





Abstract
The maintenance of DNA replication fork stability under conditions of DNA damage and at natural replication pause sites is essential for genome stability. Here, we describe a novel role for the F-box protein Dia2 in promoting genome stability in the budding yeast Saccharomyces cerevisiae. Like most other F-box proteins, Dia2 forms a Skp1-Cdc53/Cullin-F-box (SCF) E3 ubiquitin–ligase complex. Systematic analysis of genetic interactions between dia2Δ and ∼4400 viable gene deletion mutants revealed synthetic lethal/synthetic sick interactions with a broad spectrum of DNA replication, recombination, checkpoint, and chromatin-remodeling pathways. dia2Δ strains exhibit constitutive activation of the checkpoint kinase Rad53 and elevated counts of endogenous DNA repair foci and are unable to overcome MMS-induced replicative stress. Notably, dia2Δ strains display a high rate of gross chromosomal rearrangements (GCRs) that involve the rDNA locus and an increase in extrachromosomal rDNA circle (ERC) formation, consistent with an observed enrichment of Dia2 in the nucleolus. These results suggest that Dia2 is essential for stable passage of replication forks through regions of damaged DNA and natural fragile regions, particularly the replication fork barrier (RFB) of rDNA repeat loci. We propose that the SCFDia2 ubiquitin ligase serves to modify or degrade protein substrates that would otherwise impede the replication fork in problematic regions of the genome.
FAITHFUL DNA replication requires the stabilization of replication forks at natural pause sites and at sites of DNA damage (Branzei and Foiani 2005). Collapse of replication forks results in the formation of DNA double-strand breaks (DSBs) that can then lead to illegitimate recombination and genome rearrangements, both of which are thought to be underlying causes of many human cancers (Lengauer et al. 1998). Physical impediments to replication fork progression include tightly bound non-nucleosomal protein–DNA complexes, DNA secondary structures, and regions of DNA damage, whereas inadequate dNTP pools cause forks to slow and eventually stall in a non-locus-specific manner (Branzei and Foiani 2005). Accumulated DNA damage or stalled replication forks elicit a checkpoint response that results in a delay of the cell cycle, induction of damage responsive genes, and the repair or bypass of the DNA lesion (Melo and Toczyski 2002; Branzei and Foiani 2005). The DNA damage checkpoint is activated upon detection of DNA lesions in G1 and G2 phase, and in S-phase the latter sometimes is referred to as the intra-S checkpoint. A second response in S-phase, referred to as the replication checkpoint, is the response to delayed DNA synthesis as caused by lowered dNTP pools upon inhibition of ribonucleotide reductase by hydroxyurea (HU). It is likely that the replication and intra-S checkpoint pathways are integrated such that the key signal is stalled or slowed replication forks, due to either dNTP shortage or collision with DNA damage. The only essential function of the S-phase checkpoint is to stabilize the fork when cells undergo replicative stress (Tercero et al. 2003) and thereby prevent the accumulation of recombinogenic structures (Lopes et al. 2001).
In both eukaryotic and prokaryotic genomes, region-specific barriers to replication fork progression pause and maintain replication forks in problematic regions of the genome (Rothstein et al. 2000). In budding yeast, replication pause sites have been characterized at rDNA repeats, centromeres, tRNA genes, inactive origins, silent mating-type loci, telomeres, and other delimited regions along the length of chromosome (Chr) III (Brewer and Fangman 1988; Greenfeder and Newlon 1992; Cha and Kleckner 2002; Ivessa et al. 2002). These sites are likely formed by stable protein complexes, which either may be incidental or may help coordinate various cellular processes with DNA replication. In particular, a high rate of transcription impairs replication fork progression and causes transcription-associated recombination, a phenomenon that occurs both at natural loci, such as the rDNA repeats and tRNA genes, and at artificially induced high-level transcription zones (Deshpande and Newlon 1996; Ivessa et al. 2003; Prado and Aguilera 2005). The best characterized pause site is the replication fork barrier (RFB) that resides in the rDNA repeats (Brewer and Fangman 1988). The RFB establishes a polar block to replication; i.e., DNA polymerase pauses only in the direction that is convergent to transcription and is thus thought to coordinate the high rate of rDNA transcription with replication of the rDNA locus (Kobayashi et al. 1998; Takeuchi et al. 2003). RFB activity depends on Fob1, which binds a discrete sequence in the nontranscribed spacer of the rDNA repeat and is necessary for elevated rates of recombination at the rDNA locus, including extraribosomal rDNA circle (ERC) formation (Kobayashi 2003). Programmed pause sites appear to be a ubiquitous feature of the replication process; for example, polar replication barriers have been identified in bacteria (Pai et al. 1996).
When a block to replication cannot be overcome, the S-phase checkpoint machinery promotes replication fork stabilization and subsequent restart (Branzei and Foiani 2005). If this mechanism fails, ensuing fork collapse results in dissociation of the replisome and the generation of replication intermediates, including DSBs (Cha and Kleckner 2002; Sogo et al. 2002; Cotta-Ramusino et al. 2005). To resume replication, recombination proteins are recruited to sites of collapsed forks, thus permitting homologous recombination pathways to aid in the bypass of the DNA lesion (Lambert et al. 2005). While recombination in this circumstance promotes cell viability, it comes at the expense of an increase in intra- and interchromosomal recombination that leads to site-specific gross chromosomal rearrangements (GCRs) (Lambert et al. 2005). Recombination-mediated replication restart is thus a means of last resort to rescue a collapsed fork and for this reason is actively suppressed. Several parallel pathways inhibit or resolve recombination events at the replication fork, including the Sgs1 and Srs2 DNA helicases (Branzei and Foiani 2005; Chang et al. 2005; Liberi et al. 2005; Mullen et al. 2005). The replication machinery can also circumscribe regions of damaged DNA, either by fork reversal and template switching to the sister-chromatid strand or through recruitment of specialized translesion synthesis DNA polymerases, as controlled by modified forms of the processivity factor PCNA/Pol30 (Branzei and Foiani 2005).
The ubiquitin–proteasome system targets many regulatory proteins for rapid intracellular proteolysis (Hershko 1983). Ubiquitin is conjugated to target proteins by a stepwise cascade of E1, E2, and E3 enzymes, which activate and transfer ubiquitin as a thioester linkage for ultimate transfer to a lysine residue on the substrate. Reiteration of the catalytic cycle synthesizes a ubiquitin polymer that targets the substrate to the 26S proteasome, where it is rapidly unfolded and degraded. A diverse array of E3 enzymes, often also referred to as ubiquitin ligases, specifically recognize one or more cognate substrates. Ubiquitin ligases fall into general classes: the HECT domain class, which forms a catalytic thioester with ubiquitin for transfer to the bound substrate, and the RING domain class, which binds and juxtaposes both the E2 and the substrate (Pickart 2001). The archetypal and best characterized of RING domain ubiquitin ligases is the Skp1-Cdc53/Cullin-F-box (SCF) family. SCF complexes are composed of the linker protein Skp1, the cullin scaffold protein Cdc53, and the RING domain protein Rbx1/Roc1/Hrt1 and any one of a number of variable substrate recognition subunits called F-box proteins (Bai et al. 1996; Patton et al. 1998). F-box proteins contain a Skp1-binding site called the F-box and a substrate interaction domain, such as a WD40 repeat domain or a leucine rich repeat (LRR) domain, which often recognize substrates in a phosphorylation-dependent manner (Willems et al. 2004). Budding yeast contains 21 recognizable F-box proteins, but as yet only a few of these have well-characterized functions.
We describe a novel role for the F-box protein Dia2 in maintaining genome stability. DIA2 was initially identified in a screen for mutants that exhibit increased invasive growth, although dia2Δ strains are only weakly invasive (Palecek et al. 2000). It has been suggested that Dia2 mediates the degradation of Tec1, a transcription factor implicated in invasive growth (Bao et al. 2004), and of ectopically expressed human cyclin E (Koepp et al. 2001); however, it is likely that the actual F-box protein for both of these substrates is Cdc4 (Koepp et al. 2001; Chou et al. 2004).The function of Dia2 thus remains equivocal. Here, we report that the dia2Δ mutation exhibits synthetic lethal/sick interactions with a host of replication and repair mutations, as well as sensitivity to DNA-damaging agents. Concordant with these genetic interactions, dia2Δ strains are defective in S-phase progression, have a high rate of endogenous DNA damage, and constitutively activate the DNA damage checkpoint. Moreover, the rates of chromosome loss, GCRs, and ERC formation are highly elevated in the dia2Δ mutant. These dia2Δ phenotypes suggest that Dia2 normally enables the replication machinery to cope with both extrinsically damaged DNA templates and intrinsic replication barriers and that, in the absence of Dia2, accumulation of endogenous damage results from the collapse of replication forks. SCFDia2 likely mediates degradation or modification of one or more substrates that would otherwise interfere with replication fork stability in problematic regions of the genome.
MATERIALS AND METHODS
Strains and growth conditions:
Strains used in this study were constructed by standard methods and are described in Table 1.
