





Abstract
Heritable mutations, known as inborn errors of metabolism, cause numerous devastating human diseases, typically as a result of a deficiency in essential metabolic products or the accumulation of toxic intermediates. We have isolated a missense mutation in the Drosophila sugarkill (sgk) gene that causes phenotypes analogous to symptoms of triosephosphate isomerase (TPI) deficiency, a human familial disease, characterized by anaerobic metabolic dysfunction resulting from pathological missense mutations affecting the encoded TPI protein. In Drosophila, the sgk gene encodes the glycolytic enzyme TPI. Our analysis of sgk mutants revealed TPI impairment associated with reduced longevity, progressive locomotor deficiency, and neural degeneration. Biochemical studies demonstrate that mutation of this glycolytic enzyme gene does not result in a bioenergetic deficit, suggesting an alternate cause of enzymopathy associated with TPI impairment.
METABOLIC defects resulting from inherited disorders cause numerous human diseases. Glycolytic enzymopathies result from a disturbance in anaerobic metabolism; however, these diseases remain poorly understood. Familial triosephosphate isomerase (TPI) deficiency, an autosomal recessive disorder, has been reported in numerous pedigrees and results in anemia, neuromuscular wasting, and reduced longevity (Schneider et al. 1965; Valentine 1966). The relationship of anemia, neuromuscular degeneration, and glycolytic flux to disease pathogenesis is not clear, and an animal model that captures salient features of TPI deficiency has not been reported. Our studies demonstrate the utility of Drosophila sugarkill (sgk) mutants as a model of glycolytic enzymopathy.
TPI is a 26.5-kDa soluble protein responsible for the conversion of dihydroxyacetone phosphate into glyceraldehyde-3-phosphate in glycolysis (Rieder and Rose 1959). The protein's structure has been studied in yeast (Alber et al. 1981), chicken (Banner et al. 1975), bacteria (Noble et al. 1993), trypanosome (Wierenga et al. 1991), and human (Lu et al. 1984; Mande et al. 1994; Maquat et al. 1985) with a high degree of structural similarity shared between the various reported structures. TPI is considered a near perfect enzyme due to its catalytic efficiency; the rate of catalysis is diffusion controlled, suggesting the presence of strong selective pressure throughout the gene's evolution (Knowles 1977). TPI is known to exist functionally as a homodimer, and the dimer interaction sites have been well characterized (Schneider 2000).
In humans, several mutations have been reported that result in TPI deficiency, a progressive disease that eventuates in neuromuscular failure, hemolytic anemia, increased susceptibility to infection, and premature death. At least nine different TPI missense mutations that affect various positions throughout the encoded protein have been identified as the cause of glycolytic enzymopathies (Daar et al. 1986; Daar and Maquat 1988; Chang et al. 1993; Watanabe et al. 1996; Arya et al. 1997; Valentin et al. 2000) (supplemental Figure S1 at http://www.genetics.org/supplemental/). Heterozygosity of null human TPI alleles has been observed in 5% of African American and 0.5% of Caucasian populations, although the disease is typically caused by homozygous missense mutations (Mohrenweiser and Fielek 1982; Mohrenweiser 1987). Although prenatal detection is available, there is no treatment for this progressive and devastating neurological disease (Arya et al. 1996).
TPI has been studied in Drosophila and its mRNA is highly expressed during development and in adult animals (Shaw-Lee et al. 1991). Interestingly, studies of naturally occurring Drosophila populations revealed allozyme polymorphisms, including TpiS (slow) and TpiF (fast) alleles, which indicate the rate of protein anodal electrophoresis migration (Voelker et al. 1979). Research findings suggest that the TpiF polymorphism (Lys-173-Glu) may alter the “hinged lid” of the enzyme's active site; however, the functional and behavioral consequences of the fast and slow polymorphisms have not yet been characterized (Hasson et al. 1998).
We report a Drosophila mutation in the sgk gene that was originally isolated due to its temperature-sensitive locomotor impairment (Palladino et al. 2002). Our studies demonstrate that reduced longevity and progressive neural degeneration are associated with the temperature-sensitive sgk1 fly mutation. Interestingly, some of the well-characterized mutations that cause human TPI deficiency disease have been shown to be temperature sensitive, owing to thermal instability of the encoded protein (Daar et al. 1986; Chang et al. 1993). Although TPI has a well-characterized biochemical function, the mechanism of disease pathogenesis is unclear. Our biochemical studies indicate that the mutation does not cause general bioenergetic impairment, suggesting that toxicity is associated with substrate catabolism, a secondary effect of reduced glycolysis independent of total cellular energy availability, or loss of an unappreciated TPI function. The sgk mutant reported provides a useful and amenable genetic animal model to study anaerobic metabolism and human glycolytic enzymopathy in vivo.
MATERIALS AND METHODS
Drosophila stocks and culture:
Standard cornmeal molasses fly media was used. Flies were maintained at room temperature, unless otherwise noted. The sgk1 mutation is maintained as a homozygous viable ve e sgk1 strain. Wild-type controls are ve e homozygotes, unless otherwise noted. Flies were tetracycline treated to rid them of microbial pathogens, which was verified by PCR as previously reported (Celotto et al. 2006). The mutant and control strains used all contained the more common TpiS variant (Oakeshott 1984).
Life span and behavior analysis:
Life span analysis was performed as previously described (Palladino et al. 2003). Stress sensitivity (a.k.a., bang sensitivity) was assayed by vortexing flies in a standard media vial for 20 sec and measuring the length of paralysis, similar to a previously described protocol (Ganetzky and Wu 1982). Stress sensitivity (rate of recovery) was assessed by assigning a numerical score to the time in seconds to regain normal locomotion using a 10-sec incremented scale, such that 1 indicates <10 sec, 2 indicates 11–20 sec, through 20 indicating >191 sec. Temperature sensitivity was assayed as previously described at 37° (Palladino et al. 2002, 2003).
Genetics—sugarkill positional cloning, deletion, and transgenic rescue:
The sgk1 mutant, previously named ND14, was originally identified in a screen of mutants with behavioral deficits for neurodegeneration (Palladino et al. 2002). Candidate gene sequencing identified the mutation in the sgk gene. Transposon-mediated mutagenesis was utilized to delete the sgk locus, using standard procedures. Eighty revertants of PEPgy2EY03361 (located ∼100 nucleotides 5′ relative to the start of exon 1) were identified as w− Dr+ offspring of w1118;;delta2-3 Dr/PEPgy2 jump-start males. The white revertants were screened by PCR to identify deletion and precise excision events. Genomic DNA was isolated using the tissue protocol of QIAamp DNA (QIAGEN, Valencia, CA). The sgk genomic locus was amplified using a standard 50-μl PCR reaction; amplicons were resolved by electrophoresis with a 1.0% agarose gel and visualized with ethidium bromide staining. We isolated one 1.6-kb deletion (removing −319 to 1288 nucleotides relative to the start of exon 1) and numerous apparent precise excision events. Direct sequence of PCR amplicons from four homozygous viable excision events verified the precise nature of these revertants (named sgkR1–4). The deletion amplicon generated from heterozygote animals was gel purified, extracted, and directly sequenced to identify the deletion end points. Using standard P-element-mediated transgenesis, UAS− sgk transgenes were generated from EcoRI–XhoI PCR amplicons derived either from the ve e sgk+ wild type or from the ve e sgk1 genomic DNA corresponding to positions 57–1371 in FBgn0003738. The second chromosome actin∷GAL4 (P{Act5C-GAL4}25FO1) was used to drive expression in rescue experiments.
Quantitative real-time RT–PCR:
RNA was isolated from 10 flies (performed in triplicate for each genotype), using 200 μl Trizol (Invitrogen, San Diego). The chloroform-extracted sample was precipitated and the RNA pellet was resuspended in 10 μl dH2O and quantified by spectrophotometer reading at 260 nm. Five micrograms of RNA were used to perform a reverse transcription reaction (Superscript RT, Invitrogen). Quantitative real-time PCR [Bio-Rad (Hercules, CA) iCycler] was performed using standard techniques. Briefly, 2 μl of cDNA, 400 nm each of forward and reverse primers, and 12.5 μl 2× iQ SYBR Green Supermix in a total volume of 25 μl were amplified. PCR specificity was evaluated by melting-curve analysis and resolution on a 1% agarose gel. All quantitative PCR experiments were repeated three times. The data were normalized using the mRNA expression levels of RP49. Fold change (FC) was determined using the Bio-Rad equation, FC = 2−Δ(ΔCt).
Paraffin histology:
Heads were removed, probosci were dissected away, and heads were fixed overnight in Carnoy's fixative as previously described (Palladino et al. 2002). Aged specimens were obtained at the genotype's median adult age (day of 50% survivorship, see Figure 3). Bodies were dissected and fixed overnight in Carnoy's fixative. Tissue was processed into paraffin and 5-μm-thick sections were stained with hematoxylin and eosin (H&E) using previously described methods (Palladino et al. 2000).
Transmission electron microscopy:
Indirect flight muscle and brain tissue were dissected from mutant and age-matched control animals and processed similarly to methods described previously (Kawasaki et al. 1998). Briefly, fixation occurred overnight at 4° in a buffered saline solution containing 2.5% paraformaldehyde and 1.5% glutaraldehyde (primary fixative). Tissue was postfixed in 1% osmium tetroxide solution, dehydrated in a graded ethanol series, and embedded in epon resin. Sections (65 nm) were obtained from a Reichert Ultracut ultramicrotome and stained with 4% uranyl acetate and 2.7% lead citrate. The tissue was imaged on a JEOL (Akishima, Tokyo) 100CX transmission electron microscope. Stereological protocols were followed in examining tissue samples and numerous representative images were collected from muscle and central brain tissue.
Bioenergetic analysis:
Creatine (Cr), phosphocreatine (PCr), cyclocreatine (CCr), and lactic acid (LA) were measured from whole-animal extracts using high performance liquid chromatography (HPLC). This method is a modification of a previously reported method (Matthews et al. 1998). Shimadzu HPLC components were used: SIL-HTc auto sampler, LC-20AD pump, CTO-20AC column oven, and SPD-M20A diode array detector. Animals were aged at the appropriate temperature, frozen in liquid nitrogen, and kept at −20°. Whole animals (50 per preparation) were counted, weighed, and homogenized in 200 μl 0.4 m perchloric acid using an electric homogenizer. Each sample was neutralized with 25 μl 2 m K2CO3 and centrifuged (12000 × g) at 4° for 10 min. The supernatant was filtered through a PVDF 0.45-μm filter and stored at −80° until analysis. Samples were diluted (1:2 in dH2O) and 25 μl was injected and separated on a Waters Atlantis dC18 150 × 4.6-mm 3 μm column with a 4.6 × 20-mm 3-μm Guard column (changed every 100 injections). Sample separation was achieved using a 10-mm NaH2PO4 pH 2.5 mobile phase at a flow rate of 0.5ml/min and a column temperature of 30° with a detection wavelength at 190 nm. All standards were linear over a 10- to 20-fold concentration range. Retention times (minutes) were as follows: PCr, 4.2; LA, 4.4; Cr, 5.0; and CCr, 6.4.
Supplemental data:
Supplemental data are provided at http://www.genetics.org/supplemental/.
RESULTS
Positional cloning and molecular characterization of a Drosophila neurodegeneration mutant:
We have utilized forward genetic screens to identify genes required to maintain neuronal viability with age and have identified several mutants with behavioral abnormalities and neurodegeneration (Palladino et al. 2002, 2003; Celotto et al. 2006). One such neurodegenerative mutant identified in this screen, ND14, was reported to reside proximal to cytological region 3–100 (Palladino et al. 2002). All of the available transposons and mutations in the 99A–F cytological region complemented ND14 for its temperature-dependent locomotor impairment and adult longevity phenotypes. To clone the affected gene we sequenced candidate genes in the critical region including takR, axn, trp, and tpi. Only the tpi locus contained a nonpolymorphic change affecting a codon in the open reading frame of the gene (Figure 1). The tpi mutation in the ND14 strain is a T to C transition that alters a highly conserved methionine (M) to a threonine (T) codon at position 80 in the Drosophila glycolytic TPI protein. Due to the well-characterized function of the TPI protein in carbohydrate metabolism and the reduced longevity defect, the ND14 mutant was named sugarkill1 (sgk1).
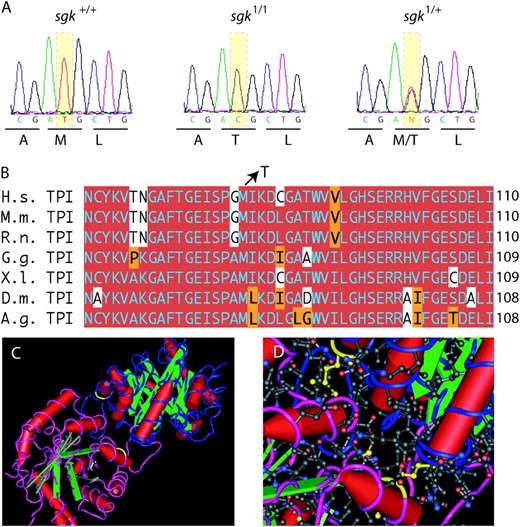
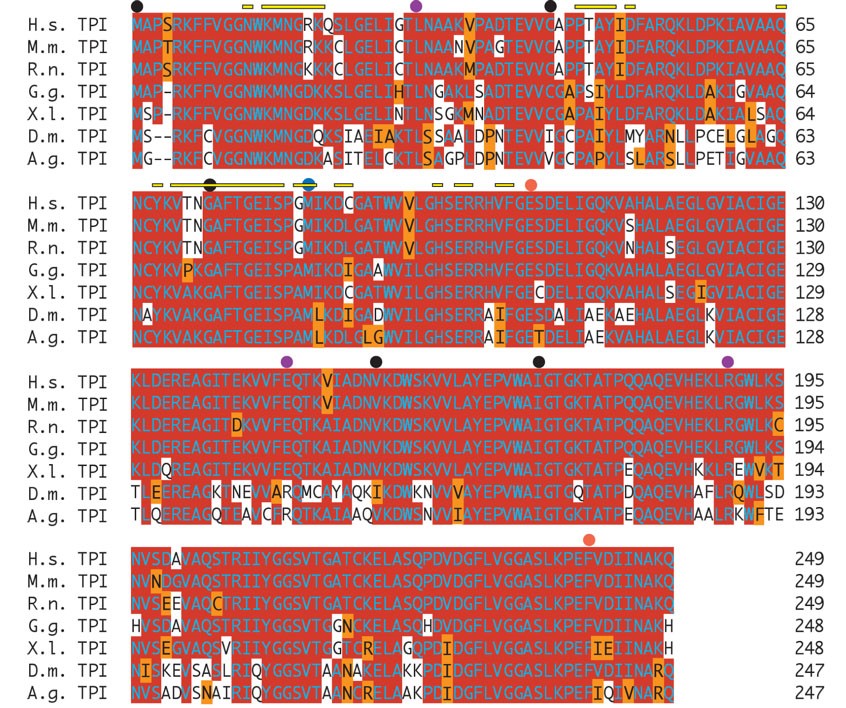
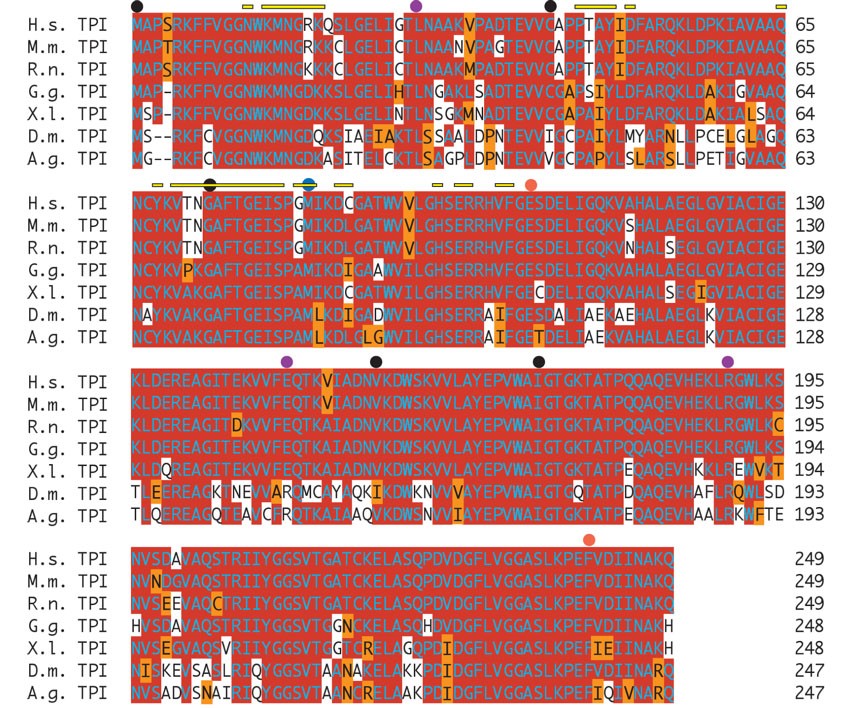
Mutation in the sugarkill gene affects a conserved methionine in the TPI protein. (A) Direct sequence analysis of genomic PCR-amplified fragments revealed exactly one mutation that was not polymorphic in the mutant strain. The mutation is a T to C transition that alters a methionine (M) codon (ATG) to a threonine (T) codon (ACG) affecting position 80 in the TPI protein (CG2171-PB). (B) Amino acid sequence alignment demonstrates the conservation of coding potential at and near the site of the mutation. Red shading indicates a match with consensus. Orange shading indicates a conservative substitution. (C) The TPI dimer depicted as a tube and worm structure on the basis of the 2.2-Å crystallographic data of the human TPI protein (Kinoshita et al. 2005). One monomer is colored blue and the other magenta. Yellow indicates the amino acid, M80, affected in each monomer by the sgk1 mutation. (D) With the amino acid side chains added (red is acidic, blue is basic) the location of the mutation at the interface between the monomers is evident. Cn3D version 4.1 was used to generate structures in C and D. Species included in the alignment are: Anopheles gambiae (A.g.), Drosophila melanogaster (D.m.), Mus musculus (M.m.), Rattus norvegicus (R.n.), Gallus gallus (G.g.), Xenopus laevis (X.l.), and Homo sapiens (H.s.). The full protein alignment is provided in supplemental Figure 1 at http://www.genetics.org/supplemental/.
To confirm that the sgk1 mutation causes a temperature-dependent defect in locomotion, we generated an independent deletion of the sgk locus (Figure 2). We isolated 80 transposase-mediated white revertants of the homozygous viable EP{gy2} P element. PCR amplification of the sgk genomic locus demonstrated numerous apparent precise excisions and a 1.6-kb deletion that removed much of the sgk genomic locus, including two of its three constitutive exons (named sgkjs10, Figure 2). Direct sequence of PCR amplicons verified four precise excisions sgkR1–R4 and the deletion end points of the sgkjs10 allele. The sgkjs10 mutation failed to complement sgk1 while all four molecularly characterized revertants and the parent EP{gy2} chromosome fully complement the sgk1 mutation and are homozygous viable. Unlike the homozygous-viable sgk1 mutation, sgkjs10 is lethal: Adult sgkjs10 homozygotes are not observed in our sgkjs10/TM3, Kr∷GAL4, UAS-GFP strain and nonfluorescent homozygotes are not identifiable beyond larval stage 2 (data not shown).
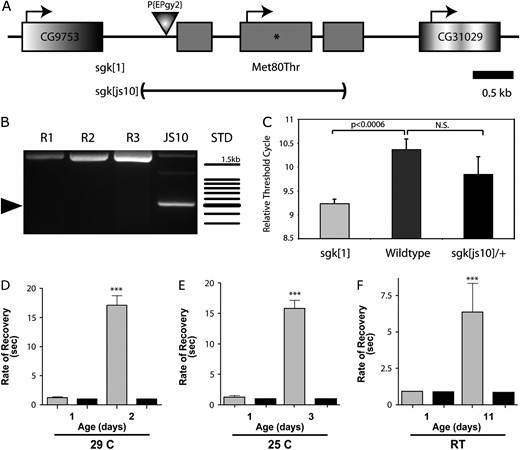
Genomic organization, expression, and locomotor function of sugarkill. (A) The sgk locus contains three constitutive exons (shaded boxes) with a predicted translational start at the beginning of exon 2. The M80T mutation resides in exon 2 (asterisks) and the js10 allele specifically removes the predicted sgk gene. (B) PCR amplicons resolved on an agarose gel show a 1.6-kb deletion resulting in a ∼500-bp product (arrowhead) in the js10 heterozygote and the normal-sized product obtained from revertant strains (R1–R3). Standard (STD) is a 100-bp ladder, and the top band is 1.5 kb. (C) mRNA expression from sgk mutants and control animals was analyzed using RT-rtPCR. Threshold cycle is the fractional cycle number at which the fluorescence reaches 10 times the standard deviation of the baseline for each genotype and is inversely related to expression. sgk mRNA expression is increased 2.2-fold in sgk1 mutants over wild-type control levels (Student's t-test). There is no significant difference between sgkjs10 and control animals. Error shown is SEM, n = 3. (D–F) Progressive locomotor impairment in sgk mutants. Locomotor impairment is quantified as the recovery time from stress-induced paralysis. Mechanical stress/hyperstimulation induces sustained paralysis in sgk1/JS10 (shaded bars) mutant strains, which increases significantly with age relative to the sgk1/R2 (solid bars) control. Locomotor impairment is progressive in flies stored at (D) 29°, (E) 25°, and (F) room temperature. A numerical score, representing time to regain normal locomotion, was assigned to each fly as described in materials and methods. sgk1/R2 and wild-type animals (data not shown) are extremely tolerant to mechanical hyperstimulation. ***, significantly different from age-matched wild-type controls (one-way ANOVA test, determined in Prism 4.0b, P < 0.001). Error is SEM. A total of 15 animals per genotype were tested. sgk1/JS10 mutants were not tested beyond the days reported due to morbidity in the populations.
Expression in sugarkill mutant strains:
The sgk1 mutation is predicted to alter a methionine (M) to threonine (T) at position 80 in the protein, which is likely the basis of the genetic defect. Alternatively, it is possible that this mutation destabilizes sgk mRNA or that a noncoding mutation causes a reduction in expression and this causes the recessive sgk1 phenotypes. Semiquantitative reverse transcription real-time PCR (RT–rtPCR) was used to determine sgk expression levels. This analysis demonstrated that sgk expression was not reduced in sgk1 homozygous animals (Figure 2C). In fact, the analysis revealed a 2.2-fold increase in sgk expression in the sgk1 mutant strain, suggesting an increase in mRNA stability or the presence of a transcriptional compensatory mechanism. No significant difference was observed in sgkjs10 heterozygote animals from wild-type animals, consistent with the recessive nature of this mutation.
Progressive dysfunction in sugarkill mutants:
Flies homozygous for sgk1 exhibit impaired locomotion that is antagonized by elevated temperature and physical stress. To quantify the stress-sensitive impairment phenotypes and determine if they are progressive, we examined young and aged sgk mutants and age-matched controls for stress-induced locomotor deficits. At all temperatures examined—room temperature (RT), 25°, and 29°—the locomotor defect arising from mechanical stress was progressive and increased significantly with the age of the animals (Figure 2, D–F). Severe stress-induced locomotor impairment is evident much earlier in sgk mutant adults when maintained at 25° and 29° (Figure 2, D–F, and data not shown). sgk1 homozygotes and sgk1/JS10 animals have a similar stress-sensitive phenotype.
Consistent with progressive decline associated with glycolytic enzymopathy, longevity is severely compromised in sgk mutants (Figure 3). Life spans of sgk1/sgkjs10 animals were compared to those of revertant and heterozygous control animals. There was no difference between the two control strains used or between sgk1/sgkjs10 and sgk1 homozygotes. Longevity was analyzed at RT, 25°, and 29°, which represent three normal physiologically relevant temperatures for Drosophila, and life span was significantly reduced at all temperatures examined (Figure 3C). Life span is also antagonized by temperature: There were 55, 89, and 92% decreases in life span from that of control strains at RT, 25°, and 29°, respectively.
![Life span analysis and transgenic rescue of sugarkill. Life span curves of sgk [1]/[js10] (A) and sgk [1]/+ (B) at room temperature (yellow), 25° (orange), and 29° (red) indicate reduced longevity in sgk mutant flies. (C) Statistical analysis of median age reveals that mutant (sgk[1]/[js10]) life spans are significantly reduced compared to heterozygote (sgk[1]/+) and revertant (sgk[R2]/[js10]) control strains (Student's t-test, **P < 0.0001). Median age is the age coincident with 50% survivorship of the genotype. Survival curves represent 4–12 independent populations per genotype. Error given is SEM. (D) Expression of wild-type TPI rescues the severely impaired longevity defect of sgk1 mutants. Transgenic UAS-sgk were generated to express wild-type or mutant sgk proteins. Ubiquitous expression of these transgenes using the actin promotor and the GAL 4 system (Act:GAL4) was used to test rescue of the sgk1 mutation. Expression using sgk+ (green) was effective at rescuing sgk1. The GAL4 control (red) and expression of a UAS-sgk1 transgene bearing the M80T mutation (blue) are insufficient to rescue sgk1. Error represents SEM from four independent longevity experiments (n > 50 animals per genotype).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/genetics/174/3/10.1534_genetics.106.063206/5/m_1237fig3.jpeg?Expires=1716735104&Signature=TgdfvIqotmaL2VggcvwwvdvNRqWp-gK4-rAOrHaH4maavUumPW7clhBrOjNfPx1~kKytXkr1YmXa~B-Nh~of6WqzZR~7McijlOdq5AdKV6uA0qI3QbipfHJfwJrdBsdm-S0q7mPS16gKexGEQV9bjbTUKtUh455Vf6YFVbghmpF8g1UAK6Csn6dmhEPdKST9nGzhWIhmP~uq-2Gqv70dBGOzvDd4whdF39SYBTmg5PJhstCC6aAXFXF9cAecN6mu-eQAE20ZXlWWkD3cgJZERRp9Z2EaX8UNe7Sbt5HvFzxdDF5XuNyafl0GcRG0emeaheVSTp56ZOgeVF4lt0T6EA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Life span analysis and transgenic rescue of sugarkill. Life span curves of sgk [1]/[js10] (A) and sgk [1]/+ (B) at room temperature (yellow), 25° (orange), and 29° (red) indicate reduced longevity in sgk mutant flies. (C) Statistical analysis of median age reveals that mutant (sgk[1]/[js10]) life spans are significantly reduced compared to heterozygote (sgk[1]/+) and revertant (sgk[R2]/[js10]) control strains (Student's t-test, **P < 0.0001). Median age is the age coincident with 50% survivorship of the genotype. Survival curves represent 4–12 independent populations per genotype. Error given is SEM. (D) Expression of wild-type TPI rescues the severely impaired longevity defect of sgk1 mutants. Transgenic UAS-sgk were generated to express wild-type or mutant sgk proteins. Ubiquitous expression of these transgenes using the actin promotor and the GAL 4 system (Act:GAL4) was used to test rescue of the sgk1 mutation. Expression using sgk+ (green) was effective at rescuing sgk1. The GAL4 control (red) and expression of a UAS-sgk1 transgene bearing the M80T mutation (blue) are insufficient to rescue sgk1. Error represents SEM from four independent longevity experiments (n > 50 animals per genotype).
Genetic rescue of sugarkill:
The sgk1 mutation causes a striking progressive locomotor impairment and marked reduction in longevity. UAS-sgk transgenes were expressed using actin∷GAL4, and animals were examined for locomotor function and life span in sgk1 homozygous mutants. Animals of the genotype UAS-sgk+; actin∷GAL4; sgk1/sgk1 did not exhibit temperature- or stress-induced locomotor impairment; however, UAS-sgk+; CyO; sgk1/sgk1 (control lacking GAL4) and UAS-sgk1; actin∷GAL4; sgk1/sgk1 (expressing mutant transgene) animals were not different from sgk1 homozygotes (data not shown).
To test the veracity of the transgenic rescue, longevity of animals bearing the UAS-sgk transgenic constructs was measured. sgk1 animals exhibit a striking 92% reduction in median life span when reared at 29°, suggesting that this would be a rigorous test of transgenic rescue. UAS-sgk+; CyO; sgk1/sgk1 (control lacking GAL4) and UAS-sgk1; actin∷GAL4; sgk1/sgk1 (expressing mutant transgene) animals had severely reduced life spans, whereas UAS-sgk+; actin∷GAL4; sgk1/sgk1 had normal longevity at 29°, demonstrating transgenic rescue of the life span defect of sgk1 with wild-type TPI expression (Figure 3D). These data demonstrate that the M80T mutation is responsible for locomotor and life span impairment in sgk1. Together with the finding that sgk1 is less severe than sgkJS10, these data demonstrate that sgk1 is a hypomorphic, loss-of-function missense mutation.
Neurodegeneration associated with sugarkill mutation:
Human TPI deficiency results in complex neuromuscular dysfunction with neurodegeneration. The sgk mutant and control flies were examined for the presence of neuropathology to determine the extent to which the fly model of TPI deficiency recapitulated this important characteristic of the human disease. The brains of young mutants were uniformly free of pathology (data not shown); however, sgk mutants developed marked neuropathology that was evident by light microscopic evaluation at their median age (Figure 4). Control flies did not exhibit neurodegeneration. Interestingly, animals reared at all three temperatures (RT, 25°, and 29°) exhibited marked neuropathology in their brain and thoracic ganglion that was not present in control tissues (Figure 4). We saw no difference in neuropathology between sgk1 homozygotes and sgk1/js10 animals (data not shown). These data demonstrate that neural dysfunction and degeneration are associated with sgk loss-of-function mutation in Drosophila.
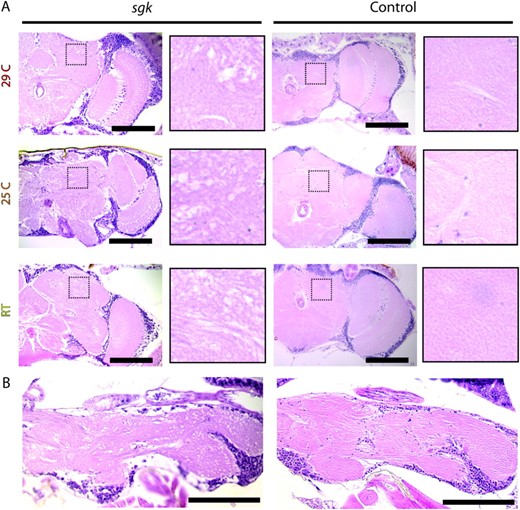
Neuropathology in Drosophila sugarkill mutants. H&E-stained neural tissue of sgk1/sgkjs10 mutants and sgk1/R2 controls is shown. (A) Midbrain frontal sections of mutant and control animals aged at 29°, 25°, and room temperature. sgk mutants show marked vacuolar pathology throughout the central brain and optic lobes. High-magnification panels are ∼4.8× higher magnification of boxed neuropil regions and demonstrate marked degeneration in sgk that is never observed in control brain tissues. (B) Thoracic ganglion sections of mutant and control flies aged at 29°. Similar degeneration was seen in animals aged at 25° and room temperature. Mutant sgk tissue is from animals aged to days 3 (29°), 7 (25°), and 22 (RT), respectively. Control tissue is from animals aged to days 37 (29°), 70 (25°), and 80 (RT), respectively. Analysis did not reveal neuropathology in aged sgk1/R2 or wild-type animals. In young animals of all genotypes there is no evidence of neural pathology (data not shown). n ≥ 15 animals per genotype. Bar, 100 μm.
Normal mitochondrial ultrastructure in sugarkill:
Previous studies have shown that stress-sensitive locomotor defects, reduced longevity, and neural dysfunction can result from mitochondrial ultrastructural defects associated with the ATP61 mutation (Celotto et al. 2006). Due to the similarity in phenotypes between ATP61 and sgk1 we hypothesized that a similar ultrastructural defect in mitochondria may underlie sgk pathogenesis, possibly resulting from inefficient glycolysis and overreliance on mitochondria. To determine whether the sgk mutation causes any obvious ultrastructural pathology, flight muscle and brain tissue were examined using transmission electron microscopy (TEM), as previously described (Celotto et al. 2006). Interestingly, muscle and brain mitochondria have normal ultrastructural morphology (Figure 5).
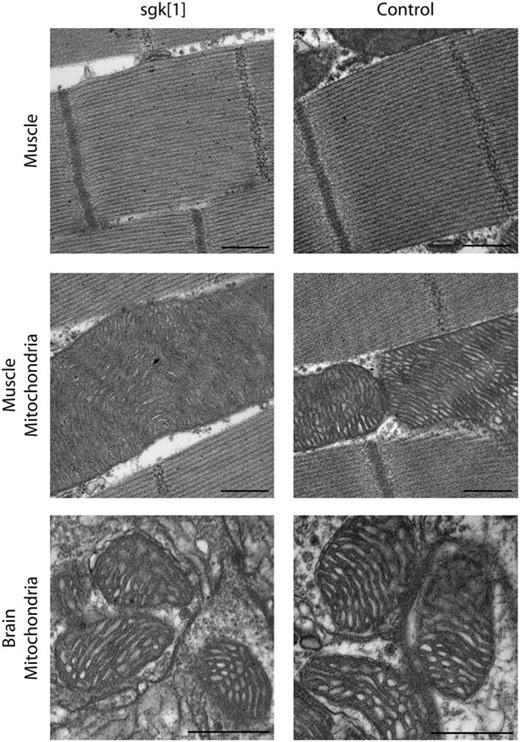
Ultrastructural analysis of sgk1. Transmission electron microscopy of sgk1 mutants reveals no morphological aberrations of indirect flight muscle structure. Myofibrillar Z and M lines appear intact and exist in consistent intervals in both mutant and wild-type control tissue. Muscle mitochondria from sgk1 do not display overt, aberrant morphology and are indistinguishable from control mitochondria. Mitochondria from brain also appear normal in mutant and control samples. Muscle micrographs are from day 5 adults. Brain micrographs are from day 3 sgk mutants and days 3–10 control adults. Bar, 500 nm. N ≥ 3, per genotype.
Bioenergetic analysis of sugarkill mutants:
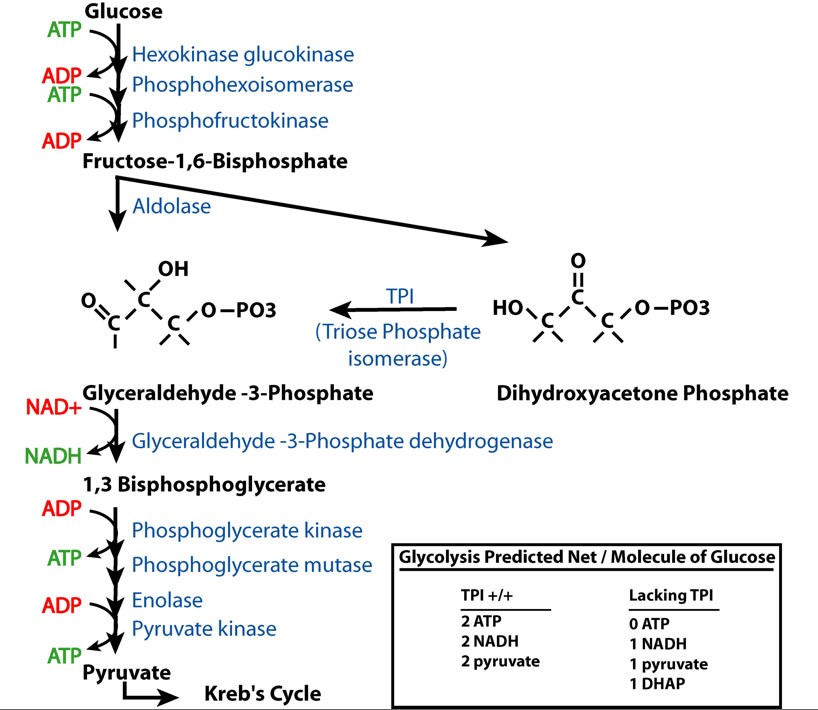
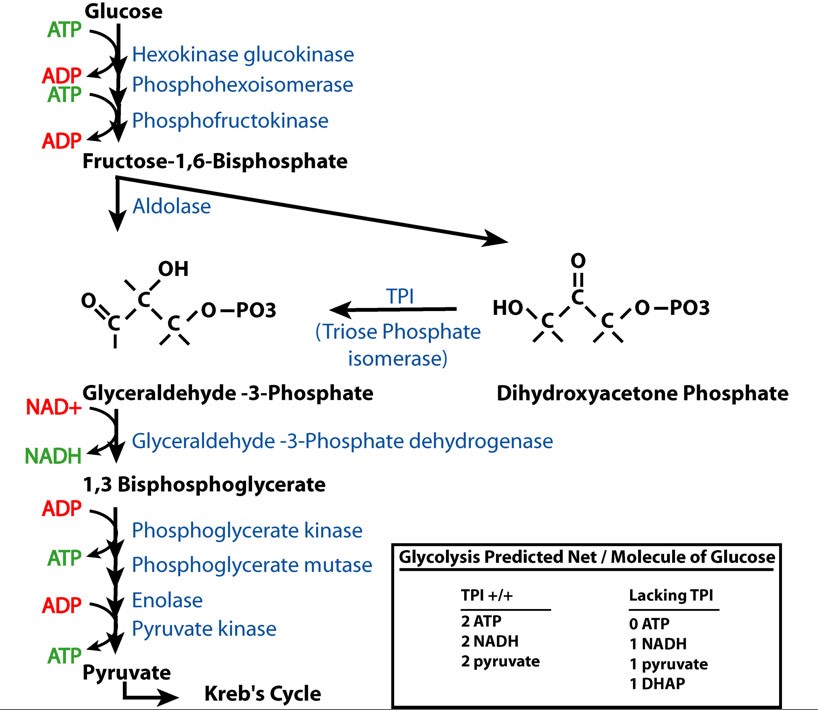
TPI has a well-described function in glycolysis as the enzyme that converts dihydroxyacetone phosphate (DHAP) to glyceraldehydes-3-phosphate (G3P). As predicted from this biochemical pathway, loss of TPI function would alter the products of glycolysis from 2 ATP, 2 NADH, and 2 pyruvate to 1 NADH, 1 pyruvate, and 1 DHAP per molecule of glucose (supplemental Figure S2 at http://www.genetics.org/supplemental/). This suggests that the basis of the pathology may be due to bioenergetic impairment, a toxic effect of DHAP accumulation or one of its catabolites, or a defect arising from reduced glycolytic flux independent of energy production.
![Bioenergetic analysis of sgk1. HPLC assay of creatine (Cr), phosphocreatine (PCr), and lactic acid (LA) in sgk1 mutants and age-matched control animals is shown. (A) There is a significant increase in the nanomoles of PCr per milligram of total protein in sgk[1] mutants. (B) A nonsignificant decrease in the nanomoles of Cr per milligram of total protein is seen in sgk[1] mutants. (C) There is a significant decrease in the nanomoles of LA per milligram of total protein in sgk[1] mutants. (D) The ratio between PCr:Cr is significantly increased in sgk[1] mutants vs. controls. The ratio in wild-type flies is similar to that seen in mice (Klivenyi et al. 2004). (E) The TCr (total nanomoles of creatine, phosphocreatine, and cyclocreatine) remains constant in both genotypes tested. N ≥ 4 for each genotype. Error shown is SEM. Significance is determined by Student's t-test, *P < 0.05, **P < 0.01.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/genetics/174/3/10.1534_genetics.106.063206/5/m_1237fig6.jpeg?Expires=1716735104&Signature=FHx19W6bc4cdIyQz81EjdgwV0WafseGSgDHVomn5uOSbr8wWrkeo~uOz71-pLlvzY57n4YXBbzDNjzMbst~fJitfPcUeCp1KSc0uRPGpii1JresxWq1sdVlJ3GRCNKHSVxSf4Ho96K7h~9M9-of0HZmugu-ByQIvPVPH-MknyN83XxxjezTYHmKcxF-bkcBYSV7IxIIpEYAWnYmX~bId-5mi1yFxw-Q6OD096BVLzH2v7oITVZjrBE5gUQ1YiwJYO9AokFP4vCs5YeqE3~Mh9cW6rktocfXAu6Z2~BtZosfNkRB9kNzH2KwP6zQ5Voogg6V~5q6Ma83XpqPBXTzDVQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Bioenergetic analysis of sgk1. HPLC assay of creatine (Cr), phosphocreatine (PCr), and lactic acid (LA) in sgk1 mutants and age-matched control animals is shown. (A) There is a significant increase in the nanomoles of PCr per milligram of total protein in sgk[1] mutants. (B) A nonsignificant decrease in the nanomoles of Cr per milligram of total protein is seen in sgk[1] mutants. (C) There is a significant decrease in the nanomoles of LA per milligram of total protein in sgk[1] mutants. (D) The ratio between PCr:Cr is significantly increased in sgk[1] mutants vs. controls. The ratio in wild-type flies is similar to that seen in mice (Klivenyi et al. 2004). (E) The TCr (total nanomoles of creatine, phosphocreatine, and cyclocreatine) remains constant in both genotypes tested. N ≥ 4 for each genotype. Error shown is SEM. Significance is determined by Student's t-test, *P < 0.05, **P < 0.01.
To directly examine the hypothesis that sgk1 mutants have impaired cellular energy stores, the bioenergetic state of sgk mutants was measured using HPLC to analyze PCr, creatine Cr, and lactic acid LA levels. Phosphocreatine is used to buffer cellular ATP concentration and is an excellent indicator of the cellular bioenergetic state (Matthews et al. 1998). Acid lactate levels were significantly lower in sgk1 mutants vs. age-matched controls, consistent with a reduced rate of anaerobic metabolism. Surprisingly, the data also revealed a significant increase in phosphocreatine in sgk1 mutants (Figure 6). These data argue that the impaired locomotion does not result from bioenergetic impairment and suggest that lack of locomotor function may be the direct cause of increased PCr and PCr:Cr ratios in sgk1 mutants. This suggests that a mechanism independent of general bioenergetic impairment is causing the poor locomotion, such as toxicity associated with DHAP or its catabolites, or poor glycolytic flux.
DISCUSSION
Forward genetic screening has been used to isolate informative mutants that model progressive neurodegenerative diseases in Drosophila (Palladino et al. 2002, 2003; Celotto and Palladino 2005; Celotto et al. 2006). Here we report the isolation of a novel mutant in Drosophila that displays reduced life span, locomotor defects, and neurodegeneration throughout the central nervous system. The affected gene was cloned and we discovered a missense mutation causing an M80T substitution in the highly conserved TPI protein. We have named this mutation sgk1 and believe it serves as a useful model of human genetic TPI deficiency disease.
Numerous mutations causing human TPI deficiency exist, including those that are temperature sensitive and interfere with the ability of the protein to form a stable homodimer (supplemental Figure S1 at http://www.genetics.org/supplemental/). The location of the sgk1 mutation affects a conserved methionine residue residing at the dimer interface. (Figure 1, supplemental Figure S1). sgk1 is thought to be a hypomorphic mutation (mild, loss of function) consistent with an amino acid substitution from an uncharged, weakly hydrophobic residue to an uncharged polar residue. The TPI protein has evolved to a state of catalytic perfection with diffusion-controlled reaction rates, suggesting a strong selective advantage associated with efficient enzyme function. Interestingly, disease results from specific recessive, loss-of-function, missense mutations that appear to be hypomorphic for protein function. Mice severely deficient for TPI function have been reported to die at early postimplantation stages of development (Merkle and Pretsch 1989), while heterozygotes of TPI mutations believed to be null do not appear to effectively model human enzymopathic disease (Zingg et al. 1995).
Our biochemical analysis indicates that bioenergetic impairment is not responsible for the decreased life span or locomotor deficits associated with sgk1. Specifically, phosophocreatine was not reduced in sgk mutants but was surprisingly found to be significantly increased, possibly resulting from reduced animal activity (Figure 6). Also, sgk1 mutants do not have aberrant mitochondrial ultrastructure (Figure 5) as observed in another stress-sensitive fly mutant (Celotto et al. 2006) and suggested by previous studies using human tissues (Bardosi et al. 1990). Lactic acid levels were found to be decreased, suggesting a reduction in the overall rate of glycolysis, but this does not result in a depletion of phosphocreatine levels that would be consistent with bioenergetic impairment (Figure 6). These data are consistent with unchanged ATP levels that were seen in erythrocytes derived from human TPI-deficient patients (Eber et al. 1991). Without TPI function there is believed to be no net ATP production from the anaerobic metabolism of glucose and production of intermediate DHAP (supplemental Figure S2 at http://www.genetics.org/supplemental/). Although increased DHAP levels have been reported in human erythrocytes (Zanella et al. 1985; Eber et al. 1991; Hollan et al. 1997; Karg et al. 2000; Olah et al. 2002, 2005), there is no direct evidence that this molecule is toxic and data suggest that it can be shunted into lipid biosynthetic pathways (Olah et al. 2002). TPI mutation resulting in reduced glycolytic flux is predicted to impair an organism's ability to function under conditions of oxygen debt and cause overreliance on mitochondrial ATP production. However, lack of ultrastructural changes in brain or muscle mitochondria suggests that pathogenesis is likely not of mitochondrial origin. The mechanistic consequences of reduced glycolytic flux and an inability to enter oxygen debt (relying on anaerobic metabolism) are not well understood but appear to underlie the pathogenesis associated with TPI impairment.
Mutations causing human TPI deficiency are complex and result in a severe disease pathogenesis marked by progressive neuromuscular impairment and reduced life expectancy. Human TPI disease conditions are typically caused by specific homozygous missense mutations that often encode thermolabile proteins (Daar et al. 1986; Chang et al. 1993). The recessive nature and temperature sensitivity of the sgk1 mutant, whose phenotypes are closely analogous to human disease symptoms, suggest that this mutant will effectively model TPI enzymopathic disease and provide an amenable genetic model system for studying the basic mechanisms of disease pathogenesis.
Footnotes
These authors contributed equally to this work.
Footnotes
Communicating editor: A. J. Lopez
Acknowledgement
We thank Sunil Iyer for assistance generating the js10 allele, Nicholas Ierovante for assistance with histology and life span analyses, University of Pittsburgh Biomedical Research Support Facilities for assistance with sequencing, the Pittsburgh Center for the Environmental Basis of Human Disease for support, Simon Watkins and the Center for Biologic Imaging for access to microscopes and histology equipment, the Bloomington Stock Center for fly strains, and Susan Amara for use of the real-time PCR machine. We thank the National Institutes of Health (NIH) National Institute of Neurological Disorders and Stroke (T32NS 07391-07) (A.M.C.), the Pittsburgh Institute for Neurodegenerative Diseases [research grant 023RA02 (M.J.P.)], the American Heart Association [award 0630344N (M.J.P.)], the Pittsburgh Foundation and Emmerling Fund [grant M2005-0068 (M.J.P.)], the NIH [grant AG025046 (M.J.P.)], and the University of Pittsburgh Department of Pharmacology and School of Medicine for financial support.
References
Chang, M. L., P. J. Artymiuk, X. Wu, S. Hollan, A. Lammi et al.,
Daar, I. O., and L. E. Maquat,
Daar, I. O., P. J. Artymiuk, D. C. Phillips and L. E. Maquat,
Eber, S. W., A. Pekrun, A. Bardosi, M. Gahr, W. K. Krietsch et al.,
Hollan, S., M. Magocsi, E. Fodor, M. Horanyi, V. Harsanyi et al.,
Kinoshita, T., R. Maruki, M. Warizaya, H. Nakajima and S. Nishimura,
Knowles, J. R.,
Mande, S. C., V. Mainfroid, K. H. Kalk, K. Goraj, J. A. Martial et al.,
Oakeshott, J., S. W. McKechnie and G. K. Chambers,
Valentin, C., S. Pissard, J. Martin, D. Heron, P. Labrune et al.,
Valentine, W. N.,
Watanabe, M., B. C. Zingg and H. W. Mohrenweiser,
Wierenga, R. K., M. E. Noble, G. Vriend, S. