





Abstract
Sustained increases in life expectancy have underscored the importance of managing diseases with a high incidence in late life, such as various neurodegenerative conditions. Alzheimer's disease (AD) is the most common among these, and consequently significant research effort is spent on studying it. Although a lot is known about the pathology of AD and the role of β-amyloid (Aβ) peptides, the complete network of interactions regulating Aβ metabolism and toxicity still eludes us. To address this, we have conducted genetic interaction screens using transgenic Drosophila expressing Aβ and we have identified mutations that affect Aβ metabolism and toxicity. These analyses highlight the involvement of various biochemical processes such as secretion, cholesterol homeostasis, and regulation of chromatin structure and function, among others, in mediating toxic Aβ effects. Several of the mutations that we identified have not been linked to Aβ toxicity before and thus constitute novel potential targets for AD intervention. We additionally tested these mutations for interactions with tau and expanded-polyglutamine overexpression and found a few candidate mutations that may mediate common mechanisms of neurodegeneration. Our data offer insight into the toxicity of Aβ and open new areas for further study into AD pathogenesis
ALZHEIMER'S disease, first described 100 years ago (reviewed in Goedert and Spillantini 2006), is a progressive neurodegenerative disease that results in gradual cognitive and behavioral changes and loss of memory (Selkoe and Podlisny 2002). Familial forms of Alzheimer's disease (AD) have been linked to mutations in the gene that encodes amyloid precursor protein (APP). Differential cleavage of APP leads to production of 40- or 42-amino-acid long amyloid peptides, designated as β-amyloid (Aβ)40 and Aβ42 (Gandy et al. 1992; Price et al. 1995). Several well-characterized familial APP missense mutations are clustered around the Aβ cleavage sites and cause an increase in either the total production of Aβ peptides or the Aβ42/Aβ40 peptide ratio. Although it is generally accepted that elevated levels of Aβ42 trigger the manifestation of disease symptoms, both peptides are found in amyloid plaques, which consist of aggregated amyloid and are one of the neuropathological hallmarks of Alzheimer's disease. However, Aβ42 is considered the more toxic form of β-amyloid, due to its hydrophobic nature and self-aggregation properties (Evin and Weidemann 2002). Recent research suggests that oligomerized, soluble Aβ42 is toxic to the cells and may also contribute to the manifestation of the disease (Walsh et al. 2002; Cleary et al. 2005). In addition to controversy regarding the toxicity of different secreted forms of Aβ, the functions of intracellular Aβ peptides and their contribution to disease manifestation are also unclear (reviewed in Golde et al. 2000). Overall, limitations in accurate detection and quantification of different pools of Aβ peptides have hindered our current understanding of mechanisms of toxicity that lead to AD.
Since production of Aβ peptides is a natural aspect of human physiology, regulation of the amounts of the peptide that are present in the human body concerns the balance of its rate of production vs. its rate of degradation. Consequently, a great amount of research effort has been devoted to the elucidation of mechanisms of Aβ metabolism. It is widely acknowledged that the proteolytic activities of β- and γ-secretases, which mediate production of Aβ from the APP precursor protein, can be modified by a variety of different pathways, including cholesterol biosynthesis and transport (reviewed in Vance et al. 2005). In addition, basal levels of neuronal activity may be responsible for the accumulation of extra amyloid in sporadic cases of Alzheimer's (Cirrito et al. 2005; reviewed in Selkoe 2006). On the basis of these studies, increased activity in brain areas performing “learned” tasks might provide protection against Alzheimer's. Upregulation of Aβ proteolysis by various enzymes is also an emerging field of potential intervention (reviewed in Eckman and Eckman 2005). Complementary to these efforts are studies of mechanisms of Aβ aggregation and the development of compounds that could inhibit it, as such compounds would potentially prevent and possibly reverse the toxic effects of aggregated peptides (reviewed in Bose et al. 2005).
To better understand the functions of Aβ42 in the cell and how these functions are altered when Aβ42 peptides are found in excess, we are using Drosophila melanogaster as a model system for analyzing the metabolism and toxic effects of Aβ through genetic screens. Drosophila has been used extensively as a model for neurodegenerative diseases, including Alzheimer's, Parkinson's and polyGlu diseases (reviewed in Cauchi and Van Den Heuvel 2006). Biochemical pathways that are affected in neurodegenerative conditions, such as detoxification, protein clearance, and immunological and stress responses, are conserved between flies and humans (reviewed in Marsh and Thompson 2006). Indeed, analysis of Drosophila models of Huntington's and other expanded polyglutamine-caused conditions (Tsuda et al. 2005; Bilen et al. 2006), as well as Parkinson's (Scherzer et al. 2003; Auluck et al. 2005; Kontopoulos et al. 2006) and tau-related diseases (Jackson et al. 2002; Mudher et al. 2004; Dias-Santagata et al. 2007; Fulga et al. 2007), has yielded important insight into pathways of neurodegeneration.
To study Aβ toxicity, we have expressed human Aβ40 and Aβ42 peptides carrying a signal sequence, in flies (Finelli et al. 2004). The APP protein and the γ-secretase activity are conserved in flies (Luo et al. 1990; Ye and Fortini 1998). However, due to lack of β-secretase activity (Fossgreen et al. 1998), to date there is no clear evidence that endogenous Aβ peptides are produced in Drosophila. In our model, neuronal expression of Aβ42, but not Aβ40, leads to phenotypes associated with neurodegeneration (Finelli et al. 2004; Iijima et al. 2004). These phenotypes are manifested in a dose- and age-dependent manner and correlate with progressive accumulation of increasingly higher levels of Aβ peptide. The progressive nature of Aβ effects, in combination with the short lifespan of flies, forms a useful system for the study of early events in the manifestation of Aβ toxicity. Indeed, the identification of such early events is important in achieving an earlier diagnosis, as well as in devising methods of earlier intervention. Complementary studies from other laboratories have also shown that expression of Aβ peptides carrying the “arctic” mutation (Nilsberth et al. 2001) also causes progressive degeneration in flies (Crowther et al. 2005). Similarly, in a triple transgenic Drosophila strain, carrying the APP, human BACE, and Drosophila presenilin genes, Aβ is produced in adequate amounts and causes aggregation and neurodegeneration (Greeve et al. 2004). These studies have established Drosophila as a suitable model for analyzing Aβ toxicity.
In our genetic interaction screens, described here, we identified Aβ phenotype-modifying mutations that participate in a number of different biochemical pathways. Although some of these candidate pathways have been previously linked to Aβ pathology, many of the specific genes that we identified have not been previously shown to have a role in AD. Notably, mutations in Drosophila homologs of carboxypeptidase D (CPD) and the δ-subunit of the AP3 adaptor complex modified Aβ phenotypes, implicating the secretory pathway in the mediation of Aβ effects. Additionally, cholesterol homeostasis and, unexpectedly, regulation of chromatin structure and function, also emerged as processes affected by Aβ overexpression. We have previously presented indirect evidence that in our Drosophila model the majority of Aβ peptides is secreted (Finelli et al. 2004). However, effects of intracellular Aβ cannot be excluded and the results from our genetic screen are highly suggestive of the importance of such effects. To evaluate the interaction of modifier mutations with Aβ peptides, we performed a detailed analysis of levels of Aβ in the presence of these mutations. In general, modifier mutations caused small changes in levels of total Aβ. In contrast, the relative levels of soluble vs. insoluble Aβ were in some cases significantly altered, suggesting that this effect is likely to be instrumental in determining the severity of Aβ toxicity. Finally, we also tested if the mutations that modified the Aβ phenotypes could also modify phenotypes caused by overexpression of tau and a polyGlu-expanded huntingtin (htt) gene fragment and discuss the existence of common pathways of neurodegeneration.
Our understanding of mechanisms of toxicity in Alzheimer's and neurodegeneration in general can be greatly aided by genetic analyses in model organisms. Our current work highlights several mutants with a role in altering Aβ42-overexpression phenotypes in Drosophila and provides a broad framework for further exploring novel pathways involved in metabolism and function of Aβ peptides as well as neurodegeneration in general.
MATERIALS AND METHODS
Drosophila strains, rearing, and phenotypic analysis:
A total of 1963 EP strains and additional candidate strains were obtained from the Bloomington (http://flystocks.bio.indiana.edu) and Szeged (http://expbio.bio.u-szeged.hu/fly/index.php) Drosophila Stock Centers. All flies were kept on yeast-containing media and were raised at 25° or 29°. Generation of the GMR-Aβ42 and UAS-Aβ42 flies is described in Finelli et al. (2004). UAS-tau flies are described in Wittmann et al. (2001) and UAS-Httex1Q93 flies are described in Steffan et al. (2001). For expression of UAS-tau we used the ey-Gal4 driver, since it gave a strong phenotype at 29° and did not cause nonspecific eye phenotypes on its own. For expression of UAS-Httex1Q93, we used the GMR-Gal4 driver, which was recombined in the UAS-Httex1Q93 flies that we obtained (Steffan et al. 2001). To eliminate the GMR-Gal4 nonspecific phenotype (caused by GMR-Gal4 at 29°), we raised these crosses at 25° and always ran a GMR-Gal4-only control in parallel. The actual genotypes of all strains are shown in the supplemental data. All flies were aged to ∼15–20 days old before being scored for eye phenotype.
Analysis of EP-induced mutations:
Using information available in FlyBase (Crosby et al. 2007), we identified the gene that is most likely affected by each of the EP insertions and the nature of the induced mutation (loss-of-function or gain-of-function). This information was confirmed by additional tests, as needed, which included reverse-transcription PCR of the candidate transcript, testing the insertion for effects on the Aβ phenotype in the absence of Gal4 activator, and testing additional mutations in the candidate gene, if available.
Protein analysis:
Protein extraction from fly heads and Western blot analysis were performed as described in Finelli et al. (2004). Where indicated, Western blot analysis was repeated two or three times with independent samples. Quantitation of Western blot analysis data was carried out using the ImageJ 1.36b tool (National Institutes of Health). Experimental samples were always compared to control samples of the same age. To account for differences in age of flies among different samples of the same genotype, we plotted the ratio of Aβ in experimental vs. control flies and grouped all measurements of a single genotype (Figure 2). We also evaluated the variability in levels of Aβ within samples carrying different mutations in the same gene. Using a two-sample T-test assuming equal variance, we carried out this evaluation for three genes, comparing two alleles for each gene [svrEP(X)0356 and svrKG02090, mubEP(3)3108 and mub04093, and EP(3)3603 and CG6767KG00420]. In all three cases, the variability we observed among the two alleles of the same gene was not statistically significant (data not shown).
For the ELISA analysis, one Drosophila head was homogenized in 10 μl ELISA buffer (Cell Lytic M; Sigma-Aldrich, St. Louis) containing 35% formic acid, after leaving it in the homogenization buffer for 20 min. The homogenate was boiled for 5 min and then centrifuged for 5 min at 10,000 rpm. A 3-μl aliquot was neutralized with 1 m Tris, pH 11, and combined with an equal volume of diluent (Sigma-Aldrich) and 1 mm serine protease inhibitor (serine protease inhibitor cocktail set 1; Calbiochem-EMD Chemicals, San Diego) in a general assay plate. Fifty microliters of each sample were loaded on an ELISA plate (Biosource-Invitrogen, Carlsbad, CA) and processed according to the manufacturer's instructions.