Strains used in this study
Strain | Genotype | Source |
|---|---|---|
| YMT235 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 | K. Nasmyth |
| YMT1448 | MATa ura3Δ0 leu2Δ0 his3Δ1 met15Δ0 | Rosetta Inpharmatics |
| YMT1738 | MATa/α ura3-1/ura3-1 leu2-3,112/leu2-3,112 his3-11 15/his3-11 15 trp1-1/ trp1-1 ade2-1/ade2-1 can1-100/can1-100 dia2Δ∷HIS3/his3-11 15 | This study |
| YMT1817 | MATa CFIII-HIS3-SUP11 dia2Δ∷URA3 | This study |
| YMT1819 | MATa CFIII-HIS3-SUP11 | This study |
| YMT1838,YMT235 | MATa dia2Δ∷HIS3 | This study |
| YMT1874 | MATα ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 rad9Δ∷URA3 | A. Amon |
| YMT1901 | MATα ura3Δ0 leu2Δ0 his3Δ1 lys2Δ0 MFA1pr-HIS3 can1Δ0 | Tong et al. (2001) |
| YMT1940, YMT1901 | MATα dia2Δ∷URA3 | This study |
| YMT2078 | MATa ura3Δ0 leu2Δ0 his3Δ1 met15Δ0 dia2Δ ∷KANR | This study |
| YMT2084 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 mec1Δ∷TRP sml1Δ∷HIS3 | R. Rothstein |
| YMT2085 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 rad53Δ∷HIS3 sml1-1 | R. Rothstein |
| YMT2545 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 RDN1∷ADE2 RAD5+ | H. Klein |
| YMT3337 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 Rad52YFP | R. Rothstein |
| YMT3398 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 rad9Δ∷HIS3 rad24Δ∷URA3 | N. Lowndes |
| YMT3401 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 chk1Δ∷TRP rad53Δ∷HIS3 sml1-1 | J. Rouse |
| YMT3415 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 Rad52YFP, dia2Δ∷TRP1 | This study |
| YMT3417 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1 100 mrc1Δ∷NAT | D. Durocher |
| YMT3420, RDKY3615 | MATa ura3-52 leu2Δ1 trp1Δ63 his3Δ200 lys2-bgl hom3-10 ade2Δ1 ade8 hxt13Δ∷URA3 | Chen and Kolodner (1999) |
| YMT3810 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 RDN1∷ADE2 RAD5+ dia2∷KANR | This study |
| YMT3812, RDKY3615 | MATa dia2Δ∷KANR | This study |
| YMT3835 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 Dia2GFP (pMT3988) | This study |
| YMT3854 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 rad9∷URA3 mrc1∷NAT (pMT2581) | This study |
| BY4741 | MATa rad14Δ∷KANR | EUROSCARF |
| BY4741 | MATa rad52Δ∷KANR | EUROSCARF |
Strain | Genotype | Source |
|---|---|---|
| YMT235 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 | K. Nasmyth |
| YMT1448 | MATa ura3Δ0 leu2Δ0 his3Δ1 met15Δ0 | Rosetta Inpharmatics |
| YMT1738 | MATa/α ura3-1/ura3-1 leu2-3,112/leu2-3,112 his3-11 15/his3-11 15 trp1-1/ trp1-1 ade2-1/ade2-1 can1-100/can1-100 dia2Δ∷HIS3/his3-11 15 | This study |
| YMT1817 | MATa CFIII-HIS3-SUP11 dia2Δ∷URA3 | This study |
| YMT1819 | MATa CFIII-HIS3-SUP11 | This study |
| YMT1838,YMT235 | MATa dia2Δ∷HIS3 | This study |
| YMT1874 | MATα ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 rad9Δ∷URA3 | A. Amon |
| YMT1901 | MATα ura3Δ0 leu2Δ0 his3Δ1 lys2Δ0 MFA1pr-HIS3 can1Δ0 | Tong et al. (2001) |
| YMT1940, YMT1901 | MATα dia2Δ∷URA3 | This study |
| YMT2078 | MATa ura3Δ0 leu2Δ0 his3Δ1 met15Δ0 dia2Δ ∷KANR | This study |
| YMT2084 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 mec1Δ∷TRP sml1Δ∷HIS3 | R. Rothstein |
| YMT2085 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 rad53Δ∷HIS3 sml1-1 | R. Rothstein |
| YMT2545 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 RDN1∷ADE2 RAD5+ | H. Klein |
| YMT3337 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 Rad52YFP | R. Rothstein |
| YMT3398 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 rad9Δ∷HIS3 rad24Δ∷URA3 | N. Lowndes |
| YMT3401 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 chk1Δ∷TRP rad53Δ∷HIS3 sml1-1 | J. Rouse |
| YMT3415 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 Rad52YFP, dia2Δ∷TRP1 | This study |
| YMT3417 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1 100 mrc1Δ∷NAT | D. Durocher |
| YMT3420, RDKY3615 | MATa ura3-52 leu2Δ1 trp1Δ63 his3Δ200 lys2-bgl hom3-10 ade2Δ1 ade8 hxt13Δ∷URA3 | Chen and Kolodner (1999) |
| YMT3810 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 RDN1∷ADE2 RAD5+ dia2∷KANR | This study |
| YMT3812, RDKY3615 | MATa dia2Δ∷KANR | This study |
| YMT3835 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 Dia2GFP (pMT3988) | This study |
| YMT3854 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 rad9∷URA3 mrc1∷NAT (pMT2581) | This study |
| BY4741 | MATa rad14Δ∷KANR | EUROSCARF |
| BY4741 | MATa rad52Δ∷KANR | EUROSCARF |
Strains used in this study
Strain | Genotype | Source |
|---|---|---|
| YMT235 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 | K. Nasmyth |
| YMT1448 | MATa ura3Δ0 leu2Δ0 his3Δ1 met15Δ0 | Rosetta Inpharmatics |
| YMT1738 | MATa/α ura3-1/ura3-1 leu2-3,112/leu2-3,112 his3-11 15/his3-11 15 trp1-1/ trp1-1 ade2-1/ade2-1 can1-100/can1-100 dia2Δ∷HIS3/his3-11 15 | This study |
| YMT1817 | MATa CFIII-HIS3-SUP11 dia2Δ∷URA3 | This study |
| YMT1819 | MATa CFIII-HIS3-SUP11 | This study |
| YMT1838,YMT235 | MATa dia2Δ∷HIS3 | This study |
| YMT1874 | MATα ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 rad9Δ∷URA3 | A. Amon |
| YMT1901 | MATα ura3Δ0 leu2Δ0 his3Δ1 lys2Δ0 MFA1pr-HIS3 can1Δ0 | Tong et al. (2001) |
| YMT1940, YMT1901 | MATα dia2Δ∷URA3 | This study |
| YMT2078 | MATa ura3Δ0 leu2Δ0 his3Δ1 met15Δ0 dia2Δ ∷KANR | This study |
| YMT2084 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 mec1Δ∷TRP sml1Δ∷HIS3 | R. Rothstein |
| YMT2085 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 rad53Δ∷HIS3 sml1-1 | R. Rothstein |
| YMT2545 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 RDN1∷ADE2 RAD5+ | H. Klein |
| YMT3337 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 Rad52YFP | R. Rothstein |
| YMT3398 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 rad9Δ∷HIS3 rad24Δ∷URA3 | N. Lowndes |
| YMT3401 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 chk1Δ∷TRP rad53Δ∷HIS3 sml1-1 | J. Rouse |
| YMT3415 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 Rad52YFP, dia2Δ∷TRP1 | This study |
| YMT3417 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1 100 mrc1Δ∷NAT | D. Durocher |
| YMT3420, RDKY3615 | MATa ura3-52 leu2Δ1 trp1Δ63 his3Δ200 lys2-bgl hom3-10 ade2Δ1 ade8 hxt13Δ∷URA3 | Chen and Kolodner (1999) |
| YMT3810 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 RDN1∷ADE2 RAD5+ dia2∷KANR | This study |
| YMT3812, RDKY3615 | MATa dia2Δ∷KANR | This study |
| YMT3835 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 Dia2GFP (pMT3988) | This study |
| YMT3854 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 rad9∷URA3 mrc1∷NAT (pMT2581) | This study |
| BY4741 | MATa rad14Δ∷KANR | EUROSCARF |
| BY4741 | MATa rad52Δ∷KANR | EUROSCARF |
Strain | Genotype | Source |
|---|---|---|
| YMT235 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 | K. Nasmyth |
| YMT1448 | MATa ura3Δ0 leu2Δ0 his3Δ1 met15Δ0 | Rosetta Inpharmatics |
| YMT1738 | MATa/α ura3-1/ura3-1 leu2-3,112/leu2-3,112 his3-11 15/his3-11 15 trp1-1/ trp1-1 ade2-1/ade2-1 can1-100/can1-100 dia2Δ∷HIS3/his3-11 15 | This study |
| YMT1817 | MATa CFIII-HIS3-SUP11 dia2Δ∷URA3 | This study |
| YMT1819 | MATa CFIII-HIS3-SUP11 | This study |
| YMT1838,YMT235 | MATa dia2Δ∷HIS3 | This study |
| YMT1874 | MATα ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 rad9Δ∷URA3 | A. Amon |
| YMT1901 | MATα ura3Δ0 leu2Δ0 his3Δ1 lys2Δ0 MFA1pr-HIS3 can1Δ0 | Tong et al. (2001) |
| YMT1940, YMT1901 | MATα dia2Δ∷URA3 | This study |
| YMT2078 | MATa ura3Δ0 leu2Δ0 his3Δ1 met15Δ0 dia2Δ ∷KANR | This study |
| YMT2084 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 mec1Δ∷TRP sml1Δ∷HIS3 | R. Rothstein |
| YMT2085 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 rad53Δ∷HIS3 sml1-1 | R. Rothstein |
| YMT2545 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 RDN1∷ADE2 RAD5+ | H. Klein |
| YMT3337 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 Rad52YFP | R. Rothstein |
| YMT3398 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 rad9Δ∷HIS3 rad24Δ∷URA3 | N. Lowndes |
| YMT3401 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 chk1Δ∷TRP rad53Δ∷HIS3 sml1-1 | J. Rouse |
| YMT3415 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 Rad52YFP, dia2Δ∷TRP1 | This study |
| YMT3417 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1 100 mrc1Δ∷NAT | D. Durocher |
| YMT3420, RDKY3615 | MATa ura3-52 leu2Δ1 trp1Δ63 his3Δ200 lys2-bgl hom3-10 ade2Δ1 ade8 hxt13Δ∷URA3 | Chen and Kolodner (1999) |
| YMT3810 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 RDN1∷ADE2 RAD5+ dia2∷KANR | This study |
| YMT3812, RDKY3615 | MATa dia2Δ∷KANR | This study |
| YMT3835 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 Dia2GFP (pMT3988) | This study |
| YMT3854 | MATa ura3-1 leu2-3,112 his3-11 15 trp1-1 ade2-1 can1-100 rad9∷URA3 mrc1∷NAT (pMT2581) | This study |
| BY4741 | MATa rad14Δ∷KANR | EUROSCARF |
| BY4741 | MATa rad52Δ∷KANR | EUROSCARF |
Except where specified, cultures were grown in rich XY medium (2% peptone, 1% yeast extract, 0.01% adenine, 0.02% tryptophan) containing 2% glucose. Cell size was determined essentially as described (Jorgensen et al. 2004) using a Coulter Channelizer Z2 (Beckman-Coulter). Cells were synchronized in G1 phase in the presence of 5 μg/ml α-factor for 2 hr at 30°. An RNR1-expressing plasmid (pMT2581) was used to ensure viability of mrc1Δ rad9Δ double mutants derived from genetic crosses.
