





Abstract
Galactose-activated transcription of the Saccharomyces cerevisiae GAL genes occurs when Gal3 binds the Gal4 inhibitor, Gal80. Noninteracting variants of Gal3 or Gal80 render the GAL genes noninducible. To identify the binding determinants for Gal3's interaction with Gal80 we carried out GAL3–GAL80 intergenic suppression analyses and selected for new GAL3 mutations that impair the Gal3–Gal80 interaction. We show that a GAL3C-D368V mutation can suppress the noninducibility due to a GAL80S-1-G323R mutation, and a GAL80-M350C mutation can suppress the noninducibility due to a gal3-D111C mutation. A reverse two-hybrid selection for GAL3 mutations that impair the Gal3–Gal80 interaction yielded 12 single-amino-acid substitutions at residues that are predicted to be surface exposed on Gal3. The majority of the affected Gal3 residues localized to a composite surface that includes D111 and a sequence motif containing D368, which has been implicated in interaction with Gal80. The striking colocalization of intergenic suppressor residues and Gal80 nonbinder residues identifies a Gal3 surface that likely interacts with Gal80.
THE GAL gene switch of Saccharomyces cerevisiae is composed of the DNA-binding transcriptional activator Gal4, its inhibitor Gal80, and the galactose sensor Gal3. These proteins provide a mechanism for swift transcriptional activation of the GAL genes in response to galactose (Lohret al. 1995). Gal4 binds to the UASGAL DNA elements in both the absence and the presence of galactose (Bram and Kornberg 1985; Ginigeret al. 1985). Without galactose the activity of Gal4 is inhibited through an interaction with Gal80 (Johnstonet al. 1987). Galactose induces an interaction between Gal3 and Gal80 that relieves Gal80 inhibition of Gal4 and allows activation of the GAL genes (Bhat and Hopper 1992; Suzuki-Fujimotoet al. 1996).
Gal3 is the galactose sensor of the GAL gene switch. Recessive gal3 mutations impair induction, whereas dominant GAL3C mutations encode proteins that bind to Gal80 independently of galactose and confer constitutive GAL gene expression (Blanket al. 1997). Gal3 is a paralogue of the Gal1 galactokinase (Bajwaet al. 1988; Wolfe and Shields 1997; Plattet al. 2000). Unlike Gal3, Gal1 is not sufficiently expressed in the absence of galactose to serve as an inducer (Tsuyumu and Adams 1974; Broach 1979; Bhatet al. 1990; Hittinger and Carroll 2007). However, when Gal1 is expressed from a surrogate promoter, it can substitute for Gal3 in activation of the GAL genes, and this does not require its galactokinase activity (Bhat and Hopper 1992). Moreover, the sequence motifs of Gal1 that bind galactose and ATP are conserved in Gal3 (Plattet al. 2000; Thodenet al. 2005). Although native Gal3 lacks galactokinase activity, the insertion of serine and alanine (Gal3-SA) at a single position within one of its galactokinase homology motifs results in the acquisition of galactokinase activity (Plattet al. 2000). It has also been shown that a D62A substitution in Gal1 can impair its ability to catalyze phosphorylation of galactose, and the corresponding amino acid substitution in Gal3 impairs its capacity to induce expression of the GAL genes in response to galactose (Sellick and Reece 2006). Thus, the binding of galactose and ATP to Gal1 or Gal3 affects their capacity to bind to Gal80 and also serves as the catalytic function of Gal1 or Gal3-SA.
Studies of the highly similar GAL gene switch of the yeast Kluyveromyces lactis (Kl) further support the notion that Gal3 is a galactose sensor (Meyeret al. 1991; Zenkeet al. 1996). K. lactis lacks a GAL3 gene (Vollenbroichet al. 1999) but expresses galactokinase (encoded by KlGAL1) at moderately high levels in the absence of galactose (Dickson and Markin 1980; Riley and Dickson 1984; Cardinaliet al. 1997). Klgal1 recessive mutations confer the noninducible phenotype and impaired binding of KlGal1 to KlGal80, whereas dominant KlGAL1 mutations confer constitutivity and galactose-independent binding of KlGal1 to KlGal80 (Vollenbroichet al. 1999; Menezeset al. 2003). Thus, KlGal1 is the galactose sensor in K. lactis.
The mechanism by which the binding of galactose and ATP triggers Gal3 interaction with Gal80 is unresolved. Gal3, Gal1, and KlGal1 belong to the GHMP superfamily of small-molecule kinases (Borket al. 1993). Crystal structures are available for several family members including galactokinase, homoserine kinase, mevalonate kinase, and phosphomevalonate kinase (Zhouet al. 2000; Krishnaet al. 2001; Romanowskiet al. 2002; Yanget al. 2002; Thoden and Holden 2003; Thodenet al. 2005). All GHMP superfamily members have conserved motifs for the binding of ATP and their specific substrates, and most have been crystallized with their substrates bound. Thus, the binding sites for galactose and ATP in galactokinase are well established.
All GHMP members share a similar overall protein fold (Andreassi and Leyh 2004). Taking advantage of this, Menezeset al. (2003) used the structure of mevalonate kinase (Yanget al. 2002) as a template to model the structure of KlGal1. On the basis of the locations of KlGAL1 mutations they proposed that the binding of galactose and ATP to a cleft in front of a hinge region brings together the upper and lower lips of KlGal1 to create a docking surface for KlGal80 (Menezeset al. 2003). Although attractive as a testable hypothesis, this proposal was based on a severely limited KlGal1 homology model due to the low level of sequence identity between KlGal1 and mevalonate kinase (13%). An additional limitation of the model of KlGal1 is that it lacks the insertion motif (97 residues in Gal3) that is common to Gal3, Gal1, and KlGal1. It is likely that the insertion motif plays an essential role in binding of Gal3 to Gal80 because Escherichia coli galactokinase, which lacks the insertion motif, provides galactokinase activity but not the Gal3-specified GAL gene induction activity in yeast cells (Bhatet al. 1990).
The crystal structure of the S. cerevisiae Gal1 bound with galactose and AMPPNP has recently been solved at 2.4-Å resolution (Thodenet al. 2005) and provides a reliable template for deriving models of Gal3 (Thodenet al. 2005; Diepet al. 2006). Because Gal3 is 72% identical and 92% similar to Gal1, these Gal3 homology models provide excellent predictive values for the structure of Gal3 and facilitate structure/function analyses.
We previously carried out intragenic suppression analyses of GAL3C mutations to address the issue of how the binding of galactose and ATP affects the binding of Gal3 to Gal80 (Diepet al. 2006). Gal3C proteins bind to Gal80 in the absence of galactose and cause constitutive expression of the GAL genes (Blanket al. 1997). A suppressor (GAL3SOC) of GAL3C mutations and four suppressible alleles (GAL3C-F237Y, -V396A, and -S509P/L) colocalize to a region in the Gal3 model that contains a putative ligand-regulated hinge region previously identified in a KlGal1 homology model (Menezeset al. 2003). In contrast, the single nonsuppressible GAL3C allele (GAL3C-D368V) is remote from the hinge region. The nonsuppressible nature of GAL3C-D368V and its location within the insertion motif suggested that D368 and other nearby residues might compose the surface that interacts with Gal80 (Diepet al. 2006).
In this study we performed a genetic analysis to identify the Gal3 surface that docks with Gal80. We found that mutations that affect Gal3's binding to Gal80 alter residues in a well-defined composite surface composed of many noncontiguous residues. We propose that this surface of Gal3 is the docking site for Gal80.
