. This was then compared to a variable l slope model, which is analogous to adding a genotype × experiment-type interaction to the model (Pinheiro and Bates 2000). The variable l slope model was a significantly better fit to the data (likelihood-ratio test comparing models: 2Δl = 30.1; d.f. = 2; P < 0.0001). The variable l slope model estimates the genetic correlation (rg) between genotypes in the two experiments. There was a slightly negative genetic correlation between genotypes in the two experiments (rg = −0.17). Although the confidence limits on this estimate are large, it is clear that any genetic correlation between the two traits is low (95% confidence intervals of rg = −0.67–0.43). The lack of genetic correlation between the two experiments is also clear from a variance-components analysis on a more conventionally structured model. The between-line variance component (σL2 = 0.000, on arcsine transformed scale) was very small compared to the line × experiment-type interaction variance component (σL×E2 = 0.044, on an arcsine-transformed scale).
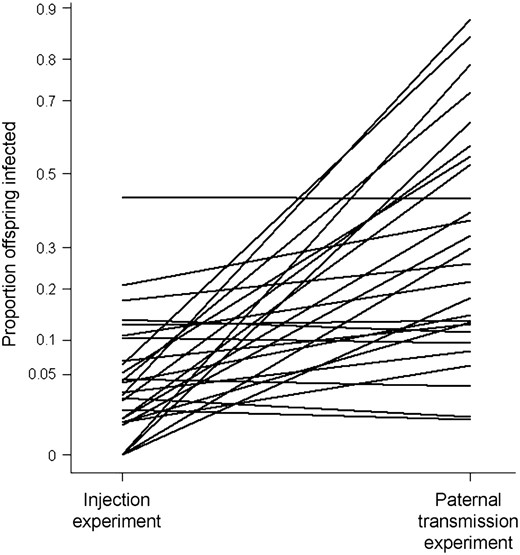
Variation in susceptibility to infection following injection and paternal transmission of the sigma virus. Showing the mean proportion of flies that were infected either by injection with the sigma virus or by vertical transmission through males. The lines join the same genotypes in the two different experiments. The proportion of infected flies is plotted on an arcsine scale.
DISCUSSION
We have found that genetic variation on chromosome 2 of D. melanogaster is associated with resistance to the sigma virus. Genetic variation affects the susceptibility of individuals injected with the virus and the transmission of the virus through both eggs and sperm. We have found that maternal transmission of the virus is strongly affected by the gene ref(2)P, whereas paternal transmission is affected by variation in both ref(2)P and other genes on the second chromosome.
The three traits we studied—infection rates following injection of the virus and transmission through eggs and sperm—are all affected by genetic variation at different loci. Maternal transmission of the virus is a simple Mendelian trait: all of the genetic variation is explained by a polymorphism in ref(2)P, a gene already well-known to affect resistance to sigma (Fleuriet 1988; Contamineet al. 1989; Druet al. 1993; Wayneet al. 1996; Banghamet al. 2007). It is possible that additional genes may cause variation among lines that carry the resistant allele of ref(2)P, but our experiments have little power to detect this. Male transmission too is affected by the ref(2)P resistance mutation, but much of the genetic variation is caused by other loci. Finally, the genetic correlation between the injection and male transmission data sets is low, indicating that they are controlled by different genes.
Why does some genetic variation affect only male, and not female, transmission of the sigma virus? One possibility might lie in differences between male and female gametes. Male gametes deliver smaller quantities of virus to offspring than females (Brun and Plus 1980) (probably because sperm are smaller than eggs). Therefore, genes that cause variation in viral titer could have a larger effect when the virus is transmitted through male gametes than through female gametes. However, if the genes blocking male transmission were simply slowing down viral replication in the gametes, then the same genes should also delay the rate at which flies become infected after injection; the injection assay is thought to primarily reflect rates of viral replication (although it may also reflect the rate that the nervous system becomes infected) (Brun and Plus 1980). This was not the case; we found a low genetic correlation between male-transmission and infection rates after injection. Therefore the genes that block male transmission are more likely to affect male-specific processes controlling the transmission of the virus into or with sperm. It is already known that in adult male flies there are specific barriers that prevent the virus from entering germ cells: if males are injected with the virus they become infected with normal viral titers but never transmit the virus to their offspring (Brun and Plus 1980). It is possible that the genes that cause variation in male transmission are affecting this process.
The resistant allele of ref(2)P reduces the replication rate of the virus (Brun and Plus 1980; Carre-Mloukaet al. 2007). We have found that it has a far greater effect on transmission rates when the offspring are homozygotes and parents heterozygotes than when the parents are homozygous and offspring heterozygous.
This pattern is seen regardless of whether the transmission occurs through eggs or sperm. Therefore, the ref(2)P polymorphism largely affects zygotic susceptibility to the sigma virus. The reasons for this are unclear, but it may simply be that there is only a very small viral titer in zygotes, so that genes that reduce viral replication can entirely cure flies of the infection at this stage. Previous studies have suggested that ref(2)P encodes a protein that functions in the Toll pathway (Avilaet al. 2002), which is an important immune pathway in D. melanogaster, although any possible anti-viral functions are still not clear.
The coefficient of genetic variation (CVg) is a useful statistic because it allows the amount of genetic variation to be compared across different traits. CVg provides an indication of a trait's “evolvability,” that is, the ability of a population to respond to natural or artificial selection (Houle 1992). Our estimates of CVg for male transmission and for infection rates after injection, were large (50 and 64%, respectively) (Table 1), relative to those described in previous studies in many different organisms and traits (Houle 1992). Although our study measured total (rather than additive) genetic variation and although we are studying only the effects of chromosome 2, it is clear that resistance to the sigma virus is a trait with unusually high levels of genetic variation. This indicates that these traits have the potential to respond to selection. Sigma virus is costly: sigma infection causes lower female fecundity and reduces overwintering survival (Fleuriet 1981), and its low prevalence, given its rate of transmission, means that it must carry a cost. This raises the question of what maintains this genetic variation in D. melanogaster populations. One possibility is that there is negative frequency-dependent selection resulting from coevolution between parasite and host.
In our experiments we used a panel of chromosome-substitution lines, where chromosomes sampled from the wild had been placed into the same genetic background. This approach is powerful and allowed us to easily infect the different lines with the sigma virus. However, it is important to bear in mind that such results might not always translate directly to average effects in the wild (MacDonald and Long 2004; Gruberet al. 2007). For example, the genetic background might affect the expression of resistance genes, and the genotype frequencies might not reflect frequencies found in the wild. In addition, in this study we have used only one viral isolate; further genetic variation in sigma virus in the wild will also influence the evolutionary dynamics of host resistance.
The D. melanogaster sigma virus system is an important model of host–parasite coevolution. However, although there is a large literature on genetic variation in resistance to sigma, these experiments have been almost exclusively designed in ways that detect only the effects of ref(2)P. Our data suggest that this is likely to have missed much of the genetic variation in natural populations that is caused by other genes. We have also found that some resistance genes block viral transmission, while others, including ref(2)P, block viral replication. This is likely to have important consequences for the evolution of virulence, analogous to the effects of different types of vaccines (Gandon 2002). Genes that block viral replication will tend to select for higher replication rates in the virus and will therefore increase the virus's virulence in susceptible hosts. In contrast, genes that specifically block paternal transmission will not have this effect.
Footnotes
Communicating editor: T. Long
Acknowledgement
Brian Lazzaro, Natasa Fytrou, and Jennifer Carpenter provided us with fly stocks. Mike Magwire and Ben Longdon helped us with experiments. Ian White and Sara Knott gave helpful advice on the statistics. This work was funded by the Royal Society, the Wellcome Trust, and the Biotechnology and Biological Sciences Research Council.
References
Bangham, J., D. J. Obbard, K. W. Kim and F. M. Jiggins,
Bell, G., and J. M. Smith,
Brun, P., and N. Plus,
Clancy, D. J., and W. J. Kennington,
Fleuriet, A.,
Fleuriet, A.,
Frank, S. A.,
Gandon, S.,
Henter, H. J.,
Hirschhorn, J. N., and M. J. Daly,
Pinheiro, J. C., and D. M. Bates,

{kind=link}
{kind=link}
{kind=link}
{kind=link}