FACS analysis:
Approximately 1 × 107 cells were collected from log-phase cultures and processed as described (Jorgensen et al. 2004). DNA was stained with Sytox Green (Molecular Probes, Eugene, OR) and profiles were analyzed using a Becton Dickinson (San Jose, CA) FACS Calibur machine and the CellQuest Pro and ModFit LT software (BD Biosciences).
DNA damage and genotoxic stress assays:
Saturated cultures were adjusted to OD600 = 0.8 and serially diluted in 10-fold steps and 4-μl volumes spotted onto untreated medium or medium containing or exposed to 0.02% (v/v) methyl methanesulfonate (MMS), 200 mm HU, 5 μg/ml camptothecin, 0.1 μg/ml 4-nitroquinolone-1-oxide (4-NQO), 200 J/m2 UV or 100 Gy X rays. Plates were incubated at 30° for 2 days. Conditions used to test for the intra-S checkpoint were as described (Paulovich and Hartwell 1995). Plasmids expressing full-length DIA2 (pMT2484) or DIA2ΔF-box (pMT2742) from the endogenous DIA2 promoter were used in MMS sensitivity complementation tests.
Protein detection:
Co-immunoprecipitation experiments with FLAG- and MYC-tagged constructs were as described previously (Ho et al. 2002). For immunoblots, proteins were separated on a 10% or 12% SDS–PAGE, transferred to PVDF membranes, and probed with 9E10 monoclonal antibody (1:10,000), anti-FLAG M2 monoclonal antibody (1:2000, Sigma, St. Louis), or anti-Skp1 polyclonal (1:5000), as indicated. Detection was with HRP-conjugated secondary antibodies at 1:10,000 dilution (Amersham, Buckinghamshire, UK) followed by Supersignal West Pico chemiluminescent substrate (Pierce, Rockford, IL) and analysis on a Fluor-S Multiimager (Bio-Rad, Hercules, CA). To detect Rad53 isoforms, log-phase cultures were arrested in G1 phase with α-factor, washed, and released into fresh media. Total protein extracts were resolved by 7.5% SDS–PAGE, transferred onto nitrocellulose, and probed with a primary rabbit anti-Rad53 antibody (1:750) and secondary donkey anti-rabbit HRP-conjugated antibody (1:10,000, Amersham). For in situ autophosphorylation (ISA) assays, log-phase cultures were either treated or not treated with 200 mm HU for 1 hr, and then processed as described (Pellicioli et al. 1999).
Synthetic genetic array screen:
A dia2Δ∷URA3 MATα strain (yMT1940) was mated to a collection of 4422 individual xxx∷kanR MATa haploid deletion mutants (Giaever et al. 2002) by manual replica pinning as described (Tong et al. 2001). Three independent screens were carried out in triplicate. All genetic interactions identified in at least two of the three screens were subsequently confirmed by tetrad analysis. Synthetic lethal interactions were defined as double mutants that failed to grow either in the original tetrad or upon restreaking onto fresh medium; synthetic sick interactions were defined as double mutants that grew notably more slowly than either single gene deletion strain. Two-dimensional hierarchical clustering of dia2Δ synthetic genetic interactions was performed against a data set of 284 synthetic genetic array (SGA) and diploid synthetic lethal analysis by microarray (dSLAM) screens (Tong et al. 2004; Pan et al. 2006; Reguly et al. 2006) using an average linkage clustering algorithm (Eisen et al. 1998) and MapleTree (http://mapletree.sourceforge.net/). Graphical representations of genetic interactions were rendered with the Osprey visualization suite (Breitkreutz et al. 2003). All data are available from the BioGRID interaction database at http://www.thebiogrid.org (Stark et al. 2006).
Microarray analysis:
Genomewide expression profiles were determined for dia2Δ (yMT2078) and isogenic wild-type (yMT1448) strains grown to early log phase (OD 0.2–0.4) at 30°. Polyadenylated RNA was fluorescently labeled with Cy3- or Cy5-conjugated dCTP (Amersham) and dia2Δ and wild-type samples were competitively hybridized against full genome 6200 feature ORF arrays (UHN Microarray Centre, Toronto). All hybridizations were replicated with fluor reversal. Arrays were scanned with a Scanarray 4000 (GSI Lumonics) and images were processed by eliminating corrupted spots, normalizing spot intensity to total Cy5 and Cy3 signal, eliminating spots with low intensity, and averaging duplicate spots (Jorgensen et al. 2004).
Genome instability assays:
Chromosome loss rate of a CFIII-SUP11-HIS reporter fragment was measured as described (Hieter et al. 1985). After growth to saturation overnight in SD–trp–his 2% glucose medium to maintain the artificial chromosome, cells were diluted into SD–trp 2% glucose media, grown for 6 hr at 30° to early log phase (2–4 × 106 cells/ml), sonicated briefly, and plated onto SD–trp–ade 2% glucose agar media supplemented with a limited amount of adenine (10 mg/liter). Chromosome loss rate per generation was calculated as the number of half-sectored colonies divided by the number of colonies. GCR rates were determined with a strain in which HXT13 (∼7.5 kb telomeric to CAN1 on Chr V) is replaced by URA3 (Chen and Kolodner 1999). Control (yMT3420) and dia2Δ (yMT3812) revertants that arose from loss of the region encompassing CAN1 and URA3 were scored on canavanine and 5-fluoroorotic acid (5-FOA)-containing medium. Rates were calculated by fluctuation analysis using the method of the median as described (Kanellis et al. 2003) and represent the average of two independent experiments using sets of five independent cultures. Mitotic recombination rates were determined by loss of an ADE2 marker integrated into the rDNA locus on Chr XII as described (Christman et al. 1988). Recombination rate per generation was calculated by dividing the number of half-sectored colonies by the total number of colonies.
Pulsed-field gel electrophoresis and hybridization:
Standard pulsed-field gel electrophoresis (PFGE) procedures were used according to the manufacturer's instructions (Bio-Rad). Agarose plugs containing 3 × 108 cells/ml were loaded onto a 1% agarose gel in 0.5 × Tris–borate EDTA (TBE) buffer and electrophoresed at 6 V at an angle of 120° for 24 hr at 14° with an initial switch time of 60 sec and a final switch time of 120 sec. Gels were stained and photographed and then transferred to nylon membranes and probed with γ-32P-ATP-labeled 35S rDNA or MCM3 fragments. To assess formation of ERCs, plugs were prepared as above and DNA was resolved on a conventional 1% agarose gel. Gels were transferred to nylon membranes and probed with a γ-32P-ATP-labeled 35S rDNA fragment.
Live cell imaging and fluorescent microscopy:
DIC and fluorescence microscopy were performed with an Eclipse E600FN microscope (Nikon) and Orca CCD camera (Hamamatsu, Bridgewater, NJ). Metamorph Software (Universal Imaging, West Chester, PA) was used to capture and process images as described (Jorgensen et al. 2004). Yellow fluorescent protein (YFP) exposure times were in the range of 150–200 msec and serial sections through live cells were set to 21 z-planes at 0.4-μm spacing.
RESULTS
Absence of Dia2 causes an S/G2/M cell cycle delay:
Unlike most F-box proteins that contain a single C-terminal protein interaction domain, Dia2 is unusual in that it contains an N-terminal tetratricopeptide repeat (TPR) domain and a C-terminal LRR domain (Willems et al. 2004). As not all F-box proteins form SCF complexes (Willems et al. 2004), we assessed whether Dia2 assembled into an SCF complex in vivo (Figure 1A). An interaction was detected between Dia2 and the SCF core subunits Cdc53 and Skp1, in accord with the previously reported in vitro reconstitution of SCFDia2 (Koepp et al. 2001; Kus et al. 2004). Strains that lacked DIA2 grew at a rate 30% slower than that of wild-type cells and exhibited a heterogeneous colony size (Figure 1B). The small-size colonies were not due to petite formation caused by loss of mitochondrial DNA because all colonies were able to grow anerobically on glycerol medium (data not shown). Intriguingly, dia2Δ strains developed an increasingly severe growth defect upon prolonged propagation (data not shown). Asynchronous populations of dia2Δ strains had a modal cell size that was twice that of wild-type strains (Figure 1C). The slow growth rate and increased cell size suggested a defect in cell cycle progression, and FACS analysis of DNA content consistently revealed an accumulation of cells in both S- and G2/M-phase (Figure 1D). The absence of Dia2 thus resulted in an S/G2/M cell cycle delay, which in other contexts often results from S-phase defects that activate the G2/M cell cycle checkpoint.