MATERIALS AND METHODS
Media and growth conditions:
Yeast strains were grown at 30° in either standard nonselective YEP medium or selective SC medium with 2% glucose (Sherman 1991). Noninduced conditions consisted of 0.05% glucose/3% glycerol/2% lactic acid (pH 5.7). For induced conditions galactose was added to the above media at a final concentration of 2%. For experiments utilizing the HIS3 reporter gene, 10 mm 3-amino-1,2,4-triazole (3-AT) (Sigma, St. Louis) was supplemented to the agar plates to inhibit low levels of His3 enzyme produced from galactose-independent HIS3 reporter expression. The chemical 5-fluoroorotic acid (5-FOA) (Toronto Research Chemicals) was added (at 0.1% w/v final concentration) to the reverse two-hybrid agar selection media after autoclaving and cooling prior to pouring the plates. The bacterial strain MG7-α (Griffith and Gietz 2003) was grown at 37° in LB medium supplemented with 50 mg/liter of ampicillin.
Yeast strains:
Yeast strains and phenotypes used in this study are listed in Table 1. ScPD2 was derived from ScVP2-103 (Pilauriet al. 2005) by disruption of the GAL1 gene. Sc750 and Sc751 were derived from Sc724 (Blanket al. 1997) by disruption of the GAL80 gene and integration of the GAL80S−0 and GAL80S−1 alleles, respectively. The GAL80S-2 mutation was not integrated into the genome, but instead was carried on a CEN plasmid (pMPW87). In this case the strain Sc787 (gal1Δgal3Δgal80Δ) was used to carry GAL80S-2 on pMPW87. Sc754 was derived from Sc723 (Blanket al. 1997) by disruption of the GAL1 gene. Further details on the construction of these strains are available upon request.
Yeast strains and genotypes
Strain | Phenotype | Study |
|---|---|---|
| ScPD2 | LYS2 can1R cyh2R | This study |
| Sc723 | MATaade1 ile leu2-3,112 ura3-52 trp1-HIII his3-Δ1 MEL1 LYS2∷ GAL1UAS-GAL1TATA-HIS3 | Blanket al. (1997) |
| Sc724 | MATaade1 ile leu2-3,112 ura3-52 trp1-HIII his3-Δ1 MEL1 LYS2∷ GAL1UAS-GAL1TATA-HIS3 gal3Δ-3∷LEU2 GAL80 WT | Blanket al. (1997) |
| Sc726 | MATaade1 ile leu2-3,112 ura3-52 trp1-HIII his3-Δ1 MEL1 LYS2∷ GAL1UAS-GAL1TATA-HIS3 gal3Δ-3∷LEU2 gal80-ΔBglII | Blanket al. (1997) |
| Sc750 | MATaade1 ile leu2-3,112 ura3-52 trp1-HIII his3-Δ1 MEL1 LYS2∷ GAL1UAS-GAL1TATA-HIS3 gal3Δ-3∷LEU2 GAL80S-0 | This study |
| Sc751 | MATaade1 ile leu2-3,112 ura3-52 trp1-HIII his3-Δ1 MEL1 LYS2∷ GAL1UAS-GAL1TATA-HIS3 gal3Δ-3∷LEU2 GAL80S-1 | This study |
| Sc754 | MATaade1 ile leu2-3,112 ura3-52 trp1-HIII his3-Δ1 MEL1 LYS2∷ GAL1UAS-GAL1TATA-HIS3 gal1Δ | This study |
| Sc781 | MATaade1 ile leu2-3,112 ura3-52 trp1-HIII his3-Δ1 MEL1 LYS2∷ GAL1UAS-GAL1TATA-HIS3 gal3Δ-3∷LEU2 gal1Δ∷ura3 | Blanket al. (1997) |
| Sc787 | MATaade1 ile leu2-3,112 ura3-52 trp1-HIII his3-Δ1 MEL1 LYS2∷ GAL1UAS-GAL1TATA-HIS3 gal3Δ-3∷LEU2 gal1Δ∷ura3 gal80-ΔBglII | Blanket al. (1997) |
Strain | Phenotype | Study |
|---|---|---|
| ScPD2 | LYS2 can1R cyh2R | This study |
| Sc723 | MATaade1 ile leu2-3,112 ura3-52 trp1-HIII his3-Δ1 MEL1 LYS2∷ GAL1UAS-GAL1TATA-HIS3 | Blanket al. (1997) |
| Sc724 | MATaade1 ile leu2-3,112 ura3-52 trp1-HIII his3-Δ1 MEL1 LYS2∷ GAL1UAS-GAL1TATA-HIS3 gal3Δ-3∷LEU2 GAL80 WT | Blanket al. (1997) |
| Sc726 | MATaade1 ile leu2-3,112 ura3-52 trp1-HIII his3-Δ1 MEL1 LYS2∷ GAL1UAS-GAL1TATA-HIS3 gal3Δ-3∷LEU2 gal80-ΔBglII | Blanket al. (1997) |
| Sc750 | MATaade1 ile leu2-3,112 ura3-52 trp1-HIII his3-Δ1 MEL1 LYS2∷ GAL1UAS-GAL1TATA-HIS3 gal3Δ-3∷LEU2 GAL80S-0 | This study |
| Sc751 | MATaade1 ile leu2-3,112 ura3-52 trp1-HIII his3-Δ1 MEL1 LYS2∷ GAL1UAS-GAL1TATA-HIS3 gal3Δ-3∷LEU2 GAL80S-1 | This study |
| Sc754 | MATaade1 ile leu2-3,112 ura3-52 trp1-HIII his3-Δ1 MEL1 LYS2∷ GAL1UAS-GAL1TATA-HIS3 gal1Δ | This study |
| Sc781 | MATaade1 ile leu2-3,112 ura3-52 trp1-HIII his3-Δ1 MEL1 LYS2∷ GAL1UAS-GAL1TATA-HIS3 gal3Δ-3∷LEU2 gal1Δ∷ura3 | Blanket al. (1997) |
| Sc787 | MATaade1 ile leu2-3,112 ura3-52 trp1-HIII his3-Δ1 MEL1 LYS2∷ GAL1UAS-GAL1TATA-HIS3 gal3Δ-3∷LEU2 gal1Δ∷ura3 gal80-ΔBglII | Blanket al. (1997) |
Yeast strains and genotypes
Strain | Phenotype | Study |
|---|---|---|
| ScPD2 | LYS2 can1R cyh2R | This study |
| Sc723 | MATaade1 ile leu2-3,112 ura3-52 trp1-HIII his3-Δ1 MEL1 LYS2∷ GAL1UAS-GAL1TATA-HIS3 | Blanket al. (1997) |
| Sc724 | MATaade1 ile leu2-3,112 ura3-52 trp1-HIII his3-Δ1 MEL1 LYS2∷ GAL1UAS-GAL1TATA-HIS3 gal3Δ-3∷LEU2 GAL80 WT | Blanket al. (1997) |
| Sc726 | MATaade1 ile leu2-3,112 ura3-52 trp1-HIII his3-Δ1 MEL1 LYS2∷ GAL1UAS-GAL1TATA-HIS3 gal3Δ-3∷LEU2 gal80-ΔBglII | Blanket al. (1997) |
| Sc750 | MATaade1 ile leu2-3,112 ura3-52 trp1-HIII his3-Δ1 MEL1 LYS2∷ GAL1UAS-GAL1TATA-HIS3 gal3Δ-3∷LEU2 GAL80S-0 | This study |
| Sc751 | MATaade1 ile leu2-3,112 ura3-52 trp1-HIII his3-Δ1 MEL1 LYS2∷ GAL1UAS-GAL1TATA-HIS3 gal3Δ-3∷LEU2 GAL80S-1 | This study |
| Sc754 | MATaade1 ile leu2-3,112 ura3-52 trp1-HIII his3-Δ1 MEL1 LYS2∷ GAL1UAS-GAL1TATA-HIS3 gal1Δ | This study |
| Sc781 | MATaade1 ile leu2-3,112 ura3-52 trp1-HIII his3-Δ1 MEL1 LYS2∷ GAL1UAS-GAL1TATA-HIS3 gal3Δ-3∷LEU2 gal1Δ∷ura3 | Blanket al. (1997) |
| Sc787 | MATaade1 ile leu2-3,112 ura3-52 trp1-HIII his3-Δ1 MEL1 LYS2∷ GAL1UAS-GAL1TATA-HIS3 gal3Δ-3∷LEU2 gal1Δ∷ura3 gal80-ΔBglII | Blanket al. (1997) |
Strain | Phenotype | Study |
|---|---|---|
| ScPD2 | LYS2 can1R cyh2R | This study |
| Sc723 | MATaade1 ile leu2-3,112 ura3-52 trp1-HIII his3-Δ1 MEL1 LYS2∷ GAL1UAS-GAL1TATA-HIS3 | Blanket al. (1997) |
| Sc724 | MATaade1 ile leu2-3,112 ura3-52 trp1-HIII his3-Δ1 MEL1 LYS2∷ GAL1UAS-GAL1TATA-HIS3 gal3Δ-3∷LEU2 GAL80 WT | Blanket al. (1997) |
| Sc726 | MATaade1 ile leu2-3,112 ura3-52 trp1-HIII his3-Δ1 MEL1 LYS2∷ GAL1UAS-GAL1TATA-HIS3 gal3Δ-3∷LEU2 gal80-ΔBglII | Blanket al. (1997) |
| Sc750 | MATaade1 ile leu2-3,112 ura3-52 trp1-HIII his3-Δ1 MEL1 LYS2∷ GAL1UAS-GAL1TATA-HIS3 gal3Δ-3∷LEU2 GAL80S-0 | This study |
| Sc751 | MATaade1 ile leu2-3,112 ura3-52 trp1-HIII his3-Δ1 MEL1 LYS2∷ GAL1UAS-GAL1TATA-HIS3 gal3Δ-3∷LEU2 GAL80S-1 | This study |
| Sc754 | MATaade1 ile leu2-3,112 ura3-52 trp1-HIII his3-Δ1 MEL1 LYS2∷ GAL1UAS-GAL1TATA-HIS3 gal1Δ | This study |
| Sc781 | MATaade1 ile leu2-3,112 ura3-52 trp1-HIII his3-Δ1 MEL1 LYS2∷ GAL1UAS-GAL1TATA-HIS3 gal3Δ-3∷LEU2 gal1Δ∷ura3 | Blanket al. (1997) |
| Sc787 | MATaade1 ile leu2-3,112 ura3-52 trp1-HIII his3-Δ1 MEL1 LYS2∷ GAL1UAS-GAL1TATA-HIS3 gal3Δ-3∷LEU2 gal1Δ∷ura3 gal80-ΔBglII | Blanket al. (1997) |
PCR mutagenesis of GAL3, gap-repair, and yeast two-hybrid selection:
Mutagenesis of GAL3 was achieved as previously described (Pilauriet al. 2005; Diepet al. 2006), using the Taq DNA polymerase (Sigma) and the manganese (Mn)-dITP error-prone PCR method (Xuet al. 1999; Fentonet al. 2002). The plasmid pCD107 was used as the template for the PCR reactions. pCD107 carries GAL3-SA fused to the N terminus of VP16. The GAL3 coding sequence was divided into three regions, using the following primers: region 1, TAMA57 (5′-TTCTGCACAATATTTCAAGCTATACCAA-3′) and DIEP43 (5′-GCAGATGAGAGTCCACCACCAGTAGG-3′); region 2, DIEP46 (5′-GCTCCGGAAAGATTTAATAATACACCC-3′) and BLNK05 (5′-GGGAAAAGTTGTCAGGTAATC-3′); region 3, BLNK03 (5′-ACGAAAACCAAGCCCAACCAT-3′) and TAMA58 (5′-TAGCGCGTCGGCATGCGCCAT-3′). A PCR product pool was generated for each region separately and was used with the cognate gapped GAL3 gene in pCD107 to reconstitute the complete plasmid using a homologous recombination-based gap-repair method (Muhlradet al. 1992). The three cognate gapped plasmids were created by the following restriction digestions: gap-I, PstI/BglII; gap-II, BglII/XhoI; and gap-III, XhoI/PshAI. Each PCR product pool and cognate gapped plasmid were combined and used to transform the ScPD2 carrying pAKS42 (DBD-GAL80) (Silet al. 1999). The reverse two-hybrid selection was carried out as previously described (Vidalet al. 1996; Pilauriet al. 2005). From ∼5000 gap-repaired colonies, ∼250 grew on plates containing 5-FOA. After screening by Western blots, 26 candidates had full-length proteins and showed repeated loss of interaction with DBD-GAL80 in both the reverse and the forward two-hybrid assays. The plasmids from these candidates were isolated and sequenced to identify the mutations. The candidate plasmids were transformed back into ScPD2 carrying DBD-GAL80 to reconfirm their phenotypes.
Plasmid construction:
The plasmid pCD107 was constructed by ligating the BglII/XhoI fragment from pAKS130 [GAL3-SA in the pTEB16 backbone (Blanket al. 1997)] into the corresponding sites of pGAL3-VP16 (Pilauriet al. 2005). GAL3 fragments in pCD107-bearing mutations were transferred into pTEB16, using the gap-repair method (Muhlradet al. 1992). This was done by cotransformation of the EcoRI fragment of pCD107 with the backbone of pTEB16 after digestion with BstBI. Mutations from pTEB16 were transferred into pMPW60 by swapping the NsiI/KpnI fragments. All mutations were verified by sequencing.
Spot assay for cell growth:
Transformed yeasts were grown to late log phase in selective medium. Each culture was adjusted to the same number of cells and serial 10-fold dilutions were made with dH2O. Six microliters of 10−1, 10−2, 10−3, and 10−4 dilution were spotted onto the appropriate dropout plates and incubated for up to 6 days.
Pull-down assay for GST-Gal3 and Gal80 interaction:
Sc787 cells cotransformed with plasmids carrying GST-GAL3 and GAL80 were grown to midlog phase and whole-cell extracts containing GST-Gal3 and Gal80 were prepared as described (Blanket al. 1997), using a modified lysis buffer (20 mm HEPES pH 7.4, 0.5% Triton X-100, 200 mm NaCl, 0.5 mm EDTA, 2 mm DTT, and 5 mm MgCl2). Protease inhibitor cocktails (PIC) (PIC-D, 88 mg/ml PMSF and 1 mg/ml pepstatin A in DMSO; PIC-W, 157 mg/ml benzamidine, 0.5 mg leupeptin, and 0.5 mg bestatin in water) were added to all lysis buffer and all subsequent solutions at 1/1000 dilutions. The whole-cell extract (∼1 mg) was brought up to a volume of 500 μl with lysis buffer containing 2 mm ATP and 25 mm galactose. Glutathione Sepharose beads (Amersham Biosciences, Arlington Heights, IL) were equilibrated and resuspended in the lysis buffer as a 50% slurry. The whole-cell extracts were then incubated with 50 μl of 50% glutathione Sepharose beads on a rotator at 4° for 2 hr. The beads were pelleted and washed three times with 500 μl of lysis buffer either with or without ATP and galactose. The beads were then boiled for 5 min in 40 μl of 1× SDS–PAGE sample loading buffer (62.5 mm Tris-HCl pH 6.8, 10% glycerol, 2% SDS, 100 mm DTT, and 0.003% Pyronin Y) before analysis by standard SDS–PAGE and Western blot. Antibodies against GST and Gal80 were used together at dilutions indicated in the figure legends.