RNA analysis:
Total RNA was extracted from 100 Drosophila heads using the RNeasy mini kit (74104) from QIAGEN (Valencia, CA), according to the manufacturer's instructions. The quality of the RNA was tested on a 1% agarose gel before it was used for the PCR reaction. The first-strand cDNA was synthesized using random hexamer primers and the MultiScribe reverse transcriptase (Applied Biosystems, Foster City, CA). Thermal cycler incubations were 10 min at 25°, 30 min at 48°, and 5 min at 95°. For the real-time PCR reaction we used SYBR Green technology (Applied Biosystems), according to the manufacturer's instructions. The following program was used on an ABI Prism 7900HT SDS machine: 2 min at 50° (1 cycle); 10 min at 95° (1 cycle); and 15 sec at 95°, 1 min at 60°, 15 sec at 95° (40 cycles). The primers for amplifying the Aβ transgene were forward 5′-GAG ACT TTG CAT CTG GCT GCT A-3′ and reverse 5′-TGC GTC TGC CTG CAC TGT A-3′. The Drosophila rp49 RNA was amplified as an internal control. The specificity of the primers was tested by dissociation analysis. Three separate reactions were performed for each sample and were used to calculate the average and the standard deviation. The ratio of Aβ over rp49 RNA levels is shown.
RESULTS
Identification of 23 modifier genes of Aβ phenotypes:
For our genetic interaction screen we used flies expressing Aβ42 peptides specifically in their eyes, through the GMR transcriptional regulatory element (Hay et al. 1994). These flies develop a rough eye phenotype, the severity of which is dose and age dependent (Finelli et al. 2004), and we used this phenotype as the basis for identifying suppressors and enhancers. Modifiers were identified from screening the collection of EP insertion-carrying transgenic strains (Rorth 1996; Rorth et al. 1998). These mutant strains are engineered to utilize the yeast UAS/Gal4 system to achieve targeted misexpression of genes (Klueg et al. 2002). Since the EP insertions are directional, they may also cause loss-of-function mutations if inserted in a direction opposite to that of the transcription of the gene they are affecting. In our Aβ screen we activated the EP elements using the eyGal4 driver, which directs expression of the Gal4 transcriptional regulator in the eye (Sheng et al. 1997; Halder et al. 1998) and does not induce nonspecific phenotypes at high temperatures (our own observations). We independently confirmed expression of the eyGal4 transgene both anterior and posterior of the morphogenetic furrow in third instar larval imaginal discs, using a UAS-nGFP reporter transgene (data not shown). Expression patterns of GMR (directing Aβ42 peptide expression) and of eyGal4 (directing EP expression) are partially overlapping in the eye tissue, indicating that we could miss some mutations that might otherwise qualify as modifiers.
From screening 1963 EP strains, we initially identified 335 modifiers. We selected 102 modifiers that had the strongest effects and screened them a second time, further selecting 23 modifiers that had reproducible effects. We also confirmed that all selected EP strains did not induce a rough eye phenotype, when coexpressed with eyGal4, in the absence of Aβ42 (data not shown). To identify which genes were responsible for the modification of the Aβ42 phenotype, we used mapping information of the EP insertions available in FlyBase (Crosby et al. 2007). In cases where the insertion might be affecting two genes, we examined whether modification of the Aβ42 phenotype was dependent on coexpression of the Gal4 protein. We supplemented this analysis by examining the RNA levels of selected candidate mutant genes by real-time PCR (rtPCR) (Table 1). In addition, we obtained and tested additional mutations in candidate genes, where available. These analyses are summarized in Table 1 and Figure 1. Below we discuss modifiers for which the identity of the affected gene is confirmed by one or more of the above assays.
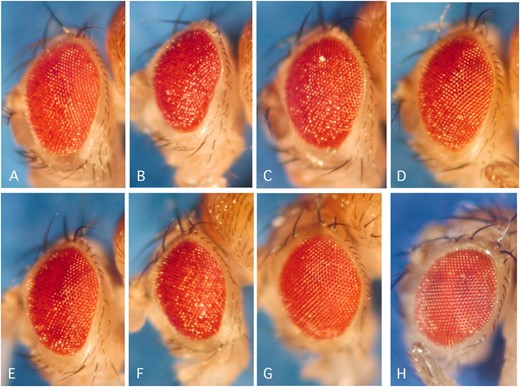
Light stereomicrographs of transgenic Drosophila eyes coexpressing Aβ42 and selected modifier mutations. (A) Aβ42-only expressing flies. (B–F) Enhancers of the Aβ42 eye phenotype. (G–H) Suppressors of the Aβ42 eye phenotype. Shown are flies coexpressing Aβ42 and Dsp1KG06700 (B), ATP7EY07895 (C), svrEP(X)0356 (D), SNFγKG10152 (E), CG6767KG00420 (F), EP(3)3603 (G), or EP(3)3348 (H).
Modifiers of Aβ phenotypes
Mutation | Direction of UASa | Predicted type of mutation | Candidate gene rtPCR | Gene name/function | Biological process | Human ortholog | Aβ eyeb |
|---|---|---|---|---|---|---|---|
| Dsp1EP(X)0355 | Same | Gofc/Gal dependent | NTd | Dsp1 corepressor | Chromatin assembly | HMG-B1 | Ee (89) |
| svrEP(X)0356 | Same | Loff/3′ exon | NT | Silver (CypD) | Secretory pathway | Carboxypeptidase D | E (59) |
| svrKG02090 | — | Lof | NT | E (61) | |||
| svr126 | — | Lof | NT | E (58) | |||
| gEP(X)0514 | Opposite | Lof/Gal independent | NT | garnet | Lysosomal transport | AP-3 subunit delta-1 | Sg (132) |
| gB166 | — | Lof | NT | S (14) | |||
| EP(X)1318 | — | — | — | Not mapped | — | — | S (33) |
| EP(X)1504 | — | Gal independent | NT | Not mapped | — | — | E (96) |
| EP(X)1596 (CG2924) | Opposite | Lof/Gal independent | NT | Ubiquitin-conjugating enzyme | Ubiquitin/proteolysis | Ubiquitin-conjugating enzyme E2Q | S (35) |
| CG2924EY02142 | Same | Unclear | NT | NEh (227) | |||
| EP(X)0308 | — | Gal dependent | — | ATP7 or HSP20 | Cu transport or protein folding | ATP7 or CML antigen 66 | S (71) |
| ATP7EY07895 | Same | Gof | 2.8-fold | ATP7 | Cu transport | Copper-transporting ATPase 1/2 | E (220) |
| EP(2)0684esg | Same | Gof/Gal dependent | NT | Escargot/transcriptional repressor | CNS development | Snail homolog 2 | E (62) |
| EP(2)0330 | No flanking sequence | Gal independent | — | Not mapped | — | — | E (68) |
| MESR4EP(2)0386 | Same | Gof/Gal dependent | NT | MESR4 misexpression suppressor of Ras | Chromatin regulation | PELP1 human isoform 2 | E (143) |
| elBEP(2)0965 | Same | Inconclusive | Multiple transcripts | Elbow (elbB) | Zn-finger/cell cycle—eye | Zn-finger protein 503 | E (21) |
| elB9 | — | Lof | NT | E (177) | |||
| EP(2)2510 (CG7231) | Opposite | Lof | NT | CG7231 | Not known | NM205548 novel protein | E |
| EP(3)0595b | Same | Two genes | CG6745 or CG6765 | FGF signaling | Pseudouridil. synthase7/no ortholog for CG6765 | E (224) | |
| EP(3)1051 | Same | Gof/Gal dependent | 4.6-fold | Toll | Immune response | Toll-like receptor 4 | E |
| SnfγEP(3)3015b | Same/3′ end | Lof? | Multiple transcripts | Lochrig/AMPγ | Energy metabolism/ cholesterol homeostasis | AMP kinase γ-subunit | E (59) |
| SnfγKG10152 | — | Lof | NT | E (111) | |||
| EP(3)3041 | Same | Gof/Gal dependent | NT | Unknown/mir282 | – | — | E (137) |
| EP(3)3099 | — | Gal dependent | NT | RhoBTB (lof) or IDE (gof) | Signal transduction or insulin degrading enzyme | Small GTPase or IDE | E (144) |
| mubEP(3)3108 | Opposite | Lof/Gal independent | NT | Mub/mushroom body | Regulation of translation | Poly(rC)-binding protein 3 | E (159) |
| mub04093 | — | Lof?(intron) | NT | NE (34) | |||
| EP(3)3348 | — | Two genes | — | SAP130 or ATG1 | Transcription regulation or autophagy | mSIN3A-associated protein or ATG1 | S (234) |
| SAP130EY12079 | Same/coding | Lof | 0.5-fold | SAP130 | Chromatin transciption regulation | mSIN3A-associated protein | E (142) |
| Rpd304556 | — | Lof | NT | Rpd3 | Chromatin transciption regulation | HDAC1 | E (56) |
| HDAC4KG09091 | — | Lof | NT | HDAC4 | Chromatin transciption regulation | HDAC4 | E (23) |
| Sin3A08269 | — | Lof | NT | Sin3A | Chromatin transcription regulation | Transcription corepressor Sin3b | E (76) |
| EP(3)3405 | Same | Gof/Gal dependent | NT | CG6175 | Unknown function | No ortholog | S (27) |
| EP(3)3470 | Same (3.6 kb) | Gof/Gal dependent | NT | Stich | Transcription regulation | hairy/enhancer of split related | E (59) |
| nep2EP(3)3549 | Same | Gof/Gal dependent | 6.5-fold | nep2 | Proteolysis | Neutral endopeptidase | S (130) |
| nep2KG05754 | — | Lof | NT | E (22) | |||
| EP(3)3603 | Opposite | Lof/Gal independent | NT | CG6767 or UbcD4 | — | — | S (233) |
| CG6767KG00420 | — | Lof | NT | CG6767 | Glial cell differentiation | Ribosyl-pyrophosphosynthetase | E (24) |
| UbcD4EY05497 | Opposite | Lof | NT | UbcD4 | Ubiquitination | NE (129) |
Mutation | Direction of UASa | Predicted type of mutation | Candidate gene rtPCR | Gene name/function | Biological process | Human ortholog | Aβ eyeb |
|---|---|---|---|---|---|---|---|
| Dsp1EP(X)0355 | Same | Gofc/Gal dependent | NTd | Dsp1 corepressor | Chromatin assembly | HMG-B1 | Ee (89) |
| svrEP(X)0356 | Same | Loff/3′ exon | NT | Silver (CypD) | Secretory pathway | Carboxypeptidase D | E (59) |
| svrKG02090 | — | Lof | NT | E (61) | |||
| svr126 | — | Lof | NT | E (58) | |||
| gEP(X)0514 | Opposite | Lof/Gal independent | NT | garnet | Lysosomal transport | AP-3 subunit delta-1 | Sg (132) |
| gB166 | — | Lof | NT | S (14) | |||
| EP(X)1318 | — | — | — | Not mapped | — | — | S (33) |
| EP(X)1504 | — | Gal independent | NT | Not mapped | — | — | E (96) |
| EP(X)1596 (CG2924) | Opposite | Lof/Gal independent | NT | Ubiquitin-conjugating enzyme | Ubiquitin/proteolysis | Ubiquitin-conjugating enzyme E2Q | S (35) |
| CG2924EY02142 | Same | Unclear | NT | NEh (227) | |||
| EP(X)0308 | — | Gal dependent | — | ATP7 or HSP20 | Cu transport or protein folding | ATP7 or CML antigen 66 | S (71) |
| ATP7EY07895 | Same | Gof | 2.8-fold | ATP7 | Cu transport | Copper-transporting ATPase 1/2 | E (220) |
| EP(2)0684esg | Same | Gof/Gal dependent | NT | Escargot/transcriptional repressor | CNS development | Snail homolog 2 | E (62) |
| EP(2)0330 | No flanking sequence | Gal independent | — | Not mapped | — | — | E (68) |
| MESR4EP(2)0386 | Same | Gof/Gal dependent | NT | MESR4 misexpression suppressor of Ras | Chromatin regulation | PELP1 human isoform 2 | E (143) |
| elBEP(2)0965 | Same | Inconclusive | Multiple transcripts | Elbow (elbB) | Zn-finger/cell cycle—eye | Zn-finger protein 503 | E (21) |
| elB9 | — | Lof | NT | E (177) | |||
| EP(2)2510 (CG7231) | Opposite | Lof | NT | CG7231 | Not known | NM205548 novel protein | E |
| EP(3)0595b | Same | Two genes | CG6745 or CG6765 | FGF signaling | Pseudouridil. synthase7/no ortholog for CG6765 | E (224) | |
| EP(3)1051 | Same | Gof/Gal dependent | 4.6-fold | Toll | Immune response | Toll-like receptor 4 | E |
| SnfγEP(3)3015b | Same/3′ end | Lof? | Multiple transcripts | Lochrig/AMPγ | Energy metabolism/ cholesterol homeostasis | AMP kinase γ-subunit | E (59) |
| SnfγKG10152 | — | Lof | NT | E (111) | |||
| EP(3)3041 | Same | Gof/Gal dependent | NT | Unknown/mir282 | – | — | E (137) |
| EP(3)3099 | — | Gal dependent | NT | RhoBTB (lof) or IDE (gof) | Signal transduction or insulin degrading enzyme | Small GTPase or IDE | E (144) |
| mubEP(3)3108 | Opposite | Lof/Gal independent | NT | Mub/mushroom body | Regulation of translation | Poly(rC)-binding protein 3 | E (159) |
| mub04093 | — | Lof?(intron) | NT | NE (34) | |||
| EP(3)3348 | — | Two genes | — | SAP130 or ATG1 | Transcription regulation or autophagy | mSIN3A-associated protein or ATG1 | S (234) |
| SAP130EY12079 | Same/coding | Lof | 0.5-fold | SAP130 | Chromatin transciption regulation | mSIN3A-associated protein | E (142) |
| Rpd304556 | — | Lof | NT | Rpd3 | Chromatin transciption regulation | HDAC1 | E (56) |
| HDAC4KG09091 | — | Lof | NT | HDAC4 | Chromatin transciption regulation | HDAC4 | E (23) |
| Sin3A08269 | — | Lof | NT | Sin3A | Chromatin transcription regulation | Transcription corepressor Sin3b | E (76) |
| EP(3)3405 | Same | Gof/Gal dependent | NT | CG6175 | Unknown function | No ortholog | S (27) |
| EP(3)3470 | Same (3.6 kb) | Gof/Gal dependent | NT | Stich | Transcription regulation | hairy/enhancer of split related | E (59) |
| nep2EP(3)3549 | Same | Gof/Gal dependent | 6.5-fold | nep2 | Proteolysis | Neutral endopeptidase | S (130) |
| nep2KG05754 | — | Lof | NT | E (22) | |||
| EP(3)3603 | Opposite | Lof/Gal independent | NT | CG6767 or UbcD4 | — | — | S (233) |
| CG6767KG00420 | — | Lof | NT | CG6767 | Glial cell differentiation | Ribosyl-pyrophosphosynthetase | E (24) |
| UbcD4EY05497 | Opposite | Lof | NT | UbcD4 | Ubiquitination | NE (129) |
The 23 EP insertions identified in the original genetic screen are underlined.
Direction of UAS element relative to transcription of candidate gene.
The number of experimental flies screened is in parentheses.
Gain-of-function mutation.
Not tested.
Enhancer.
Loss-of-function mutation.
Suppressor.
No effect.
Modifiers of Aβ phenotypes
Mutation | Direction of UASa | Predicted type of mutation | Candidate gene rtPCR | Gene name/function | Biological process | Human ortholog | Aβ eyeb |
|---|---|---|---|---|---|---|---|
| Dsp1EP(X)0355 | Same | Gofc/Gal dependent | NTd | Dsp1 corepressor | Chromatin assembly | HMG-B1 | Ee (89) |
| svrEP(X)0356 | Same | Loff/3′ exon | NT | Silver (CypD) | Secretory pathway | Carboxypeptidase D | E (59) |
| svrKG02090 | — | Lof | NT | E (61) | |||
| svr126 | — | Lof | NT | E (58) | |||
| gEP(X)0514 | Opposite | Lof/Gal independent | NT | garnet | Lysosomal transport | AP-3 subunit delta-1 | Sg (132) |
| gB166 | — | Lof | NT | S (14) | |||
| EP(X)1318 | — | — | — | Not mapped | — | — | S (33) |
| EP(X)1504 | — | Gal independent | NT | Not mapped | — | — | E (96) |
| EP(X)1596 (CG2924) | Opposite | Lof/Gal independent | NT | Ubiquitin-conjugating enzyme | Ubiquitin/proteolysis | Ubiquitin-conjugating enzyme E2Q | S (35) |
| CG2924EY02142 | Same | Unclear | NT | NEh (227) | |||
| EP(X)0308 | — | Gal dependent | — | ATP7 or HSP20 | Cu transport or protein folding | ATP7 or CML antigen 66 | S (71) |
| ATP7EY07895 | Same | Gof | 2.8-fold | ATP7 | Cu transport | Copper-transporting ATPase 1/2 | E (220) |
| EP(2)0684esg | Same | Gof/Gal dependent | NT | Escargot/transcriptional repressor | CNS development | Snail homolog 2 | E (62) |
| EP(2)0330 | No flanking sequence | Gal independent | — | Not mapped | — | — | E (68) |
| MESR4EP(2)0386 | Same | Gof/Gal dependent | NT | MESR4 misexpression suppressor of Ras | Chromatin regulation | PELP1 human isoform 2 | E (143) |
| elBEP(2)0965 | Same | Inconclusive | Multiple transcripts | Elbow (elbB) | Zn-finger/cell cycle—eye | Zn-finger protein 503 | E (21) |
| elB9 | — | Lof | NT | E (177) | |||
| EP(2)2510 (CG7231) | Opposite | Lof | NT | CG7231 | Not known | NM205548 novel protein | E |
| EP(3)0595b | Same | Two genes | CG6745 or CG6765 | FGF signaling | Pseudouridil. synthase7/no ortholog for CG6765 | E (224) | |
| EP(3)1051 | Same | Gof/Gal dependent | 4.6-fold | Toll | Immune response | Toll-like receptor 4 | E |
| SnfγEP(3)3015b | Same/3′ end | Lof? | Multiple transcripts | Lochrig/AMPγ | Energy metabolism/ cholesterol homeostasis | AMP kinase γ-subunit | E (59) |
| SnfγKG10152 | — | Lof | NT | E (111) | |||
| EP(3)3041 | Same | Gof/Gal dependent | NT | Unknown/mir282 | – | — | E (137) |
| EP(3)3099 | — | Gal dependent | NT | RhoBTB (lof) or IDE (gof) | Signal transduction or insulin degrading enzyme | Small GTPase or IDE | E (144) |
| mubEP(3)3108 | Opposite | Lof/Gal independent | NT | Mub/mushroom body | Regulation of translation | Poly(rC)-binding protein 3 | E (159) |
| mub04093 | — | Lof?(intron) | NT | NE (34) | |||
| EP(3)3348 | — | Two genes | — | SAP130 or ATG1 | Transcription regulation or autophagy | mSIN3A-associated protein or ATG1 | S (234) |
| SAP130EY12079 | Same/coding | Lof | 0.5-fold | SAP130 | Chromatin transciption regulation | mSIN3A-associated protein | E (142) |
| Rpd304556 | — | Lof | NT | Rpd3 | Chromatin transciption regulation | HDAC1 | E (56) |
| HDAC4KG09091 | — | Lof | NT | HDAC4 | Chromatin transciption regulation | HDAC4 | E (23) |
| Sin3A08269 | — | Lof | NT | Sin3A | Chromatin transcription regulation | Transcription corepressor Sin3b | E (76) |
| EP(3)3405 | Same | Gof/Gal dependent | NT | CG6175 | Unknown function | No ortholog | S (27) |
| EP(3)3470 | Same (3.6 kb) | Gof/Gal dependent | NT | Stich | Transcription regulation | hairy/enhancer of split related | E (59) |
| nep2EP(3)3549 | Same | Gof/Gal dependent | 6.5-fold | nep2 | Proteolysis | Neutral endopeptidase | S (130) |
| nep2KG05754 | — | Lof | NT | E (22) | |||
| EP(3)3603 | Opposite | Lof/Gal independent | NT | CG6767 or UbcD4 | — | — | S (233) |
| CG6767KG00420 | — | Lof | NT | CG6767 | Glial cell differentiation | Ribosyl-pyrophosphosynthetase | E (24) |
| UbcD4EY05497 | Opposite | Lof | NT | UbcD4 | Ubiquitination | NE (129) |
Mutation | Direction of UASa | Predicted type of mutation | Candidate gene rtPCR | Gene name/function | Biological process | Human ortholog | Aβ eyeb |
|---|---|---|---|---|---|---|---|
| Dsp1EP(X)0355 | Same | Gofc/Gal dependent | NTd | Dsp1 corepressor | Chromatin assembly | HMG-B1 | Ee (89) |
| svrEP(X)0356 | Same | Loff/3′ exon | NT | Silver (CypD) | Secretory pathway | Carboxypeptidase D | E (59) |
| svrKG02090 | — | Lof | NT | E (61) | |||
| svr126 | — | Lof | NT | E (58) | |||
| gEP(X)0514 | Opposite | Lof/Gal independent | NT | garnet | Lysosomal transport | AP-3 subunit delta-1 | Sg (132) |
| gB166 | — | Lof | NT | S (14) | |||
| EP(X)1318 | — | — | — | Not mapped | — | — | S (33) |
| EP(X)1504 | — | Gal independent | NT | Not mapped | — | — | E (96) |
| EP(X)1596 (CG2924) | Opposite | Lof/Gal independent | NT | Ubiquitin-conjugating enzyme | Ubiquitin/proteolysis | Ubiquitin-conjugating enzyme E2Q | S (35) |
| CG2924EY02142 | Same | Unclear | NT | NEh (227) | |||
| EP(X)0308 | — | Gal dependent | — | ATP7 or HSP20 | Cu transport or protein folding | ATP7 or CML antigen 66 | S (71) |
| ATP7EY07895 | Same | Gof | 2.8-fold | ATP7 | Cu transport | Copper-transporting ATPase 1/2 | E (220) |
| EP(2)0684esg | Same | Gof/Gal dependent | NT | Escargot/transcriptional repressor | CNS development | Snail homolog 2 | E (62) |
| EP(2)0330 | No flanking sequence | Gal independent | — | Not mapped | — | — | E (68) |
| MESR4EP(2)0386 | Same | Gof/Gal dependent | NT | MESR4 misexpression suppressor of Ras | Chromatin regulation | PELP1 human isoform 2 | E (143) |
| elBEP(2)0965 | Same | Inconclusive | Multiple transcripts | Elbow (elbB) | Zn-finger/cell cycle—eye | Zn-finger protein 503 | E (21) |
| elB9 | — | Lof | NT | E (177) | |||
| EP(2)2510 (CG7231) | Opposite | Lof | NT | CG7231 | Not known | NM205548 novel protein | E |
| EP(3)0595b | Same | Two genes | CG6745 or CG6765 | FGF signaling | Pseudouridil. synthase7/no ortholog for CG6765 | E (224) | |
| EP(3)1051 | Same | Gof/Gal dependent | 4.6-fold | Toll | Immune response | Toll-like receptor 4 | E |
| SnfγEP(3)3015b | Same/3′ end | Lof? | Multiple transcripts | Lochrig/AMPγ | Energy metabolism/ cholesterol homeostasis | AMP kinase γ-subunit | E (59) |
| SnfγKG10152 | — | Lof | NT | E (111) | |||
| EP(3)3041 | Same | Gof/Gal dependent | NT | Unknown/mir282 | – | — | E (137) |
| EP(3)3099 | — | Gal dependent | NT | RhoBTB (lof) or IDE (gof) | Signal transduction or insulin degrading enzyme | Small GTPase or IDE | E (144) |
| mubEP(3)3108 | Opposite | Lof/Gal independent | NT | Mub/mushroom body | Regulation of translation | Poly(rC)-binding protein 3 | E (159) |
| mub04093 | — | Lof?(intron) | NT | NE (34) | |||
| EP(3)3348 | — | Two genes | — | SAP130 or ATG1 | Transcription regulation or autophagy | mSIN3A-associated protein or ATG1 | S (234) |
| SAP130EY12079 | Same/coding | Lof | 0.5-fold | SAP130 | Chromatin transciption regulation | mSIN3A-associated protein | E (142) |
| Rpd304556 | — | Lof | NT | Rpd3 | Chromatin transciption regulation | HDAC1 | E (56) |
| HDAC4KG09091 | — | Lof | NT | HDAC4 | Chromatin transciption regulation | HDAC4 | E (23) |
| Sin3A08269 | — | Lof | NT | Sin3A | Chromatin transcription regulation | Transcription corepressor Sin3b | E (76) |
| EP(3)3405 | Same | Gof/Gal dependent | NT | CG6175 | Unknown function | No ortholog | S (27) |
| EP(3)3470 | Same (3.6 kb) | Gof/Gal dependent | NT | Stich | Transcription regulation | hairy/enhancer of split related | E (59) |
| nep2EP(3)3549 | Same | Gof/Gal dependent | 6.5-fold | nep2 | Proteolysis | Neutral endopeptidase | S (130) |
| nep2KG05754 | — | Lof | NT | E (22) | |||
| EP(3)3603 | Opposite | Lof/Gal independent | NT | CG6767 or UbcD4 | — | — | S (233) |
| CG6767KG00420 | — | Lof | NT | CG6767 | Glial cell differentiation | Ribosyl-pyrophosphosynthetase | E (24) |
| UbcD4EY05497 | Opposite | Lof | NT | UbcD4 | Ubiquitination | NE (129) |
The 23 EP insertions identified in the original genetic screen are underlined.
Direction of UAS element relative to transcription of candidate gene.
The number of experimental flies screened is in parentheses.
Gain-of-function mutation.
Not tested.
Enhancer.
Loss-of-function mutation.
Suppressor.
No effect.
Modifiers with a role in the secretory pathway:
Our screen identified the gene silver as a modifier of Aβ-associated toxicity. Silver encodes a Drosophila CPD homolog whose function has been implicated in pigment and wing formation, catecholamine metabolism, and mating behavior of flies in the light. Carboxypeptidase D is the only member of this gene family containing multiple carboxypeptidase domains and it has been proposed that this special structure increases the range of pH at which it can be active, ensuring activity throughout the secretory pathway (Novikova et al. 1999). The Drosophila silver protein contains three carboxypeptidase domains, a transmembrane domain, and a cytosolic tail (Sidyelyeva and Fricker 2002). We found that three independent loss-of-function mutations in the silver gene act as enhancers of Aβ-induced phenotypes. Processing of proteins in the secretory pathway is associated with their ability to assume a proper conformation and an altered conformation might change their solubility properties, leading us to hypothesize that a putative effect of CPD on Aβ might be associated with the ability of Aβ to oligomerize. Changes in the folding/aggregation state of Aβ might lead to a higher concentration of oligomers and thus to enhanced toxicity. It is worth noting that the C′-terminal amino acid of Aβ42 is Ala, found in the penultimate position of substrates for CPD (Novikova et al. 1999).
Mutations in the garnet gene were also found to modify the Aβ phenotype. Garnet is a Drosophila homolog of the δ-subunit of the AP3 adaptor complex that functions in synaptic vesicle biogenesis and vesicular transport (Boehm and Bonifacino 2002). Mutations in AP3 subunits in flies cause pigmentation phenotypes and abnormalities in pigment granule (Simpson et al. 1997). The similarity of pigment granules and lysosomes has led to the suggestion that the AP3 complex may be involved in the trafficking of proteins to the lysosomal compartment (reviewed in Boehm and Bonifacino 2002). In addition, it has been shown that mutations in AP3 components in mice cause neurological defects and it was proposed, although not directly shown, that brain-specific subunit isoforms may exist (Blumstein et al. 2001). We have found that two putative loss-of-function mutations in the garnet locus are suppressing the Aβ eye phenotype (Table 1). Although an interaction between Aβ and δ-adaptin has not been previously shown, it is possible that Aβ is sorted to the lysosomal compartment through the actions of a particular AP3 complex. Loss of function of this AP3 complex could lead to mislocalization of Aβ in the cell and could alter its toxic effects. Alternatively, loss of AP3 function might affect a downstream mediator of Aβ toxicity.
Interaction of Aβ with the cholesterol homeostasis pathway:
Mutations in the gene loechrig (loe/Snfg) were also shown to modify the Aβ eye phenotype (Table 1). Loe is a homolog of the AMP kinase γ (AMPKγ) subunit, a gene involved in energy control. In response to ATP depletion, AMPK gets activated and it negatively regulates synthesis of cholesterol, in addition to causing long-term effects on gene and protein expression (reviewed in Carling 2005). Mutant analysis of the gene loe in flies has shown that it can cause neurodegenerative phenotypes by interfering with cholesterol homeostasis (Tschape et al. 2002). Although flies do not synthesize cholesterol de novo, but rather utilize cholesterol that they ingest with their food, it was shown that loe interacts directly with the fly homolog of HMG-CoA reductase and that loe mutants cause lowering of the amounts of cholesterol ester present in fly tissue (Tschape et al. 2002). Several studies have examined the role of cholesterol in the processing of APP and the production of Aβ peptides (reviewed in Sjogren et al. 2006). More recently, a direct interaction between cholesterol and Aβ has been reported, linking the functions of Cu and cholesterol, both of which have been previously implicated in the pathology of Alzheimer's disease. Puglielli et al. (2005) have proposed that the high-affinity binding of Aβ to Cu2+ catalyzes the production of H2O2 from O2 and cholesterol, suggesting that cholesterol oxidation could be contributing to the pathology of AD. In our studies, we have shown that loss-of-function mutations in loe are enhancing the Aβ phenotype. Reduced AMPK function in these mutants is likely to affect the levels of available cholesterol. More free available cholesterol could result in increased production of H2O2, through putative oxidative functions of Aβ, causing an increase in neurodegenerative effects. Interestingly, a putative mutation in a Cu2+ transporter was also identified as an Aβ modifier (ATP7; Table 1). Since the interaction of Aβ with Cu2+ is proposed to mediate oxidative functions, further genetic analysis of ATP7 and loe double mutants in the presence of Aβ will confirm and add to our understanding of this model of action.
Effects of regulators of chromatin structure and function:
The insertion EP(3)3348 lies in the vicinity of two genes, the autophagy gene Atg1 and the Sin3A-Associated Protein (SAP130) gene. Presence of this insertion caused a strong suppression of the Aβ-induced rough eye phenotype. Although autophagy genes are also involved in modification of the Aβ phenotype (H.-J. Song, unpublished data), we showed that additional mutations in the SAP130 gene had an effect on the Aβ phenotype (Table 1), implicating processes of chromatin regulation and assembly in mediating Aβ toxicity. SAP130 is a member of the Sin3A complex, which regulates transcription and chromatin remodeling through interactions with transcription factors (reviewed in Silverstein and Ekwall 2005). The Sin3A complex is composed of DNA-binding proteins, histone modification enzymes, and other adaptor proteins and it is involved in a wide range of biological activities, including the cell cycle, DNA replication, apoptosis, and modifications of chromatin (Dannenberg et al. 2005). The histone deacetylases HDAC1 and HDAC4 are known components of the Sin3A complex. To test the involvement of other components of the complex in mediating Aβ toxicity, we tested mutations in the genes Rpd3 (Rpd304556) and HDAC4 (HDAC4KG09091), which are direct homologs of the mammalian HDAC1 and HDAC4 genes, respectively, and found that they were also enhancers of the Aβ phenotype (Table 1). A mutation in the Drosophila Sin3A gene also enhanced the Aβ phenotype, suggesting that chromatin-remodeling mechanisms are implicated in regulating Aβ effects.
In addition to components of the Sin3A complex, mutations in five other genes that are known to be involved in transcriptional regulation could modify the Aβ phenotype (Table 1). Interestingly, some of these genes have been shown to control division and differentiation of neurons. The gene escargot encodes a Zn-containing transcription factor, which is involved in the regulation of asymmetric divisions of neural progenitors (Chia and Yang 2002). In addition, escargot has been shown to regulate endoreplication (Fuse et al. 1994) and, through a distinct mechanism, cell-cycle progression in the eye (Tseng and Hariharan 2002). A second gene, elbow, acts as a regulator of cell-cycle events in the eye (Tseng and Hariharan 2002).
Other proteins with functions in the nervous system:
We found that a mutation in the gene mub can modify Aβ phenotypes. This mutation [EP(3)3108] is most likely causing loss of mub function. An additional loss-of-function mutation (mub04093) was tested but was found to have no effect on the Aβ phenotype. This could be explained by the fact that mub04093 is inserted in an intron of the mub gene and might be causing a less severe effect. Mub contains a poly(rC)-binding motif and nuclear localization signal and it has been proposed to be involved in alternative splicing (Park et al. 2004). Mub is specifically expressed in the mushroom body, the center for learning and memory in flies (Grams and Korge 1998). The targets of Mub are currently unknown but our finding suggests that manipulation of RNA processing might constitute a process that could affect Aβ toxicity.
The mutations EP(X)1318, EP(X)1504, EP(2)0330, EP(3)3041, and EP(3)3405 were also identified as Aβ42 modifiers. These mutations are not mapped on the Drosophila genome or affect genes with unknown functions.
Levels of Aβ42 peptides in the presence of modifier mutations:
We examined the levels of total and soluble Aβ in flies expressing the peptide in the presence of modifying mutations and compared them to age-matched control extracts without the modifying mutations (Table 2). Soluble Aβ was analyzed in groups of fly heads, by Western blot analysis, whereas total Aβ was analyzed in individual fly heads, using a quantitative ELISA assay. We found that eight mutations caused statistically significant changes in levels of total Aβ peptide (Figure 2). In seven of these cases, however, levels of Aβ were varied only by 20–30%. In only one case, the neprilysin-upregulating mutation EP(3)3549nep2 caused a 70% lowering in the levels of total Aβ (Table 2 and Finelli et al. 2004).
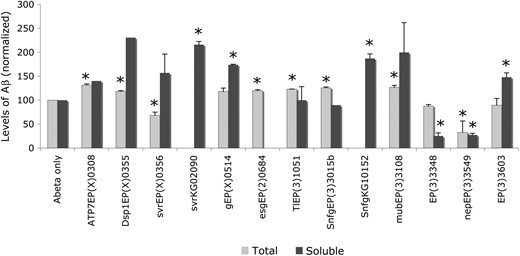
Comparison of total and soluble levels of Aβ42 peptides in transgenic flies. Measurements are normalized to the levels of Aβ42 in control flies (Aβ only). Genotypes indicate the mutation that is coexpressed with Aβ42 in each strain. Standard deviation was calculated in cases where at least three experiments of the same genotype were available. Asterisks denote statistically significant changes compared to the respective controls. We considered values with P ≤ 0.05 in measurements of soluble Aβ and P ≤ 0.005 in measurements of total Aβ.
Aβ peptide levels
Mutation | Aβ eye phenotype | Soluble Aβ | P-value (soluble) | Total Aβ | P-value (total) | Effect |
|---|---|---|---|---|---|---|
| Aβ/+ | 100 | — | 576.5 ± 22.9 | — | ||
| EP(X)0308 | S | 140 | — | 757.3 ± 18.3 | 0.001 | Total 30% up |
| ATP7EY07895 | E | 130 ± 15.5 | 0.22 | NTa | — | No change |
| Dsp1EP(X)0355 | E | 231 | — | 683.3 ± 9.3 | 0.005 | Total 30% up |
| Dsp1KG06700 | E | 157 ± 106 | 0.59 | NT | — | No change |
| svrEP(X)0356 | E | 157 ± 39.6 | 0.29 | 395.3 ± 39.3 | 0.004 | Total 30% down |
| svrKG02090 | E | 216 ± 7 | 0.03 | NT | — | Soluble 100% up |
| gEP(X)0514 | S | 174 ± 1.4 | 0.01 | 682.3 ± 42.3 | 0.011 | Soluble 75% up |
| CG2924EP(X)1596 | S | 171 | — | 662.8 ± 55.7 | 0.034 | No change |
| EP(2)0684esg | E | NT | — | 692.0 ± 12.3 | 0.003 | Total 20% up |
| EP(2)0386 | E | 200 | — | 521.2 ± 84.8 | 0.221 | No change |
| Elb9 | E | 134 | — | NT | — | — |
| CG7231EP(2)2510 | E | 110 ± 28.3 | 0.70 | NT | — | No change |
| EP(3)0595b | E | 119.7 ± 13.5 | 0.13 | 563.5 ± 96.4 | 0.796 | No change |
| EP(3)1051 | E | 100 ± 28.3 | 1.00 | 708.2 ± 4.1 | 0.002 | Total 23% up |
| SnfγEP(3)3015b | E | 90 | — | 726.6 ± 10.4 | 0.001 | Total 26% up |
| SNFγKG10152 | E | 187.5 ± 9.2 | 0.05 | NT | — | Soluble 87% up |
| EP(3)3041 | E | 83 ± 4.2 | 0.11 | NT | — | No change |
| EP(3)3099 | E | 185.5 ± 40.3 | 0.20 | 530.2 ± 60.1 | 0.192 | No change |
| mubEP(3)3108 | E | 200 ± 62.2 | 0.26 | 733.7 ± 23.2 | 0.001 | Total 27% up |
| mub04093 | NE | 123 ± 12.7 | 0.24 | NT | — | No change |
| EP(3)3348 | S | 25.5 ± 6.3 | 0.04 | 503.9 ± 19.7 | 0.027 | Soluble 75% down |
| SAP130EY10279 | E | 257 ± 39.6 | NT | — | Soluble 150% up | |
| CG2924EY02142 | NE | 123.3 ± 43 | 0.45 | NT | — | No change |
| EP(3)3405 | S | 83.5 ± 36 | 0.63 | NT | — | No change |
| EP(3)3470 | E | 63.5 ± 17.6 | 0.21 | 600.3 ± 19.3 | 0.244 | No change |
| nep2EP(3)3549 | S | 27.5 ± 3.5 | 0.02 | 190.1 ± 135.7 | 0.001 | Total/soluble 70% down |
| EP(3)3603 | S | 148 ± 9.1 | 0.01 | 516.4 ± 83.6 | 0.184 | Soluble 50% up |
| CG6767KG00420 | E | 138 ± 29.8 | 0.16 | NT | — | No change |
Mutation | Aβ eye phenotype | Soluble Aβ | P-value (soluble) | Total Aβ | P-value (total) | Effect |
|---|---|---|---|---|---|---|
| Aβ/+ | 100 | — | 576.5 ± 22.9 | — | ||
| EP(X)0308 | S | 140 | — | 757.3 ± 18.3 | 0.001 | Total 30% up |
| ATP7EY07895 | E | 130 ± 15.5 | 0.22 | NTa | — | No change |
| Dsp1EP(X)0355 | E | 231 | — | 683.3 ± 9.3 | 0.005 | Total 30% up |
| Dsp1KG06700 | E | 157 ± 106 | 0.59 | NT | — | No change |
| svrEP(X)0356 | E | 157 ± 39.6 | 0.29 | 395.3 ± 39.3 | 0.004 | Total 30% down |
| svrKG02090 | E | 216 ± 7 | 0.03 | NT | — | Soluble 100% up |
| gEP(X)0514 | S | 174 ± 1.4 | 0.01 | 682.3 ± 42.3 | 0.011 | Soluble 75% up |
| CG2924EP(X)1596 | S | 171 | — | 662.8 ± 55.7 | 0.034 | No change |
| EP(2)0684esg | E | NT | — | 692.0 ± 12.3 | 0.003 | Total 20% up |
| EP(2)0386 | E | 200 | — | 521.2 ± 84.8 | 0.221 | No change |
| Elb9 | E | 134 | — | NT | — | — |
| CG7231EP(2)2510 | E | 110 ± 28.3 | 0.70 | NT | — | No change |
| EP(3)0595b | E | 119.7 ± 13.5 | 0.13 | 563.5 ± 96.4 | 0.796 | No change |
| EP(3)1051 | E | 100 ± 28.3 | 1.00 | 708.2 ± 4.1 | 0.002 | Total 23% up |
| SnfγEP(3)3015b | E | 90 | — | 726.6 ± 10.4 | 0.001 | Total 26% up |
| SNFγKG10152 | E | 187.5 ± 9.2 | 0.05 | NT | — | Soluble 87% up |
| EP(3)3041 | E | 83 ± 4.2 | 0.11 | NT | — | No change |
| EP(3)3099 | E | 185.5 ± 40.3 | 0.20 | 530.2 ± 60.1 | 0.192 | No change |
| mubEP(3)3108 | E | 200 ± 62.2 | 0.26 | 733.7 ± 23.2 | 0.001 | Total 27% up |
| mub04093 | NE | 123 ± 12.7 | 0.24 | NT | — | No change |
| EP(3)3348 | S | 25.5 ± 6.3 | 0.04 | 503.9 ± 19.7 | 0.027 | Soluble 75% down |
| SAP130EY10279 | E | 257 ± 39.6 | NT | — | Soluble 150% up | |
| CG2924EY02142 | NE | 123.3 ± 43 | 0.45 | NT | — | No change |
| EP(3)3405 | S | 83.5 ± 36 | 0.63 | NT | — | No change |
| EP(3)3470 | E | 63.5 ± 17.6 | 0.21 | 600.3 ± 19.3 | 0.244 | No change |
| nep2EP(3)3549 | S | 27.5 ± 3.5 | 0.02 | 190.1 ± 135.7 | 0.001 | Total/soluble 70% down |
| EP(3)3603 | S | 148 ± 9.1 | 0.01 | 516.4 ± 83.6 | 0.184 | Soluble 50% up |
| CG6767KG00420 | E | 138 ± 29.8 | 0.16 | NT | — | No change |
See Table 1 footnotes for definitions of abbreviations.
Not tested.
Aβ peptide levels
Mutation | Aβ eye phenotype | Soluble Aβ | P-value (soluble) | Total Aβ | P-value (total) | Effect |
|---|---|---|---|---|---|---|
| Aβ/+ | 100 | — | 576.5 ± 22.9 | — | ||
| EP(X)0308 | S | 140 | — | 757.3 ± 18.3 | 0.001 | Total 30% up |
| ATP7EY07895 | E | 130 ± 15.5 | 0.22 | NTa | — | No change |
| Dsp1EP(X)0355 | E | 231 | — | 683.3 ± 9.3 | 0.005 | Total 30% up |
| Dsp1KG06700 | E | 157 ± 106 | 0.59 | NT | — | No change |
| svrEP(X)0356 | E | 157 ± 39.6 | 0.29 | 395.3 ± 39.3 | 0.004 | Total 30% down |
| svrKG02090 | E | 216 ± 7 | 0.03 | NT | — | Soluble 100% up |
| gEP(X)0514 | S | 174 ± 1.4 | 0.01 | 682.3 ± 42.3 | 0.011 | Soluble 75% up |
| CG2924EP(X)1596 | S | 171 | — | 662.8 ± 55.7 | 0.034 | No change |
| EP(2)0684esg | E | NT | — | 692.0 ± 12.3 | 0.003 | Total 20% up |
| EP(2)0386 | E | 200 | — | 521.2 ± 84.8 | 0.221 | No change |
| Elb9 | E | 134 | — | NT | — | — |
| CG7231EP(2)2510 | E | 110 ± 28.3 | 0.70 | NT | — | No change |
| EP(3)0595b | E | 119.7 ± 13.5 | 0.13 | 563.5 ± 96.4 | 0.796 | No change |
| EP(3)1051 | E | 100 ± 28.3 | 1.00 | 708.2 ± 4.1 | 0.002 | Total 23% up |
| SnfγEP(3)3015b | E | 90 | — | 726.6 ± 10.4 | 0.001 | Total 26% up |
| SNFγKG10152 | E | 187.5 ± 9.2 | 0.05 | NT | — | Soluble 87% up |
| EP(3)3041 | E | 83 ± 4.2 | 0.11 | NT | — | No change |
| EP(3)3099 | E | 185.5 ± 40.3 | 0.20 | 530.2 ± 60.1 | 0.192 | No change |
| mubEP(3)3108 | E | 200 ± 62.2 | 0.26 | 733.7 ± 23.2 | 0.001 | Total 27% up |
| mub04093 | NE | 123 ± 12.7 | 0.24 | NT | — | No change |
| EP(3)3348 | S | 25.5 ± 6.3 | 0.04 | 503.9 ± 19.7 | 0.027 | Soluble 75% down |
| SAP130EY10279 | E | 257 ± 39.6 | NT | — | Soluble 150% up | |
| CG2924EY02142 | NE | 123.3 ± 43 | 0.45 | NT | — | No change |
| EP(3)3405 | S | 83.5 ± 36 | 0.63 | NT | — | No change |
| EP(3)3470 | E | 63.5 ± 17.6 | 0.21 | 600.3 ± 19.3 | 0.244 | No change |
| nep2EP(3)3549 | S | 27.5 ± 3.5 | 0.02 | 190.1 ± 135.7 | 0.001 | Total/soluble 70% down |
| EP(3)3603 | S | 148 ± 9.1 | 0.01 | 516.4 ± 83.6 | 0.184 | Soluble 50% up |
| CG6767KG00420 | E | 138 ± 29.8 | 0.16 | NT | — | No change |
Mutation | Aβ eye phenotype | Soluble Aβ | P-value (soluble) | Total Aβ | P-value (total) | Effect |
|---|---|---|---|---|---|---|
| Aβ/+ | 100 | — | 576.5 ± 22.9 | — | ||
| EP(X)0308 | S | 140 | — | 757.3 ± 18.3 | 0.001 | Total 30% up |
| ATP7EY07895 | E | 130 ± 15.5 | 0.22 | NTa | — | No change |
| Dsp1EP(X)0355 | E | 231 | — | 683.3 ± 9.3 | 0.005 | Total 30% up |
| Dsp1KG06700 | E | 157 ± 106 | 0.59 | NT | — | No change |
| svrEP(X)0356 | E | 157 ± 39.6 | 0.29 | 395.3 ± 39.3 | 0.004 | Total 30% down |
| svrKG02090 | E | 216 ± 7 | 0.03 | NT | — | Soluble 100% up |
| gEP(X)0514 | S | 174 ± 1.4 | 0.01 | 682.3 ± 42.3 | 0.011 | Soluble 75% up |
| CG2924EP(X)1596 | S | 171 | — | 662.8 ± 55.7 | 0.034 | No change |
| EP(2)0684esg | E | NT | — | 692.0 ± 12.3 | 0.003 | Total 20% up |
| EP(2)0386 | E | 200 | — | 521.2 ± 84.8 | 0.221 | No change |
| Elb9 | E | 134 | — | NT | — | — |
| CG7231EP(2)2510 | E | 110 ± 28.3 | 0.70 | NT | — | No change |
| EP(3)0595b | E | 119.7 ± 13.5 | 0.13 | 563.5 ± 96.4 | 0.796 | No change |
| EP(3)1051 | E | 100 ± 28.3 | 1.00 | 708.2 ± 4.1 | 0.002 | Total 23% up |
| SnfγEP(3)3015b | E | 90 | — | 726.6 ± 10.4 | 0.001 | Total 26% up |
| SNFγKG10152 | E | 187.5 ± 9.2 | 0.05 | NT | — | Soluble 87% up |
| EP(3)3041 | E | 83 ± 4.2 | 0.11 | NT | — | No change |
| EP(3)3099 | E | 185.5 ± 40.3 | 0.20 | 530.2 ± 60.1 | 0.192 | No change |
| mubEP(3)3108 | E | 200 ± 62.2 | 0.26 | 733.7 ± 23.2 | 0.001 | Total 27% up |
| mub04093 | NE | 123 ± 12.7 | 0.24 | NT | — | No change |
| EP(3)3348 | S | 25.5 ± 6.3 | 0.04 | 503.9 ± 19.7 | 0.027 | Soluble 75% down |
| SAP130EY10279 | E | 257 ± 39.6 | NT | — | Soluble 150% up | |
| CG2924EY02142 | NE | 123.3 ± 43 | 0.45 | NT | — | No change |
| EP(3)3405 | S | 83.5 ± 36 | 0.63 | NT | — | No change |
| EP(3)3470 | E | 63.5 ± 17.6 | 0.21 | 600.3 ± 19.3 | 0.244 | No change |
| nep2EP(3)3549 | S | 27.5 ± 3.5 | 0.02 | 190.1 ± 135.7 | 0.001 | Total/soluble 70% down |
| EP(3)3603 | S | 148 ± 9.1 | 0.01 | 516.4 ± 83.6 | 0.184 | Soluble 50% up |
| CG6767KG00420 | E | 138 ± 29.8 | 0.16 | NT | — | No change |
See Table 1 footnotes for definitions of abbreviations.
Not tested.
The mutations svrKG02090 and SnfγKG10152 both increased soluble Aβ (by 100 and 87%, respectively), whereas EP(3)3348 caused a 75% decrease. The mutations in the svr and Snfγ genes enhanced the Aβ phenotype and the mutation EP(3)3348 suppressed the Aβ phenotype, suggesting that these genes may affect the Aβ phenotype by changing the solubility of the peptide. High levels of soluble Aβ may also be responsible for toxicity, possibly by mechanisms distinct from those mediating toxicity of aggregated Aβ. Two additional mutations caused notable changes in the levels of soluble Aβ peptides; however, in these cases the levels of soluble Aβ were inversely related to the phenotype. EP(X)0514g, which suppressed the Aβ phenotype, caused a 75% increase in levels of soluble Aβ. The mutation EP(3)3603 increases levels of soluble Aβ by 50% and also suppresses the Aβ phenotype. It is possible that differential subcellular localization of the peptide affects the Aβ phenotype in these cases, although additional experimentation would be required to substantiate this claim.
Several mutants of the Sin3A/Sap130/HDAC complex were analyzed for their effects on Aβ levels as well (Figure 3). Loss-of-function mutations in Sin3A, Rpd3, HDAC4, and SAP130 caused significant accumulation of soluble Aβ peptides (Figure 3A). Since Aβ RNA was not changed in the presence of SAP130 overexpression (Figure 3B), we propose that the Sin3A complex may be regulating the turnover of Aβ peptides. In support of this, it was recently shown that HDAC6 can suppress neurodegeneration caused by protein misfolding through regulation of autophagy (Pandey et al. 2007). Since several of the genes that we identified are transcriptional factors, we examined also the levels of Aβ RNA in the presence of transcriptional regulator mutations. We found that in three of five transcriptional regulators, levels of Aβ RNA were unchanged (supplemental Figure S2). In two other cases, although the Aβ RNA was increased, the total and/or soluble level of Aβ was unaffected (supplemental Figure S2 and Table 2). This suggests that the effect of these genes on Aβ toxicity is not at the level of transgene expression but rather on the metabolism or toxicity of the Aβ peptide itself.
![Effects of Sin3A-complex mutants on levels of Aβ. (A) Levels of soluble Aβ peptides were normalized relative to levels in control flies (Aβ only). Coexpression of Aβ42 with loss-of-function mutations in Sin3A, Rpd3, HDAC4, and SAP130 results in increased levels of soluble Aβ. Coexpression of a putative gain-of-function mutation in SAP130 [EP(3)3348] results in decreased levels of Aβ. (B) Levels of Aβ42 RNA, measured by quantitative PCR, in control flies (Aβ only) and in flies coexpressing a putative gain-of-function mutation in SAP130 [EP(3)3348]. The presence of the EP(3)3348 mutation does not cause a statistically significant change in levels of Aβ42 RNA.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/genetics/178/3/10.1534_genetics.107.078394/6/m_1457fig3.jpeg?Expires=1716416217&Signature=p37qI4ZiHyW2N7538imv3O7zULhFW8qnTTrAjHKgH4HnU42-zwrT-uLi5tIG5-s2UoQ-U8XASNlCBFBi7mLTJyTGLUd1steSFcqvDafqjI41jiY4pkws4aL~F-8HO9MbgDE8zJdni6ONVJwYkRMIU9xAVsLRFvdi0EIPB-GVIk-pznbpz36ffbg5foD-iWun0Et9gQTLwx-7UnE-Et3Ml35YVjLSZW3gNpoTE9xs~lC2MBFnd-mwyKIClhg6aW5Av3wVsLrSNW-GHsyYrc7eV9hoyLgA62fqYkMIjGX7b4Dwzh0tAnj6-~TUr8V~evJl1IPjTUQWfCGQUsIMEBiudg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Effects of Sin3A-complex mutants on levels of Aβ. (A) Levels of soluble Aβ peptides were normalized relative to levels in control flies (Aβ only). Coexpression of Aβ42 with loss-of-function mutations in Sin3A, Rpd3, HDAC4, and SAP130 results in increased levels of soluble Aβ. Coexpression of a putative gain-of-function mutation in SAP130 [EP(3)3348] results in decreased levels of Aβ. (B) Levels of Aβ42 RNA, measured by quantitative PCR, in control flies (Aβ only) and in flies coexpressing a putative gain-of-function mutation in SAP130 [EP(3)3348]. The presence of the EP(3)3348 mutation does not cause a statistically significant change in levels of Aβ42 RNA.
Effects of Aβ modifiers on tau- and expanded-polyglutamine-induced phenotypes:
We examined whether the EP modifiers of Aβ42 phenotypes might also alter phenotypes caused by overexpression of tau or a fragment of the huntingtin (htt) gene carrying an expanded-polyGlu tract. Drosophila modeling tau-induced toxicity express ectopic tau in the eye and develop a rough eye phenotype (Wittmann et al. 2001), which in extreme cases results in reduction of the eye tissue (Figure 4C). Drosophila modeling toxicity induced by expanded polyglutamine express the exon 1 fragment of the huntingtin (Htt) protein (Steffan et al. 2001). This fragment contains a repeat of 93 glutamines (httex1Q93) and when expressed in flies causes a rough eye phenotype (Steffan et al. 2001), accompanied by a “glassy” appearance (Figure 4D). We found that ∼50% of the EP mutations could modify the tau phenotype and 74% could modify the httex1Q93 phenotype (Table 3 and Figure 4, E–T). The effect of a particular mutant is not always consistent in the two models. The insertions EP(X)0514, EP(X)1596, EP(X)0308, EP(3)3348, and EP(3)3603 suppressed the eye phenotype in all three transgenic models. Below we review putative models regarding how these mutations might be involved in tau- and polyGlu-induced toxicity.
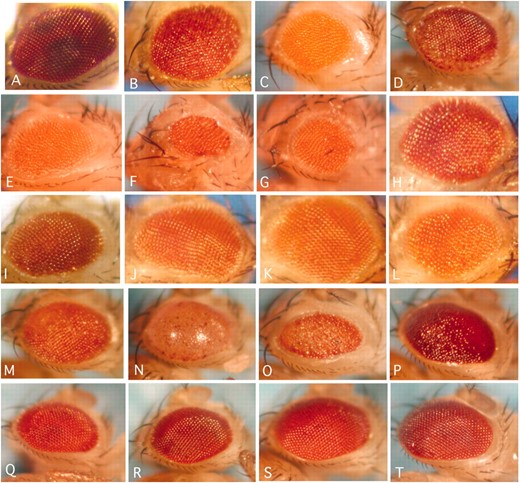
Modifiers of the tau- and httex1Q93-induced eye phenotypes. (A–D) Comparison of eye phenotypes caused by expression of different disease proteins. (A) Wild-type flies, (B) flies expressing Aβ42, (C) flies expressing tau, (D) flies expressing httex1Q93. (F and G) Enhancers of the tau phenotype. (H–L) Suppressors of the tau phenotype. Shown are flies expressing only tau (E) or coexpressing tau and EP(X)1318 (F), EP(3)3470 (G), mubEP(3)3108 (H), EP(3)3405 (I), svrEP(X)0356 (J), EP(3)3348 (K), or EP(X)0308 (L). (N–P) Enhancers of the httex1Q93 phenotype. (Q–T) Suppressors of the httex1Q93 phenotype. Shown are flies expressing only httex1Q93 (M) or coexpressing httex1Q93 and Dsp1EP(X)0355 (N), EP(3)595b (O), SNFγEP(3)3015b (P), EP(X)0308 (Q), EP(2)0330 (R), MESR4EP(2)0386 (S), or CG2924EP(X)1596 (T).
Effect of Aβ modifiers on tau and httex1Q93
Mutation | tau eye | Q93 eye | Aβ eye | Aβ life | Gene/function |
|---|---|---|---|---|---|
| Dsp1EP(X)0355 | La | Eb (102) | E | E | Dsp1 |
| svrEP(X)0356 | Sc (186) | S (72) | E | E | silver |
| gEP(X)0514 | S (12) | S (70) | S | NEd | garnet |
| EP(X)1318 | E (12) | NE (49) | S | L | Unknown |
| EP(X)1504 | S (19) | NE (37) | E | E | Unknown |
| EP(X)1596 | S (41) | S (99) | S | E | NICE5(Ubc) or CG2918 (stress response) or CG3071 (vesicular transport) |
| CG2924EY02142 | NT | NE (62) | NE (227) | NT | |
| EP(X)0308 | S (38) | S (189) | S | E | ATP7 or HSP20 |
| ATP7EY07895 | NT | S (40) | E (220) | NT | ATP7 |
| EP(2)0684esg | L | L | E | L | escargot |
| EP(2)0330 | S (60) | S (90) | E | E | Unknown |
| MESR4EP(2)0386 | S (50) | S (53) | E | NE | MESR4 |
| elBEP(2)0965 | NT | NT | E | E | elbB |
| CG7231EP(2)2510 | NT | NT | E | L | Unknown |
| EP(3)0595b | S (44) | E (25) | E | E | FGF signal or CG6745 (unknown) |
| EP(3)1051 | L | E (53) | E | E | Toll |
| SnfγEP(3)3015b | L | E (22) | E | NE | Snfγ |
| EP(3)3041 | L | NE (98) | E | E | Unknown |
| EP(3)3099 | L | S (94) | E | E | RhoBTB or IDE |
| mubEP(3)3108 | S (116) | S (83) | E | E | mub |
| EP(3)3348 | S (125) | S (116) | S | E | SAP130 or ATG1 |
| SAP13006924 | NT | NE (88) | NE | NT | SAP130 |
| EP(3)3405 | S (14) | NE (50) | S | E | Unknown |
| EP(3)3470 | L | NE (54) | E | E | CG17100 or Rfx |
| nep2EP(3)3549 | L | NE (64) | S | S | nep2 |
| EP(3)3603 | S (17) | S (23) | S | E | Glial cell differentiation or UbcD4 |
| UbcD4EY05497 | NT | S (118) | NE (129) | NT | UbcD4 |
Mutation | tau eye | Q93 eye | Aβ eye | Aβ life | Gene/function |
|---|---|---|---|---|---|
| Dsp1EP(X)0355 | La | Eb (102) | E | E | Dsp1 |
| svrEP(X)0356 | Sc (186) | S (72) | E | E | silver |
| gEP(X)0514 | S (12) | S (70) | S | NEd | garnet |
| EP(X)1318 | E (12) | NE (49) | S | L | Unknown |
| EP(X)1504 | S (19) | NE (37) | E | E | Unknown |
| EP(X)1596 | S (41) | S (99) | S | E | NICE5(Ubc) or CG2918 (stress response) or CG3071 (vesicular transport) |
| CG2924EY02142 | NT | NE (62) | NE (227) | NT | |
| EP(X)0308 | S (38) | S (189) | S | E | ATP7 or HSP20 |
| ATP7EY07895 | NT | S (40) | E (220) | NT | ATP7 |
| EP(2)0684esg | L | L | E | L | escargot |
| EP(2)0330 | S (60) | S (90) | E | E | Unknown |
| MESR4EP(2)0386 | S (50) | S (53) | E | NE | MESR4 |
| elBEP(2)0965 | NT | NT | E | E | elbB |
| CG7231EP(2)2510 | NT | NT | E | L | Unknown |
| EP(3)0595b | S (44) | E (25) | E | E | FGF signal or CG6745 (unknown) |
| EP(3)1051 | L | E (53) | E | E | Toll |
| SnfγEP(3)3015b | L | E (22) | E | NE | Snfγ |
| EP(3)3041 | L | NE (98) | E | E | Unknown |
| EP(3)3099 | L | S (94) | E | E | RhoBTB or IDE |
| mubEP(3)3108 | S (116) | S (83) | E | E | mub |
| EP(3)3348 | S (125) | S (116) | S | E | SAP130 or ATG1 |
| SAP13006924 | NT | NE (88) | NE | NT | SAP130 |
| EP(3)3405 | S (14) | NE (50) | S | E | Unknown |
| EP(3)3470 | L | NE (54) | E | E | CG17100 or Rfx |
| nep2EP(3)3549 | L | NE (64) | S | S | nep2 |
| EP(3)3603 | S (17) | S (23) | S | E | Glial cell differentiation or UbcD4 |
| UbcD4EY05497 | NT | S (118) | NE (129) | NT | UbcD4 |
The number of flies screened is shown in parentheses.
Lethal.
Enhancer.
Suppressor.
No effect.
Effect of Aβ modifiers on tau and httex1Q93
Mutation | tau eye | Q93 eye | Aβ eye | Aβ life | Gene/function |
|---|---|---|---|---|---|
| Dsp1EP(X)0355 | La | Eb (102) | E | E | Dsp1 |
| svrEP(X)0356 | Sc (186) | S (72) | E | E | silver |
| gEP(X)0514 | S (12) | S (70) | S | NEd | garnet |
| EP(X)1318 | E (12) | NE (49) | S | L | Unknown |
| EP(X)1504 | S (19) | NE (37) | E | E | Unknown |
| EP(X)1596 | S (41) | S (99) | S | E | NICE5(Ubc) or CG2918 (stress response) or CG3071 (vesicular transport) |
| CG2924EY02142 | NT | NE (62) | NE (227) | NT | |
| EP(X)0308 | S (38) | S (189) | S | E | ATP7 or HSP20 |
| ATP7EY07895 | NT | S (40) | E (220) | NT | ATP7 |
| EP(2)0684esg | L | L | E | L | escargot |
| EP(2)0330 | S (60) | S (90) | E | E | Unknown |
| MESR4EP(2)0386 | S (50) | S (53) | E | NE | MESR4 |
| elBEP(2)0965 | NT | NT | E | E | elbB |
| CG7231EP(2)2510 | NT | NT | E | L | Unknown |
| EP(3)0595b | S (44) | E (25) | E | E | FGF signal or CG6745 (unknown) |
| EP(3)1051 | L | E (53) | E | E | Toll |
| SnfγEP(3)3015b | L | E (22) | E | NE | Snfγ |
| EP(3)3041 | L | NE (98) | E | E | Unknown |
| EP(3)3099 | L | S (94) | E | E | RhoBTB or IDE |
| mubEP(3)3108 | S (116) | S (83) | E | E | mub |
| EP(3)3348 | S (125) | S (116) | S | E | SAP130 or ATG1 |
| SAP13006924 | NT | NE (88) | NE | NT | SAP130 |
| EP(3)3405 | S (14) | NE (50) | S | E | Unknown |
| EP(3)3470 | L | NE (54) | E | E | CG17100 or Rfx |
| nep2EP(3)3549 | L | NE (64) | S | S | nep2 |
| EP(3)3603 | S (17) | S (23) | S | E | Glial cell differentiation or UbcD4 |
| UbcD4EY05497 | NT | S (118) | NE (129) | NT | UbcD4 |
Mutation | tau eye | Q93 eye | Aβ eye | Aβ life | Gene/function |
|---|---|---|---|---|---|
| Dsp1EP(X)0355 | La | Eb (102) | E | E | Dsp1 |
| svrEP(X)0356 | Sc (186) | S (72) | E | E | silver |
| gEP(X)0514 | S (12) | S (70) | S | NEd | garnet |
| EP(X)1318 | E (12) | NE (49) | S | L | Unknown |
| EP(X)1504 | S (19) | NE (37) | E | E | Unknown |
| EP(X)1596 | S (41) | S (99) | S | E | NICE5(Ubc) or CG2918 (stress response) or CG3071 (vesicular transport) |
| CG2924EY02142 | NT | NE (62) | NE (227) | NT | |
| EP(X)0308 | S (38) | S (189) | S | E | ATP7 or HSP20 |
| ATP7EY07895 | NT | S (40) | E (220) | NT | ATP7 |
| EP(2)0684esg | L | L | E | L | escargot |
| EP(2)0330 | S (60) | S (90) | E | E | Unknown |
| MESR4EP(2)0386 | S (50) | S (53) | E | NE | MESR4 |
| elBEP(2)0965 | NT | NT | E | E | elbB |
| CG7231EP(2)2510 | NT | NT | E | L | Unknown |
| EP(3)0595b | S (44) | E (25) | E | E | FGF signal or CG6745 (unknown) |
| EP(3)1051 | L | E (53) | E | E | Toll |
| SnfγEP(3)3015b | L | E (22) | E | NE | Snfγ |
| EP(3)3041 | L | NE (98) | E | E | Unknown |
| EP(3)3099 | L | S (94) | E | E | RhoBTB or IDE |
| mubEP(3)3108 | S (116) | S (83) | E | E | mub |
| EP(3)3348 | S (125) | S (116) | S | E | SAP130 or ATG1 |
| SAP13006924 | NT | NE (88) | NE | NT | SAP130 |
| EP(3)3405 | S (14) | NE (50) | S | E | Unknown |
| EP(3)3470 | L | NE (54) | E | E | CG17100 or Rfx |
| nep2EP(3)3549 | L | NE (64) | S | S | nep2 |
| EP(3)3603 | S (17) | S (23) | S | E | Glial cell differentiation or UbcD4 |
| UbcD4EY05497 | NT | S (118) | NE (129) | NT | UbcD4 |
The number of flies screened is shown in parentheses.
Lethal.
Enhancer.
Suppressor.
No effect.
Previous studies have suggested that unexpanded huntingtin has a role in vesicle trafficking and that polyGlu-expanded huntingtin blocks axonal transport (Velier et al. 1998). In Drosophila, expression of polyGlu-expanded huntingtin led to disruption of axonal transport in larval neurons, resulting in synaptic defects. These defects were attributed to the presence of cytoplasmic inclusions of expanded-polyGlu proteins and were distinct from apoptosis attributed to nuclear aggregates (Gunawardena et al. 2003; Lee et al. 2004). Loss of vesicular components such as adaptins might reduce trafficking in the cell, minimizing the effects of polyGlu-huntingtin and causing the suppression of the phenotype that we observed with the gEP(X)0514 mutation. Our findings strengthen the proposed involvement of expanded polyGlu proteins in impairing vesicular traffic. Normal function of tau has also been linked with axonal transport. Neuronal cells transfected with tau show disruptions of organelle movement and elimination of neurites (Mandelkow et al. 2003). In Drosophila, overexpression of tau in motor neurons was shown to induce stalling of synaptic vesicle movement, resulting in defective axonal transport and locomotor phenotypes (Mudher et al. 2004). Here we show that a mutation in garnet (δ-adaptin) causes suppression of a tau-overexpression phenotype. Recently, tau protein has been shown to mediate the association of Golgi vesicles with microtubules (Farah et al. 2006). On the basis of these data, loss of garnet would either minimize the association of Golgi vesicles with microtubules or result in a reduced number of vesicles, alleviating the “clogging” effect caused by excess tau protein and causing suppression of the tau phenotype. Detailed morphological analysis of larval neurons lacking adaptins would be needed to corroborate this model of action. In conclusion, our data suggest that δ-adaptin mutations can change phenotypes induced by toxic aggregates in neurons, possibly through effects on vesicular transport. These data are supported by recent suggestions that defective vesicular trafficking is a hallmark of Alzheimer's disease (reviewed in Keating et al. 2006) and other neurodegenerative conditions.
EP(X)1596 may be affecting one of the genes NICE5, a ubiquitin ligase, CG2918, a stress response protein, or CG3071, a protein involved in vesicular transport. Although we have independently confirmed that NICE5 modifies the Aβ phenotype, the identity of the gene involved in polyGlu and tau phenotypic modifications is less clear. However, all processes putatively affected by this insertion have been implicated in tau and polyGlu neurodegeneration (Petrucelli et al. 2004; Gong et al. 2005; Jana et al. 2005; Al-Ramahi et al. 2006). The mutation EP(X)0308 may be affecting two different genes, the copper-transporter ATP7 or a Drosophila gene of unknown function (CG10347) that is homologous to the human chronic myelogenous leukemia antigen 66. We tested a gain-of-function mutation in ATP7 and found that it modified the polyGlu phenotype. Although we cannot assess the contribution of each of these different mutations on the tau and httex1Q93 phenotypes, various aspects of metal homeostasis have been implicated in Alzheimer's disease (reviewed in Adlard and Bush 2006) and in neurodegeneration in general (Mattson 2004). The mutation EP(3)3348 may also be affecting two genes, the transcriptional corepressor SAP130 or the autophagy gene ATG-1. We found that a loss-of-function mutation in SAP130 had no effect on the polyGlu phenotype, suggesting that the effect of EP(3)3348 may be mediated by ATG-1. Loss-of-function mutations in Drosophila Sin3A and Rpd3 (HDAC1) were shown to enhance polyGlu-containing sca1 phenotypes (Fernandez-Funez et al. 2000). Although we have not confirmed the specific involvement of ATG-1 mutations in modifying toxicity in the three models, autophagy is a global mechanism by which the cell recycles misfolded and aggregated proteins and has been extensively linked with neurodegeneration (Martinez-Vicente and Cuervo 2007). The mutation EP(3)3603 may affect two genes, CG6767, which encodes a kinase involved in purine/pyrimidine metabolism, or UbcD4. Although we showed that the CG6767 gene, which is needed for glial cell differentiation (Egger et al. 2002), modifies the Aβ phenotype, we found that the polyGlu phenotype is suppressed by a loss-of-function mutation in UbcD4 (Table 3).
We have compared the results from previous genetic screens in Drosophila, for modifiers of polyGlu phenotypes. Although there is not extensive overlap with our analysis, it is worth noting that overexpression of the RNA-binding gene mub enhances the Htt128Q phenotype whereas it suppresses the Ataxin-182Q phenotype (Branco et al. 2008). Our data, in which a loss-of-function mub mutation suppresses the httex1Q93 phenotype, support this analysis.
DISCUSSION
Biochemical pathways that affect Aβ toxicity:
Several transcriptional regulators were identified as modifiers of Aβ phenotypes. Although Aβ is a secreted peptide, it has been proposed that intracellular Aβ pools may regulate the p53 gene (Ohyagi et al. 2005). Several studies have also reported RNA expression differences in a variety of models that are associated with overexpression of Aβ peptides, ranging from lymphocytes (Kalman et al. 2005) or other cell types from AD patients (Kim et al. 2003; Maes et al. 2007) to brains from APP transgenic mice (Reddy et al. 2004) and Caenorhabditis elegans expressing Aβ (Wu and Luo 2005). These studies have proposed that Aβ expression causes misregulation of stress-response genes, cytoskeletal components, DNA repair, and transcription. On the basis of the wide range of pathways that are responsive to overexpression of Aβ, it seems reasonable to propose that Aβ may directly or indirectly titer out components of global regulatory networks that act at the chromatin level, such as the Sin3A complex. Further reduction of the function of these complexes caused by mutations in their components would be expected to enhance the Aβ effects, which is what was observed in our experiments.
Throughout its production, intracellular trafficking, and turnover, Aβ is associated with parts of the secretory pathway, making this cellular compartment critical for Aβ function and ultimately the development of Alzheimer's disease. In our studies we have identified carboxypeptidase D, which belongs to a family of enzymes that proteolytically process peptides in the secretory pathway (Arolas et al. 2007), as a modifier of Aβ toxicity. Previous reports have identified carboxypeptidase B as an enzyme that may degrade Aβ intracellularly (Matsumoto et al. 2001) by processing its carboxy terminus (Hamazaki 1998). The C terminus of Aβ plays an important role in the formation of Aβ fibrils (Cerda-Costa et al. 2007), suggesting that the processing by carboxypeptidases may have an important function in the toxicity of Aβ. In support of this, we found that in the presence of the loss-of-function mutants, the levels of soluble Aβ were higher and in one of them (svrKG02090, Figure 2) the difference was statistically significant. Thus, a possible cellular mechanism that is recruited to compensate for Aβ toxicity is carboxy-terminal proteolytic processing that reduces the concentration of fibril formation-competent Aβ species.
Effects on levels of Aβ protein and RNA:
The amyloid cascade hypothesis considers that accumulation of Aβ is the primary event in triggering Alzheimer's pathology. Consequently, approaches toward the development of disease-modifying therapies have focused on halting the production of Aβ (reviewed in Hull et al. 2006) or lowering its levels in the brain. This can be achieved either by manipulating the activity of Aβ-specific proteases (reviewed in Higuchi et al. 2005; Nixon and Cataldo 2006) or by administering passive or active immunization against Aβ (reviewed in Dasilva et al. 2006; Solomon 2007). Great effort is invested in exploring these approaches but their implementation is still at the experimental stage.
Although high levels of Aβ are responsible for its neurotoxic properties, a few recent studies have indicated that it is possible to ameliorate its toxic effects by pharmacological approaches that do not change the overall levels of the peptide. Some of these treatments address the inflammatory response that is known to be elicited by Aβ overexpression. Administration of minocycline, an anti-inflammatory drug, did not change the levels of total, soluble, or insoluble Aβ, while ameliorating behavioral deficits in transgenic mice (Fan et al. 2007). Similarly, treatment with dithiocarbamates, which can inhibit the preinflammatory agent NFkB, also improved AD pathology without affecting Aβ burden (Malm et al. 2007). In addition to anti-inflammatory treatments, the cholesterol-lowering drug simvastatin induced cognitive improvement without changing levels of Aβ in transgenic mice (Li et al. 2006). All of these studies indicate that it is possible to change the toxic effects of Aβ without lowering the levels of the peptide. Supported by these findings, the modifiers of Aβ phenotypes that we identified in our genetic interaction analyses either left total levels of Aβ unchanged or caused a 20–30% change. Since this small change in levels of total Aβ did not always correlate with the type of phenotypic modification that the mutation caused, we hypothesize that these changes of total Aβ do not account for the modification of the Aβ phenotype. In contrast, four modifiers caused significant changes in the levels of soluble Aβ. Of those, the mutants in the silver and Snfγ genes increased the levels of soluble Aβ while causing an enhancement of the Aβ phenotype. These results suggest that an increase of the levels of soluble Aβ, possibly inside the cell, could account for the more severe Aβ phenotype. Similarly, the mutation EP(3)3348, which is likely to affect the SAP130 transcriptional coregulator, reduced levels of soluble Aβ, while suppressing the phenotype. These results indicate that soluble intracellular Aβ may contribute to Aβ toxicity. The effects of two additional modifiers, EP(X)0514g and EP(3)3603, are more difficult to explain, since the change in the levels of soluble Aβ does not correlate with the modification of the Aβ phenotype. Histological analyses in these flies might help us understand the mechanism of these particular interactions. In conclusion, we propose that small changes (20–30%) in levels of Aβ may not be causative in the modification of Aβ phenotypes. Rather, changes in solubility or subcellular sequestering of Aβ, or interactions with downstream effectors of Aβ toxicity, are possible mechanisms of action of the modifiers that we identified.
Common pathways in neurodegeneration:
Although significant advances have been made toward understanding the production of Aβ peptides, less is known about the mechanisms that regulate its turnover or how it becomes toxic to the cells. Analogous mechanisms regulating toxicity of other neurodegenerative agents such as tau- and polyGlu-containing proteins are also less well understood. Neurodegeneration is in general considered an “aggregation disease” and is associated with elevated levels of peptides or proteins. The induction of proteolytic pathways is a consequence of such accumulation and is considered a natural protective response of the cell. It can be envisioned that a common “aggregation sensor” operates within cells, identifying the abnormal accumulation of proteins or peptides and activating the ubiquitination machinery. Specific reverse genetic screens using ubiquitination pathway components will help us conclude on the existence of common ubiquitination responses in neurodegenerative conditions.
Despite the common phenotype of protein aggregation, the accumulating proteins and peptides in different neurodegenerative conditions are occupying distinct subcellular localizations. Biogenesis and metabolism of Aβ peptides is closely linked to the various compartments of the secretory pathway, whereas tau is a cytoplasmic protein that associates with the cytoskeleton and was recently also shown to interact with actin (Fulga et al. 2007). Proteins that contain extended polyglutamine stretches are found normally in the cytoplasm but upon polyglutamine expansion they are translocated to the cell nucleus where they interfere with transcriptional functions. Given the diverse localization of these aggregates, it is challenging to propose common mechanisms of toxicity. Our studies suggest that several processes, including ubiquitination, vesicular transport, and chromatin regulation, may be common in either mediating or confining toxicity in neurodegenerative conditions. It can be envisioned that such involvement can result either as a primary or as a secondary response. Accumulation of extra Aβ peptides in secretory compartments might affect vesicular trafficking, which might subsequently turn on ubiquitination. In the case of cytoplasmically localized toxic agents, ubiquitination might be induced first, followed by disruption of the secretory pathway and possibly of neurotransmission. Interfering with ubiquitination might result in misregulation of chromatin structure.
Although Drosophila models of neurodegeneration are faithfully reproducing several aspects of the human condition, they do not fully reflect the complexity and cell-type specificity of the human brain. This extra level of diversity can account for the differences in symptoms, which correspond to the biochemical pathways that are affected in each case. However, as shown here, Drosophila is instrumental in guiding the identification, through unbiased genetic screens, of pathways involved in neurodegeneration and in comparing these pathways among models of different diseases.
Footnotes
Communicating editor: T. Schüpbach
Acknowledgement
We thank Larry Marsh, Mel Feany, and the Bloomington and Szeged Stock Centers and the Harvard Exelixis Stock Collection for providing Drosophila strains. We also thank Marie-Agnes Michellod and Dewey Royal for critical reading of the manuscript. This work was supported by a grant from the American Health Assistance Foundation to M.K.
References
Arolas, J. L., J. Vendrell, F. X. Aviles and L. D. Fricker,
Fossgreen, A., B. Bruckner, C. Czech, C. L. Masters, K. Beyreuther et al.,
Gong, C. X., F. Liu, I. Grundke-Iqbal and K. Iqbal,
Iijima, K., H. P. Liu, A. S. Chiang, S. A. Hearn, M. Konsolaki et al.,
Lee, W. C., M. Yoshihara and J. T. Littleton,
Ohyagi, Y., H. Asahara, D. H. Chui, Y. Tsuruta, N. Sakae et al.,
Pandey, U. B., Z. Nie, Y. Batlevi, B. A. Mccray, G. P. Ritson et al.,
Park, J. W., K. Parisky, A. M. Celotto, R. A. Reenan and B. R. Graveley,
Reddy, P. H., S. Mcweeney, B. S. Park, M. Manczak, R. V. Gutala et al.,
Rorth, P.,
Solomon, B.,
Vance, J. E., H. Hayashi and B. Karten,

{kind=link}
{kind=link}
{kind=link}
{kind=link}