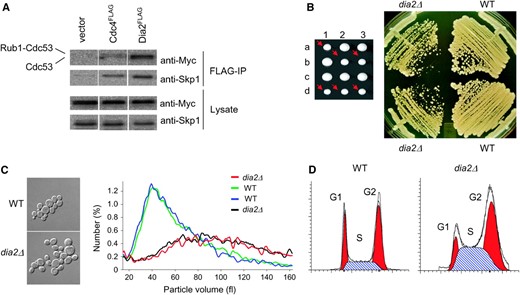
The F-box protein Dia2 forms an SCF complex and is required for normal cell cycle progression. (A) Physical interactions between Dia2Flag and core components of the SCF complex. Cells were transformed with an empty vector or plasmids encoding Cdc4FLAG or Dia2FLAG expressed from the GAL1 promoter, in conjunction with a CEN plasmid that expressed Cdc53MYC from the CDC53 promoter. Immunoblots of whole-cell lysates and anti-Flag immunoprecipitations were probed anti-Skp1 and anti-MYC antibodies. (B) Growth defect of dia2Δ strains. The dia2Δ spore clones from a sporulated heterozygous diploid DIA2/dia2Δ strain (yMT1738) are indicated by arrows. Cells from a representative tetrad were restreaked onto rich medium and grown at 30o for 2 days. (C) Cell size distribution of dia2Δ strains. Spore clones from a DIA2/dia2Δ tetrad were grown to early log phase in liquid medium, analyzed on a Coulter channelizer, and visualized by DIC microscopy at ×100 magnification. (D) DNA content of asynchronous wild-type (WT) and dia2Δ populations. FACS profiles were deconstructed into G1, S, and G2/M components using ModFit LT software.
Genetic interactions of DIA2 with DNA replication, repair, and checkpoint pathways:
To delineate Dia2 function, we employed SGA technology to construct a collection of double mutants between dia2Δ and an ordered array of 4422 viable single gene deletion strains (Tong et al. 2001). Double-mutant combinations that result in inviability (synthetic lethality) or in reduced fitness (synthetic sickness) identify genes that either converge on an essential biological process or exhibit a damage-response relationship (Hartman et al. 2001). The SGA screen identified 55 deletion mutants that exhibited overt synthetic genetic interactions with dia2Δ, all of which were confirmed by tetrad analysis. Of the 55 double-mutant interactions, 19 were synthetic lethal (Figure 2). Many of the interactions occurred with genes implicated in DNA replication (e.g., RRM3, PIF1, RAD27, RNR4), recombination (e.g., RAD52, MRE11, XRS2), chromosome segregation (e.g., BFA1, MAD1, CIK1, KAR3), and the DNA damage checkpoint (PPH3). Significantly, DIA2 also exhibited strong interactions with a spectrum of genes that promote replication fork restart, namely SGS1 and TOP3, SLX1 and SLX4, SLX5 and SLX8, and MUS81 and MMS4; each of these gene pairs encode protein complexes that play different roles in fork restart (Fabre et al. 2002; Fricke and Brill 2003; Zhang et al. 2006). In addition, DIA2 displayed synthetic interactions with genes required for oxidative stress metabolism (e.g., SOD1, LYS7), transcription (e.g., CTK1, SWI4), and chromatin structure (e.g., SWR1, HST4). Recently, a cohort of dSLAM screens focused on DNA replication and chromosome segregation pathways recovered 112 synthetic lethal/synthetic sick interactions with a dia2Δ query strain, and an additional 9 dia2Δ interactions with other query strains (Pan et al. 2006). Of the 55 confirmed hits in our dia2Δ screen, only 23 overlapped with the Pan et al. (2006) data set, indicating that neither experimental approach exhaustively identified dia2Δ genetic interactions. The myriad interactions recovered by each systematic dia2Δ screen suggested that Dia2 function is required to cope with a host of defects in normal replication, repair, and recombination processes, as caused either directly or indirectly by various mutations.
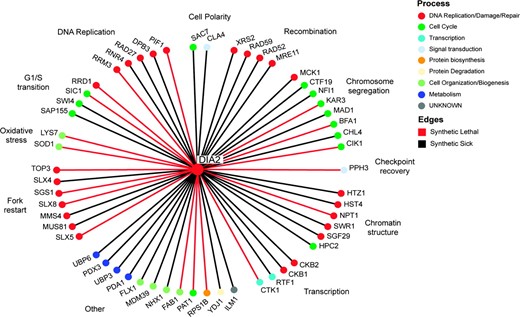
Systematic analysis of dia2Δ synthetic genetic interactions. A total of 55 synthetic lethal (red edges) and synthetic sick genetic interactions (black edges) detected in triplicate SGA screens were confirmed by tetrad analysis. Interactions are grouped according to indicated cellular functions and individual nodes are colored by a reduced hierarchy of gene ontology (GO) biological processes ranked in the order shown in the color key.
Genes that perform related functions tend to cluster together in genomewide synthetic lethal interaction profiles (Tong et al. 2004). We therefore performed two-dimensional hierarchical clustering of the dia2Δ genetic interaction profile in the context of a large combined data set of 10,334 synthetic lethal interactions (Figure 3A), as derived from 284 systematic SGA and dSLAM screens reported in the primary literature (Tong et al. 2004; Pan et al. 2006; Reguly et al. 2006). As revealed by the dominant central cluster, many of the systematic screens carried out to date have focused on DNA replication, DNA damage, and chromosome segregation pathways (Tong et al. 2004; Pan et al. 2006). Within this large data set, the dia2Δ profile clustered most closely with query mutations that disrupt aspects of DNA replication and repair, namely homologous recombination (rad51Δ, rad52Δ, rad54Δ, rad57Δ, hpr5Δ/srs2Δ), the replication checkpoint (csm3Δ, tof1Δ, mrc1Δ), an alternative replication factor C complex implicated in establishment of sister-chromatid cohesion (dcc1Δ, ctf8Δ, ctf18Δ), the Mre11–Rad50–Xrs2 complex that mediates the DNA damage response and facilitates DSB repair (mre11Δ, rad50Δ, xrs2Δ), and postreplicative repair (rad5Δ, rad18Δ). Overall, the correlated genetic profiles revealed by hierarchical clustering suggested a role for Dia2 in some aspect of DNA replication.
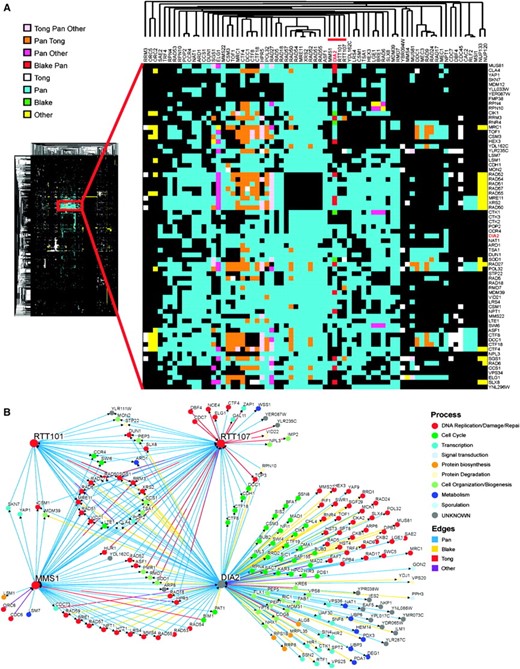
Two-dimensional hierarchical clustering of synthetic genetic interaction profiles. (A) A combined unique set of 144 genetic interactions from dia2Δ query screens reported in this study and in Pan et al. (2006) were clustered against 284 systematic genetic screens curated from the primary literature (Reguly et al. 2006). A locally dense region of interactions that contains the dia2Δ profile is expanded and immediate dia2Δ neighbors are indicated by the red bar. The source of each genetic interaction is indicated by the color key. (B) Shared interactions among dia2Δ, rtt101Δ, rtt107Δ,, and mms1Δ. Network shows all interactions retrieved from the full 284-screen data set for each of the four nodes. Edges are colored by interaction source; nodes are colored by the same GO biological processes as in Figure 2.
In addition to the above well-characterized replication and repair functions, dia2Δ clustered immediately adjacent to rtt101Δ, rtt107Δ/esc4Δ, and mms1Δ/rtt108Δ (Figure 3A, red bar). The RTT genes were isolated in a screen for regulators of Ty1 transposition, which uncovered many pathways that dictate genome stability (Scholes et al. 2001). Rtt101 is the cullin subunit of an SCF-like ubiquitin ligase that has recently been implicated in replication fork progression through natural pause sites and damaged DNA (Luke et al. 2006); Rtt107/Esc4 is a BRCT repeat-containing protein that is required for replication restart after DNA damage (Rouse 2004); Mms1 is required for resistance to MMS and other genotoxins (Hryciw et al. 2002) and appears to operate in the same pathway as Mms22 (Araki et al. 2003). Direct inspection of all genetic interactions in this cohort revealed that 21 of 29 interactions with rtt101Δ, 34 of 55 hits with rtt107Δ and 38 of 49 hits with mms1Δ also interact with dia2Δ; in addition, many other cross-interactions were evident, including all possible pairwise synthetic lethal interactions (Figure 3B). These similar genetic profiles suggest that Dia2, Rtt101, Rtt107, and Mms1 functions converge at the replication fork, particularly under adverse circumstances. Because the dia2Δ mutant exhibited a larger and more diverse set of interactions than other members of the cohort (Figure 3B), Dia2 likely performs additional functions as well.
Dia2 is required for resistance to some but not all DNA-damaging agents:
Mutations that compromise S-phase progression often cause sensitivity to DNA-damaging agents or replication stress. Prompted by the synthetic genetic interactions identified in the SGA screen, we tested the sensitivity of a dia2Δ strain to various genotoxic agents, using appropriate checkpoint (mec1Δ sml1Δ), nucleotide excision repair (rad14Δ), and homologous recombination (rad52Δ) mutants as controls (Figure 4A). Plating efficiency of the dia2Δ strain was severely inhibited by the potent alkylating agent MMS. This sensitivity to MMS was most likely due to defective SCFDia2 activity because a dia2Δ strain bearing a version of Dia2 that lacked the F-box domain was as susceptible as the dia2Δ strain itself (Figure 4B). Intermediate growth inhibition was caused by 4-NQO, which forms bulky adducts that are substrates for nucleotide excision repair, and by camptothecin, a poison that traps covalent topoisomerase I/DNA intermediates. A high concentration of the replication inhibitor HU modestly reduced the plating efficiency of a dia2Δ strain. The sensitivity of the dia2Δ mutant to MMS, HU, and camptothecin has recently been reported in other studies (Ohya et al. 2005; Koepp et al. 2006; Pan et al. 2006). To determine if these defects reflected a general sensitivity to DNA damage, we also tested sensitivity of the dia2Δ strain to UV light, which induces thymidine dimers and other photo products, and to X rays, which produce DSBs. Unlike many other replication and DNA damage response mutants, the dia2Δ strain was insensitive to these agents (Figure 4A). Dia2 thus does not appear to have a role in nucleotide excision repair or homologous recombination per se, but rather seems to aid in replication fork progression in the presence of certain DNA adducts.
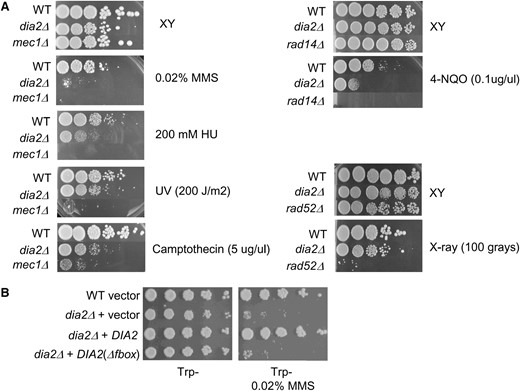
Dia2 is required for resistance to agents that generate DNA adducts. The indicated strains were serially diluted in 10-fold steps and plated onto rich medium (A) or synthetic medium lacking tryptophan (B) in either the presence or the absence of 0.02% (v/v) MMS, 200 mm HU, 5 μg/ml camptothecin, 0.1 μg/ml 4-NQO, 200 J/m2 of UV, or 100 Gy of X rays. Photographs were taken after 2 days at 30°.
The DNA damage checkpoint is essential in the absence of DIA2:
As cells lacking Dia2 exhibited a pronounced S and G2/M cell cycle delay, reliance on nonessential checkpoint proteins recovered in the SGA screen, and hypersensitivity to certain DNA-damaging agents, we determined in further detail if the DNA damage checkpoint was necessary for viability of a dia2Δ strain (Figure 5). Both Mec1 and Rad53 are essential proteins that transduce signals in the DNA damage checkpoint. Viability of rad53Δ and mec1Δ mutant strains, but not their checkpoint deficiencies, is rescued by deleting the ribonucleotide reductase inhibitor SML1 (Zhao et al. 1998). Triple mutants of the genotype dia2Δ mec1Δ sml1Δ and dia2Δ rad53Δ sml1-1 were inviable, indicating that Dia2 normally prevents some form of endogenous DNA damage that invokes a necessary checkpoint response.
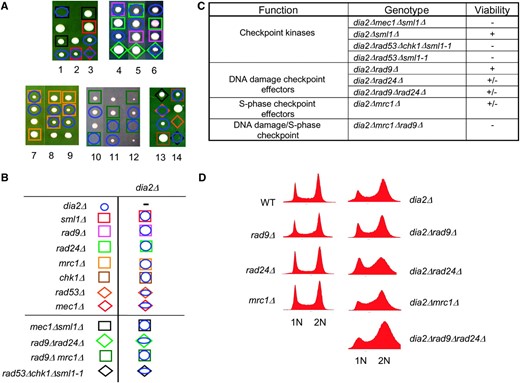
The DNA checkpoint is required for viability of a dia2Δ strain. (A) Representative tetrads were dissected for dia2Δ mec1Δ sml1Δ, dia2Δ rad53Δ chk1Δ sml1-1, dia2Δ rad9Δ rad24Δ, and dia2Δ mrc1Δ rad9Δ heterozygous diploids. Wild-type spores are unmarked, as are inviable spores whose genotype could not be inferred. The dia2Δ rad53Δ chk1Δ cross carries an unmarked sml1-1 allele to allow viability of the rad53Δ mutation. Note that, in the last tetrad shown, rad53Δ alone is inviable because sml1-1 did not segregate to this spore clone; however, specific inviability of dia2Δ rad53Δ sml1-1 was inferred from the viability of rad53Δ chk1Δ sml1-1 in the adjacent cross, as well as from other tetrads not shown. The mrc1Δ rad9Δ cross carries a <RNR1 LEU2 2μm> plasmid to maintain viability of the mrc1Δ rad9Δ double mutant. (B) Legend for determined genotypes. (C) Summary of genetic interactions. Minus sign indicates full inviability; plus sign indicates full viability. (D) Failure of various DNA damage checkpoint mutants to bypass the G2/M cell cycle delay of dia2Δ strains. DNA content of asynchronous cultures of the indicated strains was determined by FACS analysis.
To delineate the checkpoint pathway(s) that maintain dia2Δ viability, we assessed genetic interactions between dia2Δ and mutations in the S-phase (mrc1Δ) and DNA damage (rad9Δ, rad24Δ) branches of the checkpoint. Absence of the fork-associated checkpoint protein Mrc1 moderately reduced the growth rate of the dia2Δ mutant, whereas elimination of the checkpoint adaptor Rad9 and/or the clamp loading factor Rad24 only slightly impaired growth rate (Figure 5, A–C). Loss of both the Mrc1 and the Rad9 branches results in synthetic lethality, but this lethality can be suppressed by overexpression of RNR1 (Alcasabas et al. 2001). As expected, a mrc1Δ rad9Δ dia2Δ triple mutant was inviable despite overexpression of RNR1. These results suggested that while either of the S-phase and DNA damage branches of the DNA checkpoint suffice to allow viability of the dia2Δ mutant, the S-phase branch is the more important of the two arms. Given that checkpoint activation is usually accompanied by a cell cycle delay, we anticipated that the accumulation of cells in G2/M in the dia2Δ mutant might be dependent on Mrc1 and/or Rad9/Rad24. However, abrogation of either of the main checkpoint branches failed to alter the G2/M cell cycle delay in the dia2Δ strain, as shown by FACS analysis (Figure 5D). This delay thus either requires both arms of the checkpoint or is mediated by an as yet uncharacterized checkpoint effector.
Interestingly, as recovered in the SGA screen, the dia2Δ strain also requires the phosphatase Pph3 for viability (Figure 2). Because Pph3 is required for dephosphorylation of γ-H2Ax and recovery from the checkpoint response (Keogh et al. 2006), persistent DNA replication defects in dia2Δ cells may cause permanent checkpoint arrest in the absence of desensitization.
Dia2 prevents endogenous DNA damage in S-phase:
The requirement for Mec1, Rad53, and Mrc1 for viability of the dia2Δ mutant and the accumulated S-phase fraction in asynchronous dia2Δ cultures (Figure 1D) suggested a possible S-phase progression defect in dia2Δ strains. We therefore assessed the kinetics of S-phase progression in synchronous cultures released from a mating pheromone-induced G1 phase arrest. While most of the dia2Δ population appeared to progress from G1 phase to G2/M phase with wild-type kinetics, a significant subpopulation accumulated in S-phase (Figure 6A). Signals that trigger the replication checkpoint result in Mec1-dependent activation of Rad53 (Sanchez et al. 1996; Sun et al. 1996), which can be monitored by an ISA (Pellicioli et al. 1999). Given that dia2Δ cells have a cell cycle delay, we tested whether this defect correlates with activation of Rad53 kinase activity. As expected, upon treatment of wild-type cells with 200 mm HU, Rad53 was activated, as revealed by its autophosphorylation in vitro (Figure 6B). However, in dia2Δ strains, Rad53 exhibited marked activation in untreated asynchronous cultures. Exposure to 200 mm HU further induced Rad53 autophosphorylation in dia2Δ cells, possibly because fewer cycling cells are in S-phase as compared to cultures arrested in HU. Because MMS and 4-NQO also further activated Rad53 in the dia2Δ strain (data not shown), Dia2 is not required for maximal activation of the checkpoint. To determine whether checkpoint activation in dia2Δ cells required S-phase progression, we monitored Rad53 phosphorylation in synchronous cultures by virtue of its phosphorylation-dependent mobility shift (Figure 6C). In the absence of genotoxic stress, wild-type cells passed through S into G2 without Rad53 phosphorylation. In contrast, while Rad53 was unphosphorylated in G1 phase in the dia2Δ strain, as cells progressed through S-phase and into G2 phase, slower migrating Rad53 isoforms became apparent. This result suggested that Dia2 normally prevents endogenous DNA damage from arising in S-phase.
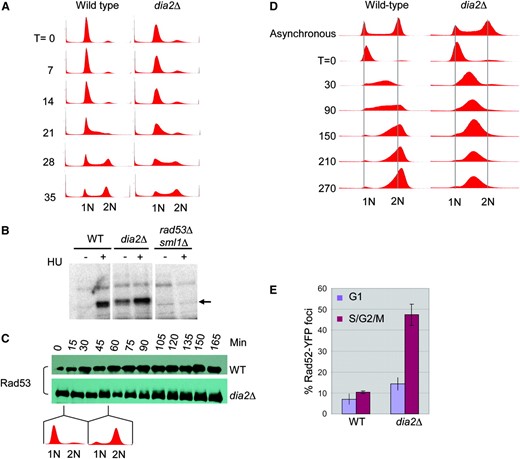
Activation of the DNA damage response in the dia2Δ strain. (A) Cell cycle progression of unperturbed wild-type and dia2Δ strains. Cells were arrested in G1 phase with α-factor and released into rich medium. DNA content was assessed by FACS at the indicated time points in minutes. (B) Rad53 kinase activity was detected using an ISA assay on lysates of the indicated strains grown in either the presence or the absence of 200 mm HU for 1 hr. Arrow indicates in situ 32P incorporation into Rad53. (C) Cell cycle dependence of Rad53 activation. The indicated strains were arrested with α-factor and released into rich medium and lysates were prepared at the indicated time points. Immunoblots were probed with a polyclonal Rad53 antibody. (D) Cell cycle progression of MMS-treated wild-type and dia2Δ strains. Cells were arrested in G1 phase with α-factor, released into rich medium containing 0.033% MMS, and assessed for DNA content at the indicated time points. (E) DNA damage repair foci in a dia2Δ strain. Rad52YFP was detected by fluorescence microscopy in wild-type and dia2Δ cells. A composite of 21 collapsed Z-sections was taken for a minimum of 300 cells/strain, which were classified as unbudded (G1) and budded (S/G2/M) cells. Bars indicate standard error.
Despite the ability of a dia2Δ strain to activate Rad53 in response to HU, MMS, and 4-NQO, the strain is nevertheless sensitive to MMS. To examine whether this sensitivity might result from a partially defective S-phase checkpoint, G1 phase cultures were released into medium containing a low dose of MMS, which activates the intra-S-phase checkpoint but not the G1 DNA damage checkpoint (Sidorova and Breeden 1997). Wild-type cells required at least 210 min to complete replication in the presence of MMS, ∼175 min longer than without DNA damage. In contrast, MMS-treated dia2Δ cells were unable to progress past mid-S-phase for the duration of the 270-min experiment (Figure 6D). As checkpoint defective cells normally progress through S-phase faster than wild-type cells (Paulovich and Hartwell 1995), sensitivity of the dia2Δ strain to MMS was not a consequence of a defective intra-S checkpoint, but rather due to an inability to recover from MMS-induced DNA damage.
The S-phase-dependent activation of Rad53 suggested that dia2Δ cells accumulate endogenous DNA damage. This idea is also supported by the genetic result that RAD52-dependent DSB repair is required for viability of dia2Δ strains (Figure 2), whereas in direct tetrad analysis, the Ku70/Ku80 nonhomologous end joining factors required to repair breaks in G1 phase are not required (data not shown). To directly assess the occurrence of DNA strand breaks in dia2Δ cells, we measured the occurrence of DNA repair foci in live cells using YFP fusions to DNA-damage-responsive proteins (Lisby et al. 2001; Melo et al. 2001). As expected, Rad52YFP foci formed at a low frequency in log-phase cultures of wild-type cells. In contrast, the dia2Δ mutant showed a significant increase in foci formation of 2- and 4.5-fold in unbudded and budded cells, respectively (Figure 6E). A similar increase was seen by both a TUNEL assay for DNA strand breaks and a Ddc1YFP damage foci reporter (data not shown). This increase in repair foci implies that DNA lesions, most likely DSBs, accumulate in dia2Δ cells and activate the DNA checkpoint response. The slight increase of Rad52 foci in G1 phase dia2Δ cells suggested an additional defect, perhaps due to incomplete repair of lesions before desensitization of the G2/M checkpoint arrest. However, we note that the extent of G1 phase damage foci in dia2Δ cells is modest compared to other DNA repair/checkpoint mutants, such as rmi1Δ (Chang et al. 2005).
Dia2 ensures high-fidelity chromosome segregation:
We examined the dia2Δ phenotype by genomewide expression profiles, which can often be used to deduce gene function (Hughes et al. 2000a). The overall expression profile of the dia2Δ strain was highly similar to that of wild-type cells, with the notable exception of a 1.5- to 3.5-fold increase in ribonucleotide reductase (RNR) transcripts in the dia2Δ mutant (Figure 7A). RNR induction is a hallmark checkpoint response to replication stress (Elledge and Davis 1990). In addition, several general stress responsive genes, including XBP1 and HSP26, were elevated in the dia2Δ strain. Aside from the obvious transcriptional difference in RNR transcript levels, comparison of chromosome-wide mean expression levels revealed that two independent dia2Δ isolates, one haploid and one diploid, exhibited significantly higher levels of gene expression from Chr I and Chr X, respectively (Figure 7B). This result indicates that these dia2Δ isolates were aneuploid, consistent with previous detection of aneuploidy in other dia2Δ isolates (Hughes et al. 2000b).
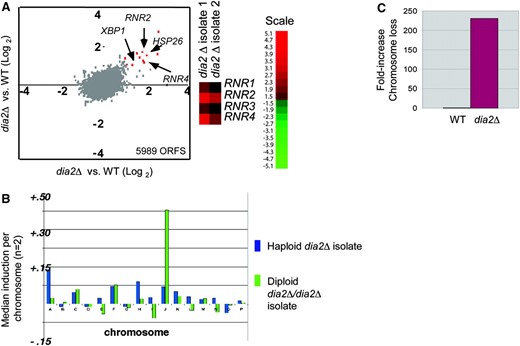
Dia2 is required to ensure faithful chromosome transmission. (A) Induction of RNR transcripts in a dia2Δ strain. Two replicate dia2Δ haploid isolates were competitively hybridized against wild-type control mRNA on genomewide microarrays (5989 ORFs detected) and plotted against one another. The top 10 induced genes in the dia2Δ strain (fold increase indicated in parentheses) were MET17 (6.5×), GPH1 (4.2×), HSP26 (3.4×), RNR4 (2.9×), YGP1(3.0×), RNR2 (3.4×), HSP12 (3.0×), YMR250w (3.1×), XBP1 (2.3×), and LAP4 (2.1×). Signals for RNR genes in replicate dia2Δ hybridizations are shown separately. (B) Aneuploidy in dia2Δ mutants detected by microarray analysis. Median gene induction per chromosome was calculated for independent haploid and homozygous diploid dia2Δ isolates. (C) Chromosome loss rates. Wild-type and dia2Δ strains bearing an artificial test chromosome (CFIII-SUP11-HIS3) were plated onto nonselective media for 2 days and scored for half red/white colonies (i.e., first division missegregation events). At least 2400 colonies were scored per strain.
To directly test whether dia2Δ plays a role in chromosome segregation, the rate of chromosome loss was measured by a colony-sectoring assay (Hieter et al. 1985). In this assay, cells carry a reporter chromosome fragment with the SUP11 (ochre-suppressing tRNA) gene, which is used to complement an ade2-101 (ochre) mutation. Colonies that retain the test chromosome are white, whereas cells that undergo a loss event accumulate a red adenine biosynthesis intermediate. The dia2Δ mutation caused an ∼200-fold increase in chromosome loss rate relative to wild type (Figure 7C). In comparison, the spindle checkpoint mutants bub1Δ and bub3Δ exhibit only a 50-fold increase in chromosome loss rate under identical conditions (Warren et al. 2002). Dia2 thus plays a significant role in ensuring the fidelity of chromosome transmission, likely through suppression of replication-induced DSBs, which can lead directly to aneuploidy through missegregation events (Kaye et al. 2004).
Dia2 suppresses GCRs:
DNA double strand breaks can lead to whole or partial chromosome loss (Kaye et al. 2004). We therefore tested whether dia2Δ cells are prone to genomic alterations by using a mutator assay that measures the incidence of GCRs (Chen and Kolodner 1999). Rearrangements are detected in haploid cells by the simultaneous loss of CAN1 and URA3 from the left arm of Chr V, such that cells lacking CAN1 and URA3 are able to form colonies on medium containing 5-FOA and canavanine (can). Strikingly, deletion of dia2Δ in this reporter strain caused a 117-fold increase in GCR rate (Figure 8A). This rate is comparable to that reported for S-phase checkpoint mutants (Myung et al. 2001).
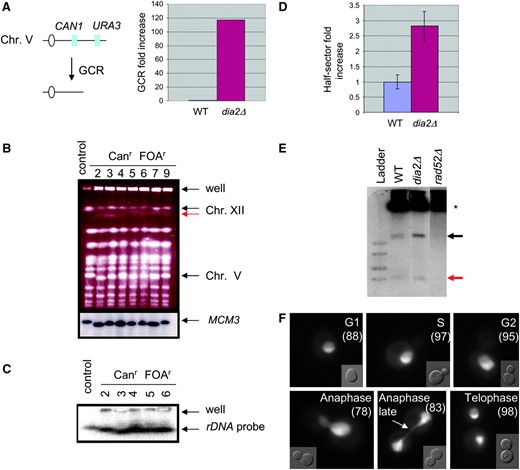
Increased GCR rate is correlated with hyperrecombination at the rDNA locus in dia2Δ cells. (A) Assay used to detect gross chromosomal rearrangements. Sensitivity to canavanine (can) and 5-FOA selects for colonies that have undergone simultaneous loss of both genes via a GCR event. GCR rates were from two independent experiments; fold induction was calculated as the mean of dia2Δ GCR rate divided by the mean GCR rate of the parental control strain. (B) Chromosomal DNA from a control and seven independent canr 5-FOAr dia2Δ isolates was resolved by PFGE, stained with ethidium bromide, and probed with a Chr V-specific MCM3 sequence. (C) Chromsomal DNA from five of the same isolates was rerun and probed with a 35S rDNA sequence. (D) Recombination at the rDNA locus. Wild-type and dia2Δ strains bearing an ADE2 marker at the rDNA locus were plated onto rich medium and grown for 3 days. Recombination rates were calculated from counting first division missegregation events for at least 20,000 colonies. Bars indicate standard error. (E) Accumulation of ERCs. Genomic DNA isolated from each of the indicated strains was resolved by electrophoresis and probed with rDNA sequences. Asterisk indicates chromosomal rDNA and arrows point to mono- and multimeric ERCs (red and black arrows, respectively). (F) Subcellular localization of Dia2GFP. A wild-type strain expressing Dia2GFP was grown to log phase and visualized by fluorescence microscopy. Dia2GFP signal that segregates late in anaphase as a string of fluorescence stretched across the bud neck is indicated by the arrow. Numbers in parentheses indicate the percentage of cells that show nucleolar Dia2 localization at each cell cycle stage.
Three classes of rearrangements are detected by the GCR assay: interstitial deletions, nonreciprocal translocations, and partial deletions of Chr V with de novo telomere addition (Kolodner et al. 2002). To characterize the spectrum of GCRs that occur in dia2Δ strains, we analyzed Chr V structure by PFGE. Chromosomal DNA from the control parental strain and seven independent 5-FOAr canr dia2Δ isolates was resolved and probed with a Chr V-specific MCM3 sequence (Figure 8B). In six of the seven dia2Δ GCR isolates, MCM3 sequences migrated slightly faster than in the control lane, presumably due to a partial chromosome deletion. In addition to these discrete chromosomal rearrangements, each dia2Δ strain tested retained a significant amount of chromosomal DNA in the well, suggestive of unresolved replication and recombination structures (Desany et al. 1998).
Dia2 suppresses recombination at the rDNA locus:
Each of the seven dia2Δ GCR isolates tested above had genomic rearrangements in addition to Chr V. In particular, we observed substantial mobility disparities between Chr IV and Chr XII, which normally comigrate (Figure 8B). Chr XII harbors the rDNA locus as a tandem array of ∼100–200 repeats (Petes 1979). The rDNA array is inherently unstable and undergoes cycles of expansion/contraction through intra- and interchromosomal recombination (Keil and Roeder 1984; Kobayashi et al. 1998). We thus tested if the observed rearrangements were due to alterations in Chr XII. Chromosomal DNA for five of the GCR isolates was resolved by PFGE and blotted with a 35S rDNA probe. In the dia2Δ strains, a faster migrating Chr XII was evident, confirming that this chromosome had indeed undergone gross rearrangements (Figure 8C). Furthermore, cross-hybridizing signals were detected in the wells of these lanes, indicative of unresolved rDNA replication intermediates (Desany et al. 1998).
To assess whether rearrangement of Chr XII in dia2Δ cells was a consequence of increased rDNA recombination, we measured recombination rates by scoring the loss of an rDNA ADE2 marker (Merker and Klein 2002). dia2Δ mutants lost ADE2 at a rate 2.8-fold higher than that of wild-type cells (Figure 8D), an increase comparable to those reported for hpr1Δ (3.1-fold) and sir2Δ (4.8-fold) strains (Merker and Klein 2002), both of which have defects in rDNA repeat maintenance. Consistently, the rDNA hyperrecombination phenotype in dia2Δ cells correlated with an accumulation of both mono- and multimeric ERCs (Figure 8E), which are known to arise by recombination (Sinclair and Guarente 1997).
The specific defects in rDNA repeat maintenance in the dia2Δ mutant suggested a particularly acute role for Dia2 in this problematic region of the genome. To investigate whether Dia2 might function directly at the rDNA locus, we examined the localization of a GFP fusion protein in live cells. Dia2GFP was detected in the nucleus with substantial enrichment in the nucleolus (Figure 8F). This GFP pattern was unaltered during mitosis, except for a period during late anaphase. At this stage, Dia2GFP was evident in both mother and daughter nuclei with a string of fluorescence stretched across the bud neck. Because the rDNA locus has been shown to segregate later than the rest of the genome (D'Amours et al. 2004), this result demonstrates that Dia2 is tightly colocalized with the rDNA locus. Taken together, these observations suggest that Dia2 has an important, but probably not exclusive, role in maintaining rDNA integrity.
DISCUSSION
The ubiquitin system plays an important and conserved role in the control of DNA replication and the DNA damage response (Branzei and Foiani 2005). Here, through characterization of the genetic and cell biological attributes of the dia2Δ mutant, we have uncovered a new role for the ubiquitin system in the suppression of genome instability associated with replication fork collapse. In the absence of Dia2, cells exhibit an S/G2/M cell cycle delay, greatly increased sensitivity to damage-induced replication blocks, and an increase in chromosomal lesions. As a consequence of accumulated endogenous DNA damage, the dia2Δ mutant constitutively activates the DNA damage checkpoint. Activation of the checkpoint may account for the weakly invasive phenotype of the dia2Δ strains (Kang et al. 2003). The viability of dia2Δ strains depends on the central checkpoint kinases Mec1 and Rad53, as well as on the collective activity of factors that mediate the DNA damage and replication arms of the checkpoint (i.e., Mrc1 and Rad9). Because the pre-anaphase cell cycle delay in dia2Δ strains does not solely depend on Mrc1 or Rad9, it may be imposed in part by the spindle checkpoint pathway, as observed in cells treated with DNA-damaging agents and a variety of replication mutants (Garber and Rine 2002; Collura et al. 2005). Consistently, our genetic analysis indicates that viability of the dia2Δ strain requires full spindle function and an intact spindle assembly checkpoint. Although not recovered in our SGA screen, dia2Δ strains are also severely compromised in the absence of the anaphase inhibitor Pds1 (Sarin et al. 2004), which both mediates aspects of the DNA damage response and couples the completion of replication to the onset of mitosis (Clarke et al. 1999). Despite constitutive Rad53 activation, dia2Δ cells exhibit highly elevated chromosome loss and GCR rates. Unrepaired replication-induced DSBs not only can cause catastrophic genome rearrangements (Chen and Kolodner 1999; Myung et al. 2001; Kolodner et al. 2002) but also can directly lead to failures in chromosome segregation (Kaye et al. 2004). Multiple aberrations in chromosome replication and segregation pathways thus appear to underlie the high rate of genome instability of the dia2Δ mutant.
What might be the nature of DNA damage in dia2Δ cells? When a replication fork stalls, it may be processed into toxic replication intermediates, or alternatively, it may simply collapse, which produces DSBs (Branzei and Foiani 2005). In either case, cells accumulate repair foci, as observed in dia2Δ cells. Most significantly, the genetic result that dia2Δ is synthetic lethal with rad52Δ strongly argues that DSBs accumulate in the dia2Δ mutant and thus must be repaired by homologous recombination. In addition, dia2Δ displays synthetic genetic interactions with sgs1Δ and top3Δ, as well as with slx4Δ, slx5Δ, slx8Δ, mus81Δ, and mms4Δ. The SLX genes (SLX1, -4, -5, -8, MUS81, and MMS4) are required in the absence of the Sgs1 helicase (Mullen et al. 2001), which helps resolve replication intermediates (Chang et al. 2005; Liberi et al. 2005; Mullen et al. 2005). The Slx1/4 and Mus81/Mms4 complexes are endonucleases that cleave structures generated from stalled and collapsed forks, respectively (Bastin-Shanower et al. 2003; Fricke and Brill 2003). Given its synthetic interactions with slx mutations, it is likely that the dia2Δ mutant accumulates both stalled and collapsed forks and therefore requires the Slx products for replication restart.
In wild-type cells, MMS treatment retards fork progression and inhibits late origin firing, but these effects are transient and reversed once MMS-induced damage is repaired (Santocanale and Diffley 1998; Tercero and Diffley 2001). In contrast, dia2Δ mutants completely fail to resume replication following MMS-induced DNA damage. Dia2 thus may be required for passage of replication forks through damaged DNA templates, which rapidly accumulate large protein structures as part of the repair process. The inviability of dia2Δ sod1Δ and dia2Δ lys7Δ double mutants is also consistent with a role for Dia2 in aiding replication through damaged templates, as the Sod1 superoxide dismutase and its copper chaperone Lys7 normally prevent DNA damage from endogenous oxidative stress (Pan et al. 2006). Because dia2Δ strains appear to progress through S-phase with near wild-type kinetics, it seems unlikely that Dia2 is a component of the general replication machinery. While a significant subpopulation of dia2Δ cultures are delayed for completion of S-phase, this effect probably arises from checkpoint-mediated inhibition of late origins (Santocanale and Diffley 1998; Tercero and Diffley 2001) and/or from an inability to replicate problematic regions (Cha and Kleckner 2002; Lemoine et al. 2005; Admire et al. 2006). Consistent with this interpretation, we have been unable to detect genetic interactions between dia2Δ and mutant components of DNA polymerase α-primase complex encoded by pri1-M4 or pri2-1, implying that Dia2 does not facilitate either replication initiation or lagging-strand DNA synthesis (data not shown).
Our systematic genetic analysis strongly suggests that Dia2 enables the replication machinery to cope with natural replication slow zones, in particular the RFB of the rDNA repeat. In budding yeast, replication pause sites occur at well-defined regions throughout the genome, including the rDNA locus (Rothstein et al. 2000; Cha and Kleckner 2002; Ivessa et al. 2002; 2003; Torres et al. 2004a). The two related helicases Rrm3 and Pif1 have opposing effects on rDNA breakage and recombination via their ability to regulate RFB activity (Ivessa et al. 2000). Rrm3 suppresses fork stalling by driving fork progression through non-nucleosomal protein–DNA complexes (Ivessa et al. 2003; Torres et al. 2004a), whereas Pif1 appears to promote fork arrest, in addition to its role in suppression of de novo telomere addition (Ivessa et al. 2000; Pennaneach et al. 2006). The synthetic lethal interaction between dia2Δ and rrm3Δ suggests that Dia2 and Rrm3 may function redundantly to displace protein–DNA complexes, at least under some circumstances. However, because rrm3Δ strains neither are sensitive to DNA-damaging agents nor require the Mus81/Mms4 or Slx1/4 complexes for viability (Torres et al. 2004b), Dia2 may play a more general role than Rrm3 in facilitating replication fork progression through problematic regions. Given the opposing effects of Rrm3 and Pif1 at the RFB, the synthetic lethal interaction between dia2Δ and pif1Δ is enigmatic. This interaction may reflect either the accumulation of aberrant replication-associated structures in the absence of Pif1 or, alternatively, a requirement for Pif1 to cope with DNA-damaging events that arise in the absence of Dia2. Regardless of these more general replication effects, the preferential localization of Dia2 to the nucleolus and the increased recombination at the rDNA locus in dia2Δ strains, as well as the synthetic lethal interactions between dia2Δ and both rrm3Δ and pif1Δ, clearly implicates Dia2 as a regulator of the RFB and rDNA replication.
RNA Pol II-associated protein complexes are also intrinsic barriers to replication fork progression (Deshpande and Newlon 1996; Aguilera 2002). Head-on collision between the transcriptional machinery and the replication fork results in a replication block that is resolved by Rrm3 (Prado and Aguilera 2005). The synthetic genetic interactions between dia2Δ and mutations in various elongation factors, including ctk1Δ, rtf1Δ, and cdc73Δ (Jona et al. 2001; Squazzo et al. 2002), thus may be explained by an inability of the replication machinery to transit past stalled transcriptional complexes. A variety of mutations that disrupt different aspects of chromatin structure, including hpc2Δ, htz1Δ, swr1Δ, chl4Δ, npt1Δ, and hst4Δ, also exhibit synthetic genetic interactions with dia2Δ. Defects in chromatin architecture might engender stalled or defective transcriptional complexes that impede the replication machinery in the absence of Dia2; alternatively, chromatin remodeling may be required for recovery from intrinsic DNA damage in the dia2Δ strain (Vidanes et al. 2005). While the dia2Δ mutant is defective in both telomeric and rDNA silencing (data not shown), these effects likely arise from relocalization of silencing factors to sites of endogenous DNA damage (Martin et al. 1999). Although Dia2 might conceivably regulate higher-order chromatin structure and thereby indirectly affect replication and genome stability, we favor a more direct role for Dia2 in allowing the replication machinery to cope with proteinaceous barriers.
Further insight into possible DIA2 functions comes from its overlapping genetic interactions with RTT101, RTT107/ESC4, and MMS1/RTT108. The pairwise synthetic lethal interactions between these four genes suggest convergence on an essential process in DNA replication. Rtt101 and Rtt107 share similar DNA damage and replication phenotypes as Dia2 (Rouse 2004; Luke et al. 2006), and both physically interact with Mms22 (Ho et al. 2002), a protein that operates in the same pathway as Mms1 (Hryciw et al. 2002; Araki et al. 2003). Intriguingly, the likely human counterpart of Rtt101, called Cul4, forms a ubiquitin–ligase complex with the Ddb1 protein and targets a variety of repair- and replication-associated proteins for degradation (Willems et al. 2004). It is thus possible that Dia2 acts in a redundant fashion with an Rtt101-based ubiquitin ligase to modify or eliminate one or more substrates at the replication fork.
Two recent reports have suggested replication-associated functions for Dia2 (Koepp et al. 2006; Pan et al. 2006). On the basis of apparent premature S-phase entry of synchronous cultures of a dia2Δ strain and detection of replication origin sequences in crosslinked Dia2 immunoprecipitates, Koepp et al. (2006) proposed that Dia2 prevents precocious firing of replication origins. Dia2 thus might function in a manner analogous to the CDK inhibitor Sic1, which is known to prevent Clb5/6-Cdc28 activation and subsequent origin firing (Lengronne and Schwob 2002). However, a number of our results are inconsistent with a role for Dia2 in replication initiation. We did not detect overt premature S-phase entry in the dia2Δ strain, at least as judged by bulk DNA replication in synchronous populations. Unlike the rescue of the sic1Δ phenotype by codeletion of clb5Δ clb6Δ (Lengronne and Schwob 2002), we did not observe amelioration of the dia2Δ cell size and G2/M cell cycle delay phenotypes in a clb5Δ clb6Δ background (data not shown). Moreover, in contrast to dia2Δ strains, the replication checkpoint is not activated in a sic1Δ strain (Lengronne and Schwob 2002). The markedly different spectrum of genetic interactions exhibited by sic1Δ and dia2Δ mutations also implies different functions (in the combined data set used to generate Figure 3A, of 153 total dia2Δ interactions and 62 total sic1Δ interactions, only 20 overlap). Finally, the inability of dia2Δ cells to resume replication following MMS treatment suggests a role for Dia2 beyond replication initiation.
A second recently proposed function for Dia2 is elimination of the replication- and cohesion-associated factor Ctf4 (Pan et al. 2006), both on the basis of the spectrum of replication and checkpoint genes recovered with dia2Δ in a dSLAM genetic network (Pan et al. 2006) and on the basis of a putative interaction detected between Dia2 and Ctf4 in a high-throughput study (Ho et al. 2002). Consistently, overexpression of CTF4 is toxic in wild-type cells and a cft4Δ mutation restores viability to a dia2Δ hst3Δ double mutant (Pan et al. 2006). However, we have been unable to detect a Dia2–Ctf4 physical interaction in direct co-immunoprecipitation tests, nor does ctf4Δ appear to suppress the phenotype of a dia2Δ single mutant (data not shown). In addition, endogenous Ctf4 levels are not altered in a dia2Δ strain (J. Boeke and X. Pan, personal communication). The precise role of Dia2 in DNA replication thus remains a mystery.
One obvious route to understanding Dia2 function is the identification of SCFDia2 substrates. While a number of interacting partners for Dia2, including Ctf4, have been reported in high-throughput studies (Ho et al. 2002), to date we have been unable to convincingly recapitulate any of these physical interactions. The identification of ubiquitin–ligase substrates in general remains a difficult problem. The only clear Dia2 homolog in other species is fission yeast Pof3, which, like Dia2, is distinguished from other F-box proteins by the presence of both TPR and LRR repeats. The constellation of phenotypes in the pof3Δ mutant partially parallels that of dia2Δ: each accumulates DNA damage, exhibits checkpoint activation and G2/M delay, and displays high chromosome loss rates (Katayama et al. 2002). However, disparities in UV tolerance and sensitivity to microtubule-destabilizing drugs suggest that these two F-box proteins may not have entirely analogous functions, perhaps because of divergence in checkpoint responses between budding and fission yeast (Melo and Toczyski 2002). To date, no Pof3 substrates have been reported and so no further insight can be gleaned from the fission yeast homolog of Dia2. While it seems reasonable that the complex dia2Δ phenotype might reflect deregulation of multiple substrates, it seems likely that the primary function of Dia2 is to facilitate DNA replication through taxing regions of the genome.
Genomic regions prone to replication delays and chromosomal rearrangements have been termed replication slow zones or fragile sites (Cha and Kleckner 2002; Lemoine et al. 2005; Admire et al. 2006). Such sites often coincide with non-nucleosomal protein–DNA complexes, as, for example, at rDNA repeats, tRNA genes, telomeres, and loci with high rates of transcription (Ivessa et al. 2003; Prado and Aguilera 2005). In addition, endogenous and exogenous genotoxins can burden the genome with protein–DNA complexes that arise in the course of normal repair processes. These various forms of replication impedance, when combined with defects either in the replication machinery itself or in checkpoint pathways, can lead to rampant genome instability (Kolodner et al. 2002). Fragile sites in both yeast and mammalian cells are susceptible to enfeebled DNA replication and diminished checkpoint responses (Richards 2001; Casper et al. 2002; Lemoine et al. 2005; Admire et al. 2006). A key feature of fragile sites in yeast is persistent, and even enhanced, fragility after an initial rearrangement has occurred (Admire et al. 2006). Moreover, many human tumors are often marked by rearrangements at common fragile site breakpoints (Glover and Stein 1988; Wang et al. 1997). Understanding the pathways that enable the cell to cope with replication impedance at fragile regions will provide critical insight into the genesis of cancer and other genetic disorders.
Footnotes
Present address: Swiss Institute for Cancer Research, ISREC, Case postale CH-1066 Epalinges, Lausanne, Switzerland.
Present address: Department of Systems Biology, Harvard Medical School, Boston, MA 02115.
Present address: Ontario Cancer Institute and Campbell Family Institute for Breast Cancer Research, Toronto, ON M5G 2C1, Canada.
Present address: TM Bioscience, Toronto, ON M5G 1Y8, Canada.
Footnotes
Communicating editor: A. P. Mitchell
Acknowledgement
We thank Jef Boeke, Xuewen Pan, Grant Brown, and Patrick Paddison for helpful discussions and communication of unpublished results; Rodney Rothstein, Charlie Boone, Hannah Klein, and Howard Bussey for providing reagents; and Nizar Batada and Jan Wildenhain for advice on data analysis. D.B. was supported in part by a Canadian Institutes of Health Research (CIHR) Doctoral Award. P.K. is a recipient of a studentship from the National Cancer Institute of Canada. D.D. and M.T. are supported by grants from the CIHR and are holders of Canada Research Chairs. M.P. was supported by grants from the Swiss Federal Institute of Technology and the Swiss National Science Foundation.
References
Chen, C., and R. D. Kolodner,
Eisen, M. B., P. T. Spellman, P. O. Brown and D. Botstein,
Fabre, F., A. Chan, W. D. Heyer and S. Gangloff,
Glover, T. W., and C. K. Stein,
Greenfeder, S. A., and C. S. Newlon,
Lisby, M., R. Rothstein and U. H. Mortensen,
Ohya, Y., J. Sese, M. Yukawa, F. Sano, Y. Nakatani et al.,
Petes, T. D.,
Torres, J. Z., S. L. Schnakenberg and V. A. Zakian,
Zhang, C., T. M. Roberts, J. Yang, R. Desai and G. W. Brown,

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}