Homology model manipulation and bioinformatics:
Derivation of the Gal3 homology model was previously described in detail (Thodenet al. 2005; Diepet al. 2006). Visualization of the model was carried out using PyMOL. The multiple sequence alignment was generated using the TCoffee web server (3DCoffee mode) and modified by GeneDoc and Microsoft Word.
RESULTS
Allele-specific intergenic suppression of GAL80S-1 by GAL3C-D368V:
Previously identified dominant GAL80S mutations (GAL80S-0, GAL80S-1, and GAL80S-2) were shown to produce a noninducible phenotype because they encode variant proteins that are impaired in their interaction with Gal3 (Douglas and Hawthorne 1972; Nogiet al. 1977; Yano and Fukasawa 1997). GAL3 mutations that restore its interaction with one or more of the Gal80S proteins could identify residues of Gal3 and Gal80 that are important for the interaction. We reasoned that such suppressors of GAL80S alleles might be among the previously identified GAL3C mutations that are gain-of-function alleles that cause Gal3 to interact with Gal80 in the absence of galactose and ATP (Blanket al. 1997; Diepet al. 2006). Therefore we used a PGALHIS3 reporter-based colony-growth assay to evaluate Gal4-mediated activation of the GAL genes for each of the three GAL80S mutations in combination with each of the five previously isolated GAL3C alleles (Blanket al. 1997; Diepet al. 2006).
In the presence of wild-type GAL80, wild-type GAL3 conferred GAL gene activation only in the presence of galactose as expected, whereas all five GAL3C mutations showed constitutive activation (Figure 1A). Of all the GAL80S and GAL3C combinations, only one showed intergenic suppression: GAL80S-1-G323R and GAL3C-D368V. Cells carrying both of these mutations exhibited GAL gene activation only in the presence of galactose. The GAL80S-1 mutation was epistatic to the remaining four GAL3C alleles, whereas the GAL80S-0 and GAL80S-2 mutations were epistatic to all five GAL3C alleles. This allele-specific interaction between the GAL80S-1-G323R and GAL3C-D368V mutations suggests that the Gal80-G323 and Gal3-D368 residues affect galactose-dependent interaction of these proteins.
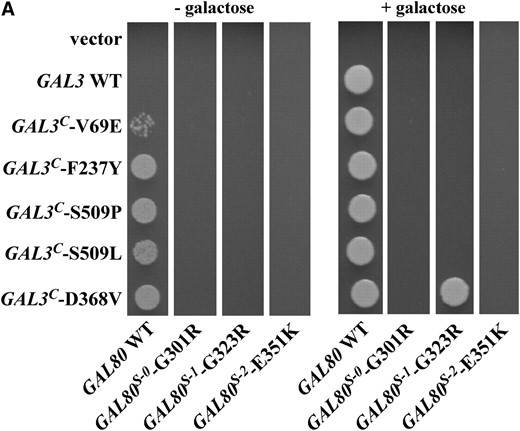
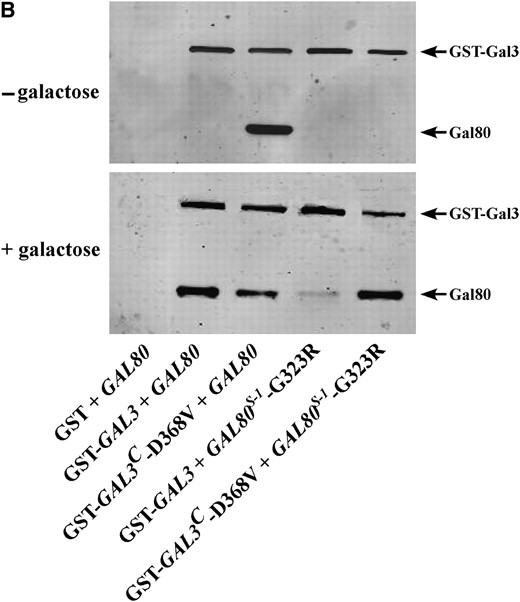
Allele-specific intergenic suppression of GAL80S-1-G323R by GAL3C-D368V. (A) Colony-growth assay. The strains Sc724 (genomic GAL80 WT), Sc750 (integrated GAL80S-0), Sc751 (integrated GAL80S-1), and Sc787 (GAL80S-2 carried on pMPW87) were transformed with the empty vector (pRS414), GAL3 WT (pTEB16), GAL3C-V69E (pCD71), GAL3C-F237Y (pCL-3C-311), GAL3C-S509P (pCL-3C-362), GAL3C-S509L (pCL-3C-371), or GAL3C-D368V (pCL-3C-322). Cells were grown in selective liquid media until late log phase, spotted onto the appropriate selective agar plates with or without galactose, and incubated for 4 days. Intergenic suppression was determined by colony growth directly coupled to expression of the HIS3 reporter gene driven by the GAL1 promoter integrated in the genome of each strain (PGALHIS3). (B) GST pull-down assay. Protein extracts were prepared from the strain Sc787 (gal1Δgal3Δgal80Δ) bearing GST (pMPW61), GST-GAL3 (pMPW60), or GST-GAL3C-D368V (pCLAGC-322) plus either GAL80 (pMPW82) or GAL80S-1-G323R (pMPW86). One milligram of protein extract was incubated with GT–Sepharose for 2 hr at 4° either in the absence or in the presence of galactose and ATP, washed three times, and then boiled and resolved on standard SDS–PAGE and analyzed by Western blot. GST-Gal3 and Gal80 were detected on the same blot by using a mixture of rabbit polyclonal anti-Gal80 (1:300 dilution) and rabbit polyclonal anti-GST (1:3000 dilution).
The intergenic suppression of GAL80S-1-G323R by GAL3C-D368V was also tested for physical interaction in vitro by a GST pull-down assay. As expected, GST-Gal3C-D368V was able to interact with the wild-type Gal80 in the absence of galactose and ATP (Figure 1B). Consistent with the PGAL1HIS3 reporter growth assay (Figure 1A), the physical interaction of Gal80S-1-G323R with GST-Gal3 was dramatically reduced compared to that with wild-type Gal80 in the presence of galactose and ATP. When Gal80S-1-G323R was combined with GST-Gal3C-D368V, the interaction was completely restored. Therefore, both the PGAL1HIS3 reporter growth assay and the GST pull-down assay demonstrated that the noninducibility of GAL80S-1-G323R was suppressed by GAL3C-D368V. These results suggest that the Gal80-G323 and Gal3-D368 residues interact directly or indirectly to modulate the docking surfaces of these proteins.
The noninducible phenotype due to gal3-D111C is suppressed by GAL80-M350C:
Intergenic suppression of a Klgal1 noninducible mutation by a KlGAL80 mutation was reported for the K. lactis GAL gene switch (Menezeset al. 2003). To test if a similar suppression could be established in S. cerevisiae, we changed the corresponding residues in the S. cerevisiae Gal3 and Gal80 to cysteines and expressed the genes on plasmids (gal3-D111C on pCD123 and GAL80-M350C on pCD125). The gal3-D111C mutation combined with the wild-type GAL80 causes the GAL genes to be noninducible as determined by a colony-growth assay with the PGALHIS3 reporter (Figure 2, row 5), but this phenotype was suppressed by GAL80-M350C (Figure 2, row 6) and was indistinguishable from cells carrying wild-type GAL3 (pMPW66) and GAL80 (pMPW82) (Figure 2, row 2). The suppression of gal3-D111C by GAL80-M350C mirrors the Klgal1/KlGAL80 intergenic suppression and provides further support for the similarity of the pairwise interactions.
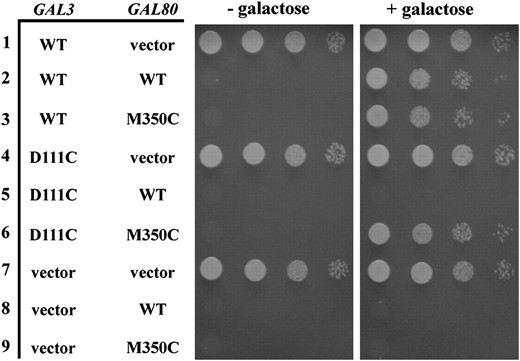
Intergenic suppression of a gal3-D111C noninducible mutation by a GAL80-M350C mutation. The strain Sc787 carrying GAL3 and GAL80 on separate plasmids was grown in selective liquid media until late log phase and 10-fold dilutions were spotted on selective agar plates with or without galactose and incubated up to 6 days. Intergenic suppression was determined by colony growth coupled to PGALHIS3 expression. Row 1, GAL3 WT (pMPW66) + vector (pRS416); row 2, GAL3 WT + GAL80 WT (pMPW82); row 3, GAL3 WT + GAL80-M350C (pCD125); row 4, GAL3-D111C (pCD123) + vector; row 5, GAL3-D111C + GAL80 WT; row 6, GAL3-D111C + GAL80-M350C; row 7, vector (pRS414) + vector; row 8, vector + GAL80 WT; row 9, vector + GAL80-M350C.
We next tested for the suppression of physical interaction between Gal3-D111C and Gal80-M350C. However, we were unable to further characterize these interactions because tagging gal3-D111C with GST at the N terminus caused it to interact with Gal80 independent of galactose in vitro and produce the constitutive phenotype in vivo (data not shown). This is another example where tagging a variant form of the Gal3 protein with an epitope changes its properties. We observed previously (Diepet al. 2006) and also here (Figure 4) that fusion of particular Gal3 variant proteins with GST grossly alters stability or overall behavior.
Isolation and characterization of gal3 missense mutations that impair binding to Gal80:
To identify amino acids of Gal3 that are involved in interaction with Gal80, we isolated GAL3 mutations that impair the Gal3–Gal80 interaction. Three Gal3 variants defective in binding to Gal80 were previously reported but the causal amino acid substitutions were not identified (Suzuki-Fujimotoet al. 1996). We used the yeast reverse two-hybrid selection in conjunction with error-prone PCR mutagenesis to isolate gal3 mutations. We employed the GAL3-SA allele as the bait, which encodes a Gal3 protein with an insertion of serine and alanine between residues 164 and 165. The Gal3-SA protein has galactokinase activity that can complement a gal1-deletion (gal1Δ) mutant for growth on galactose (Plattet al. 2000). We used this in vivo assay to screen the isolates for those that are impaired in Gal80 interaction but retain the ability to complement a gal1Δ, as those would likely be folded properly. GAL3-SA was fused to the VP16 activation domain to form the prey construct (pCD107). In the forward two-hybrid assay, Gal3-SA-VP16 interacted with the DBD-Gal80 bait (pAKS42) in a galactose-dependent manner, indicating that the SA insertion and the VP16 fusion did not perturb the galactose-induced interaction between Gal3 and Gal80 (data not shown). The interaction was also detected in the reverse two-hybrid assay, as the strain ScPD2 carrying both GAL3-SA-VP16 and DBD-GAL80 was unable to grow in the presence of galactose and 0.1% 5-FOA (data not shown).
Mutagenesis of GAL3-SA-VP16 yielded 26 5-FOA-resistant single-amino-acid missense variants (Table 2). All 26 expressed full-length proteins at levels similar to wild-type Gal3-SA-VP16 (data not shown). The prey plasmids were isolated from these colonies and their DNA sequence was determined. All mutations resulted in single-amino-acid substitutions (Table 2). These plasmids were then reintroduced into strain ScPD2 carrying the DBD-GAL80 bait to determine their forward and reverse two-hybrid interactions in the presence of galactose. With the exception of three variant proteins (M71V, H199R, and N278Y), which were only mildly impaired, all of the remaining 23 mutations completely prevented Gal3 interaction with DBD-Gal80 in the forward two-hybrid assay (Figure 3). For most of the isolates the relative magnitudes of the interaction in the forward and reverse two-hybrid assays were similar. However, some mutations caused a complete loss of growth in the forward two-hybrid assay but allowed mild growth in the reverse two-hybrid assay (such as C429Y). When the forward two-hybrid assay plates were incubated longer (6 days, data not shown), these isolates eventually exhibited weak growth, consistent with retention of some interaction with DBD-Gal80. Importantly, all 26 Gal3 variant proteins were at least partially impaired in interaction with DBD-Gal80.

Impaired two-hybrid interaction of the Gal3-SA variants with Gal80. Yeast reverse and forward two-hybrid interactions of Gal3 variants with Gal80 are shown. The strain ScPD2 expressing DBD-GAL80 (pAKS42) and VP16 (pVP16), GAL3-SA-VP16 (pCD107), or the GAL3 mutant alleles in the GAL3-SA-VP16 background was grown in selective liquid media until late log phase and 10-fold dilutions were spotted on selective agar plates containing galactose and 0.1% 5-FOA (reverse two-hybrid) or galactose without 5-FOA (forward two-hybrid).
Isolated gal380NB mutations
Amino acid change | Nucleotide change |
|---|---|
| C58R | TGC → CGC |
| M71V | ATG → GTG |
| L103S | TTA → TCA |
| D111N | GAT → AAT |
| E116G | GAA → GGA |
| L162P | CTC → CCC |
| H199R | CAC → CGC |
| TCT → TCC (silent) | |
| G216E | GGG → GAG |
| TTT → TTC (silent) | |
| L268I | TTA → ATA |
| TCA → TCT (silent) | |
| N278Y | AAC → TAC |
| CTA → CTG (silent) | |
| V286E | GTG → GAG |
| R330C | CGT → TGT |
| L337W | TTG → TGG |
| CAA → CAG (silent) | |
| CGA → CGT (silent) | |
| CAG → CAA (silent) | |
| S341P | TCT → CCT |
| TTT → TTC (silent) | |
| F342Y | TTC → TAC |
| Y369D | TAC → GAC |
| R376C | CGC → TGC |
| GAT → GAC (silent) | |
| H388Y | CAC → TAC |
| Y390H | TAC → CAC |
| TTC → TTT (silent) | |
| CTC → CTG (silent) | |
| C429R | TGT → CGT |
| C429Y | TGT → TAT |
| L432P | CTT → CCT |
| G461D | GGT → GAT |
| TTA → CTA (silent) | |
| GTA → GTG (silent) | |
| ATT → ATC (silent) | |
| W462G | TGG → GGG |
| G463D | GGC → GAC |
| L469H | CTT → CAT |
Amino acid change | Nucleotide change |
|---|---|
| C58R | TGC → CGC |
| M71V | ATG → GTG |
| L103S | TTA → TCA |
| D111N | GAT → AAT |
| E116G | GAA → GGA |
| L162P | CTC → CCC |
| H199R | CAC → CGC |
| TCT → TCC (silent) | |
| G216E | GGG → GAG |
| TTT → TTC (silent) | |
| L268I | TTA → ATA |
| TCA → TCT (silent) | |
| N278Y | AAC → TAC |
| CTA → CTG (silent) | |
| V286E | GTG → GAG |
| R330C | CGT → TGT |
| L337W | TTG → TGG |
| CAA → CAG (silent) | |
| CGA → CGT (silent) | |
| CAG → CAA (silent) | |
| S341P | TCT → CCT |
| TTT → TTC (silent) | |
| F342Y | TTC → TAC |
| Y369D | TAC → GAC |
| R376C | CGC → TGC |
| GAT → GAC (silent) | |
| H388Y | CAC → TAC |
| Y390H | TAC → CAC |
| TTC → TTT (silent) | |
| CTC → CTG (silent) | |
| C429R | TGT → CGT |
| C429Y | TGT → TAT |
| L432P | CTT → CCT |
| G461D | GGT → GAT |
| TTA → CTA (silent) | |
| GTA → GTG (silent) | |
| ATT → ATC (silent) | |
| W462G | TGG → GGG |
| G463D | GGC → GAC |
| L469H | CTT → CAT |
Isolated gal380NB mutations
Amino acid change | Nucleotide change |
|---|---|
| C58R | TGC → CGC |
| M71V | ATG → GTG |
| L103S | TTA → TCA |
| D111N | GAT → AAT |
| E116G | GAA → GGA |
| L162P | CTC → CCC |
| H199R | CAC → CGC |
| TCT → TCC (silent) | |
| G216E | GGG → GAG |
| TTT → TTC (silent) | |
| L268I | TTA → ATA |
| TCA → TCT (silent) | |
| N278Y | AAC → TAC |
| CTA → CTG (silent) | |
| V286E | GTG → GAG |
| R330C | CGT → TGT |
| L337W | TTG → TGG |
| CAA → CAG (silent) | |
| CGA → CGT (silent) | |
| CAG → CAA (silent) | |
| S341P | TCT → CCT |
| TTT → TTC (silent) | |
| F342Y | TTC → TAC |
| Y369D | TAC → GAC |
| R376C | CGC → TGC |
| GAT → GAC (silent) | |
| H388Y | CAC → TAC |
| Y390H | TAC → CAC |
| TTC → TTT (silent) | |
| CTC → CTG (silent) | |
| C429R | TGT → CGT |
| C429Y | TGT → TAT |
| L432P | CTT → CCT |
| G461D | GGT → GAT |
| TTA → CTA (silent) | |
| GTA → GTG (silent) | |
| ATT → ATC (silent) | |
| W462G | TGG → GGG |
| G463D | GGC → GAC |
| L469H | CTT → CAT |
Amino acid change | Nucleotide change |
|---|---|
| C58R | TGC → CGC |
| M71V | ATG → GTG |
| L103S | TTA → TCA |
| D111N | GAT → AAT |
| E116G | GAA → GGA |
| L162P | CTC → CCC |
| H199R | CAC → CGC |
| TCT → TCC (silent) | |
| G216E | GGG → GAG |
| TTT → TTC (silent) | |
| L268I | TTA → ATA |
| TCA → TCT (silent) | |
| N278Y | AAC → TAC |
| CTA → CTG (silent) | |
| V286E | GTG → GAG |
| R330C | CGT → TGT |
| L337W | TTG → TGG |
| CAA → CAG (silent) | |
| CGA → CGT (silent) | |
| CAG → CAA (silent) | |
| S341P | TCT → CCT |
| TTT → TTC (silent) | |
| F342Y | TTC → TAC |
| Y369D | TAC → GAC |
| R376C | CGC → TGC |
| GAT → GAC (silent) | |
| H388Y | CAC → TAC |
| Y390H | TAC → CAC |
| TTC → TTT (silent) | |
| CTC → CTG (silent) | |
| C429R | TGT → CGT |
| C429Y | TGT → TAT |
| L432P | CTT → CCT |
| G461D | GGT → GAT |
| TTA → CTA (silent) | |
| GTA → GTG (silent) | |
| ATT → ATC (silent) | |
| W462G | TGG → GGG |
| G463D | GGC → GAC |
| L469H | CTT → CAT |
To test for physical interaction with Gal80 by a pull-down assay we reconstituted the Gal3 variant proteins as GST-Gal3-SA fusion proteins and expressed them in the strain Sc787 that carried a wild-type GAL80. In the presence of galactose, GST-Gal3-SA interacted with Gal80 as expected (Figure 4). Two of the 26 Gal3 variants (M71V and R330C) retained partial interactions with DBD-Gal80 in the reverse two-hybrid assay (Figure 3) and this was mirrored in the pull-down assay results. We were unable to detect 6 Gal3 variants (V286E, L337W, S341P, Y369D, C429R, and L469H) as GST fusion proteins. This was not surprising because all 6 represent drastic amino acid changes that could affect protein stability, reminiscent of what we have observed previously (Diepet al. 2006). Three Gal3 variants (L103S, N278Y, and H388Y) showed a partial loss of physical interaction with Gal80, while the remaining 15 variants were completely impaired in physical interaction with Gal80. In summary, of the 20 Gal3 variants that produced detectable GST fusion proteins, 18 showed defective interaction with Gal80. We refer to these proteins as Gal3 Gal80-nonbinder (Gal380NB) variants.
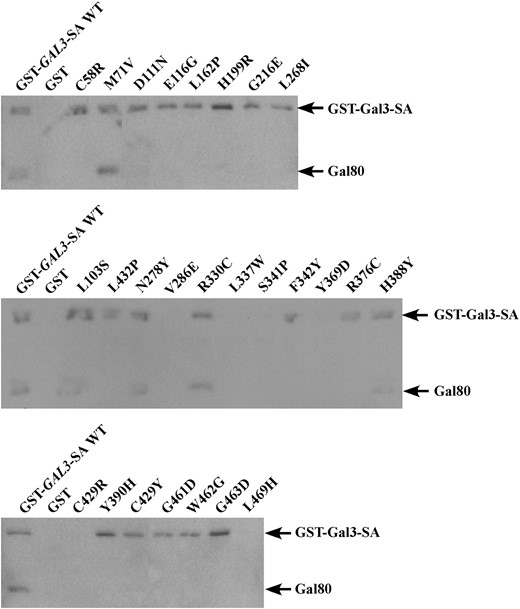
Physical interaction of GST-Gal3-SA variant proteins with Gal80 in vitro. Protein extracts from the strain Sc787 expressing GAL80 (pMPW82) and GST (pMPW61), GST-GAL3-SA (pCD121), or the GAL3 mutant alleles in the GST-GAL3-SA background were prepared. Approximately 1 mg of protein extracts was incubated with GT–Sepharose for 2 hr at 4°, washed three times, and then boiled and resolved on standard SDS–PAGE and analyzed by Western blot using antibodies against GST and Gal80 together at a 1:200 dilution.
The Gal380NB variants are defective in activation of a GAL gene promoter:
The capacity of the Gal380NB variants to activate the GAL gene switch was determined by assaying growth of cells containing the PGALHIS3 reporter gene on plates lacking histidine. Each gal380NB mutation in cis with the GAL3-SA mutation was expressed in a gal1Δgal3Δ strain (Sc781), and the cells were spotted on a plate containing galactose. Although cells with GAL3-SA showed appreciable activation of the GAL promoter, the level was reduced compared to cells with wild-type GAL3 (Figure 5), as might be expected since the SA insertion occurs at the highly conserved P-loop motif found within the active site where galactose and ATP bind (Plattet al. 2000; Thodenet al. 2005). The SA insertion could cause a decrease in galactose affinity and/or impair the conformational change thought to be induced by the ligands and required for binding to Gal80. Most of the gal380NB-SA derivatives showed complete loss of GAL gene activation while some showed only weak to moderate levels of activation. For example, the M71V and R330C variants supported a weaker activation of the GAL gene promoter than did Gal3-SA. This was consistent with the relative levels of their physical and two-hybrid interactions with Gal80. In summary, our yeast reverse two-hybrid selection yielded Gal3 variants showing impaired interaction with Gal80 and impaired GAL gene induction.

Impaired activation of the GAL promoter by the gal380NB mutant alleles. The strain Sc781 carrying the vector (pRS414), GAL3 WT (pTEB16), GAL3-SA (pAKS130), or the gal380NB mutant alleles in the GAL3-SA background was grown in selective liquid media until late log phase and 10-fold dilutions were spotted on selective agar plates. Activation of the GAL promoter was determined by colony growth coupled to PGALHIS3 expression.
Identification of gal380NB mutations that retain galactokinase-complementing activity in the GAL3-SA background:
Each gal380NB mutation in cis with the GAL3-SA mutation was tested for its ability to complement a gal1Δ for galactokinase activity. Those gal380NB mutations that lose galactokinase activity might affect either the global integrity or the local conformation of the protein or both. Strain Sc754 (gal1Δ) carrying a gal380NB mutation in cis with the GAL3-SA mutation was spotted onto plates with galactose as the sole carbon source. GAL3-SA, but not wild-type GAL3, was able to support cell growth on this medium, consistent with the observed galactokinase activity of the Gal3-SA protein (Figure 6) (Plattet al. 2000). Two of the 26 gal380NB mutations complemented the gal1Δ (L103S and D111N, Figure 6). The remaining 24 gal380NB mutations did not support growth in this assay (data not shown). We discuss below how these 26 Gal380NB variants might be defective in binding to Gal80.
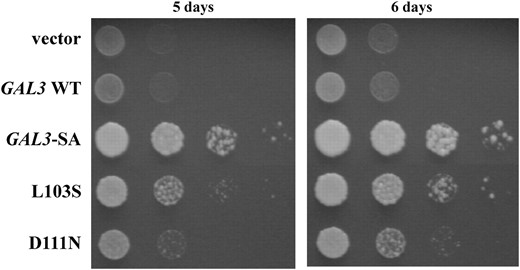
Capacity of gal380NB mutant alleles to complement the gal1Δ. The strain Sc754 (gal1Δ) carrying the vector (pRS414), GAL3 WT (pTEB16), GAL3-SA (pAKS130), or the gal380NB mutant alleles in the GAL3-SA background was grown in selective liquid media until late log phase and 10-fold dilutions were spotted on selective agar plates. Complementation of the gal1Δ was determined by colony growth on agar plates that contained galactose as the sole carbon source.
Localization of Gal3 variants on a Gal3 homology model implicates a surface in docking with Gal80:
With the ligand-bound crystal structure of Gal1 available (Thodenet al. 2005), it was possible to generate highly representative Gal3 homology models (Thodenet al. 2005; Diepet al. 2006) since the Gal1 and Gal3 proteins are highly similar. We mapped the various Gal3 amino acid variants onto a Gal3 homology model that we previously derived (Figure 7) (Diepet al. 2006). Fourteen of the Gal380NB variants affected residues that are buried within the hydrophobic core (C58R, M71V, L162P, G216E, L337W, F342Y, Y369D, H388Y, Y390H, C429R/Y, G461D, G463D, and L469H, red spheres in Figure 7). These amino acid substitutions could be acting to impair the binding of Gal3 to Gal80 through either direct or indirect effects on the allosteric communication, but due to their buried location, they are unlikely to act directly at Gal3's surface for docking with Gal80. The L162P variant is likely to affect the binding of ATP because it is found within the highly conserved ATP-binding P-loop motif (Plattet al. 2000). The 12 remaining Gal380NB variants (L103S, D111N, E116G, H199R, L268I, N278Y, V286E, R330C, S341P, R376C, L432P, and W462G) were localized on the surface of the model (green spheres in Figure 7). Two of these surface variants (L103S and D111N) retained the capacity to complement the gal1Δ for galactokinase activity and thus strongly implicate the L103 and D111 residues directly in either the allosteric communication or the docking surface. Remarkably, even though the remaining10 surface variants showed concordant loss of galactokinase-complementing activity and Gal80 binding, they colocalized in the same region of the homology model as L103 and D111 (Figure 7, green spheres). Moreover, this is the region that also includes D368, the site of the GAL3C-D368V suppressor of GAL80S-1-G323R (see above). This striking spatial colocalization of these 10 Gal380NB variant residues with L103, D111, and D368 highlights this region as a likely candidate for interaction with Gal80. The putative Gal80-interaction surface is localized on the Gal3 homology model on the opposite side of the hinge region (Figure 7, cyan spheres). We discuss possible implications of these observations and propose a mechanistic model for explaining how the binding of galactose and ATP induces Gal3 to interact with Gal80.
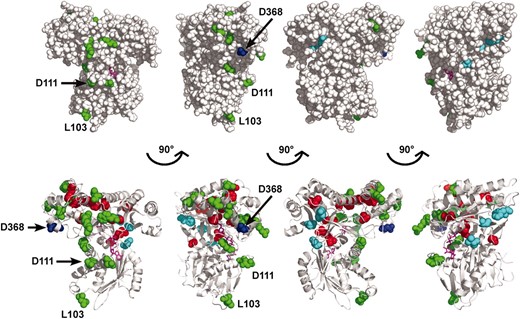
Structural interpretation of the Gal380NB variants. Amino acids corresponding to the Gal380NB variants were mapped onto the Gal3 homology model (previously derived) using the program PyMOL. The model was rendered to represent spheres (top) or spheres and ribbons (bottom), with each model having a 90° rotation to the right. Green, amino acids of Gal380NB variants that are exposed on the surface of the model; red, amino acids of Gal380NB variants that are buried in the hydrophobic core of the model; blue, D368, a residue within the insertion motif; cyan, V69, F237, and S509, residues within the hinge region; magenta, galactose and AMPPNP.
DISCUSSION
It is well established that the galactose-dependent interaction of Gal3 and Gal80 causes a relief of Gal80 inhibition of Gal4 and allows Gal4 to activate transcription of the GAL genes. Yet, knowledge of the Gal3 residues and structural elements that contribute to its galactose- and ATP-responsive interaction with Gal80 is lacking. We can anticipate the occurrence of three classes of residues that are directly involved in the conversion of Gal3 to a form active in binding to Gal80: (1) residues involved in forming the ligand-binding sites, (2) residues that participate in the allosteric communication between ligand-binding and the Gal80-docking determinants, and (3) residues that lie within the docking surface and directly contribute to binding energy.
In our selection for Gal3 variants defective for binding to Gal80 we identified the L162P substitution that is likely to affect ligand binding since L162 resides within the P-loop, a conserved nucleotide-binding motif within the GHMP superfamily. Thirteen other substitutions affect residues buried in the hydrophobic core of the Gal3 homology model. Such buried residues could participate directly in allosteric communication between ligand binding and articulation of the Gal80-binding surface or could act indirectly by altering the overall fold or local conformation. The remaining 12 substitutions show striking colocalization to structural elements that form a single composite surface on a Gal3 homology model. This composite surface is localized opposite to a previously identified hinge region that has been implicated in the allosteric communication effected by ligand binding (Menezeset al. 2003; Diepet al. 2006).
Our intergenic suppression studies of GAL3 and GAL80 mutations strongly implicate the importance of the affected amino acids in the mutual docking of Gal3 and Gal80. Four observations strengthen this notion. First, the inability of Gal3 to bind to the Gal80S-1-G323R protein is restored with the Gal3-D368V substitution variant, thereby confirming the physical basis of genetic suppression observed with the GAL3C-D368V/GAL80S-1-G323R suppression pair. Second, the Gal3-D368 residue (of which D368V suppresses GAL80S-1-G323R) lies within one of the four contiguous helices that constitute a 97-residue insertion motif that is common to the Gal80-binding proteins, Gal3, Gal1, and KlGal1 (Figure 8A, blue helices). The E. coli galK protein, which is unable to bind to Gal80, lacks two of the four helices including the Gal3-D368 helix. Third, the fact that the Gal3-D111N and Gal3-L103S variants are defective in binding to Gal80 but retain the capacity to complement the gal1Δ strongly argues for the direct involvement of residues D111 and L103 in galactose-mediated interaction with Gal80. Furthermore, the overall colocalization of residues D368, D111, and L103 to a single composite face of the Gal3 homology model (Figures 7 and 8B) suggests that this is the Gal80-binding surface. Fourth, on the basis of the crystal structure of KlGal80, it was suggested that a large disordered region of KlGal80 consisting of residues 328–362 becomes ordered when bound with KlGal1 (Thodenet al. 2007). Importantly, this region is surrounded by KlGal80 residues equivalent to variant residues of the S. cerevisiae Gal80 that are defective in interaction with Gal3. Thus, Thoden et al. proposed that the KlGal80 residues 328–362 mark the binding surface for KlGal1. The corresponding disordered region of the S. cerevisiae Gal80 consists of residues 327–346, which are flanked by residues G323 and M350 that were identified in our intergenic suppression. This is consistent with our proposal that the Gal80-G323 and -M350 residues directly interact with Gal3 and in turn implicate the Gal3-D111 and -D368 residues as constituents of the Gal80-binding surface. On the basis of the convergence of evidence presented above, we refer to the identified Gal3 surface as a putative Gal80-docking surface.

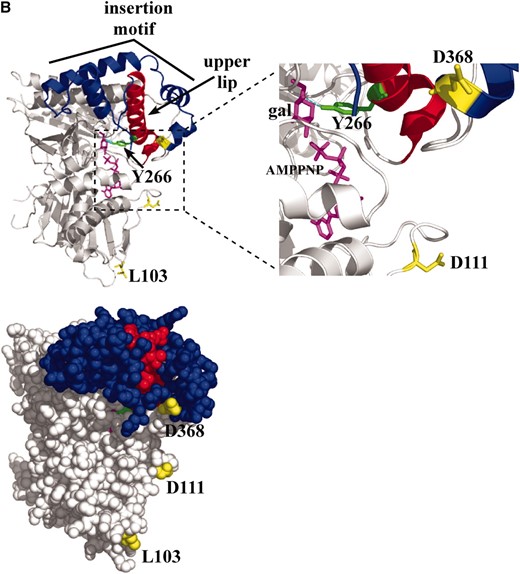
The upper lip and insertion motif of Gal3. (A) Multiple-sequence alignment. The alignment was performed using the TCoffee web server under the 3DCoffee mode, which considered secondary structural elements. Green, Gal1-Y274 (hydrogen bonding to galactose) and Gal3-Y266 (corresponds to Gal1-Y274); black bar, upper lip helix of HSK; red, corresponding upper lip helix of Gal1 and Gal3; blue, four helices of the insertion motif; yellow, Gal3-D368 (a residue within the insertion motif). Protein Data Bank codes: 1H72, HSK of Methanococcus jannaschii; 1PIE, galactokinase of Lactococcus lactis; 1S4E, galactokinase of Pyrococcus furiosus; 2AJ4, galactokinase (Gal1) of Saccharomyces cerevisiae. (B) Ribbon and sphere rendering of the Gal3 homology model. Green, Gal3-Y266; red, upper lip helix; blue, insertion motif helices; yellow, Gal3-L103, -D111, and -D368; magenta, galactose and AMPPNP.
The candidate Gal80-docking surface overlaps with the surface of Gal3 that is implicated in Gal80 binding on the basis of our reverse two-hybrid selection. All 12 of the surface-exposed residues affected by the gal380NB mutations colocalize to the Gal80-docking surface on the homology model. Moreover, they populate a composite surface that includes D368 (Figure 7, green spheres). Some of these may represent residues that do not participate directly in allosteric communication but rather have indirect nonspecific influences on Gal80 binding. However, here we have shown that 2 of these 12 substitutions (L103S and D111N) were impaired in Gal80 interaction but retained complementation of a gal1Δ, indicating that the mutations at these residues specifically affected the Gal80-binding activity.
How might the binding of galactose and ATP to Gal3 allosterically modulate the presentation of its Gal80-docking face? Gal3 is a member of the GHMP superfamily consisting of several members, including Gal1, whose crystal structures are known. The only member to have its apo- and holo-structures determined is homoserine kinase (HSK) (Zhouet al. 2000; Krishnaet al. 2001). Superimposition of the two structures identified conformational changes upon binding of ligands (homoserine and AMPPNP) (Krishnaet al. 2001). Those studies revealed that a local conformational change occurred at the upper lip helix (residues 181–189) of HSK and predicted a direct contact between the side chain of the R187 residue and the substrate homoserine by hydrogen bonding (Zhouet al. 2000; Krishnaet al. 2001). The corresponding upper lip helix in Gal1 (residues 271–292) also contains a side chain (Y274) (Figure 8A, green) that could make direct contact with galactose by hydrogen bonding (Figure 8B, cyan) (Thodenet al. 2005). The corresponding upper lip helix of Gal3 (263–284) (Figure 8A, red helix) and Gal1 immediately precedes the insertion motif (Figure 8A, blue helices), which is topologically linked to the upper lip helix by folding around it and forming a cap-like structure (Figure 8B). The last two helices of the four-helical insertion motif contain residue D368 and are absent in bacterial galactokinases (Figure 8A). Thus, we propose that binding of galactose and ATP induces a local conformational change at the Gal3 upper lip helix that in turn directly modulates the conformation of the insertion motif and thereby articulates presentation of the candidate Gal80-binding face.
We speculate that in addition to the insertion motif other residues proximal to the insertion motif also contribute to the interaction. Specifically, the 12 surface-exposed Gal380NB variants that localize predominantly around the insertion motif could contribute to the interaction with Gal80. Thus, we propose that the Gal80-binding surface of Gal3 is composed of noncontiguous residues including L103, D111, D368, and residues represented by the surface-exposed Gal380NB variants.
In summary, we identified Gal3 residues that participate in intergenic suppression of impaired Gal3–Gal80 binding (D368 and D111) and Gal3 residues that are important for binding to Gal80 (Gal380NB variants). These distinct data sets converge to reveal a single composite surface of Gal3 as a likely Gal80-binding surface. We presented a testable hypothesis of how the various structural elements of Gal3 and specific residues within the candidate docking surface might constitute an allosteric mechanism. Deciphering this mechanism and obtaining physical/chemical evidence for which residues directly contribute to the binding energy of the Gal3–Gal80 interaction await detailed biochemical studies.
Footnotes
Present address: Center for Regenerative Medicine, Massachusetts General Hospital, Harvard Medical School, 185 Cambridge St., CPZN-4265A, Boston, MA 02114.
Present address: Department of Biochemistry, Ohio State University, Room 233, Biological Sciences Bldg., 484 W. 12th Ave., Columbus, OH 43210.
Present address: Department Hematology and Oncology, Mount Sinai School of Medicine, Basic Sciences Bldg., 10 E. 101st St., Room 340, New York, NY 10029.
Present address: Bacteriology Division, USAMRIID, 1425 Porter St., Fort Detrick, MD 21702-5011.
Footnotes
Communicating editor: M. Johnston
Acknowledgement
This work was supported by the National Institutes of Health grant GM27975 to J.E.H.
References
Bajwa, W., T. E. Torchia and J. E. Hopper,
Bhat, P. J., and J. E. Hopper,
Bram, R. J., and R. D. Kornberg,
Fenton, C., H. Xu, E. I. Petersen, S. B. Petersen and M. R. el-Gewely,
Meyer, J., A. Walker-Jonah and C. P. Hollenberg,
Platt, A., H. C. Ross, S. Hankin and R. J. Reece,
Vidal, M., R. K. Brachmann, A. Fattaey, E. Harlow and J. D. Boeke,
Yano, K.-i., and T. Fukasawa,

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}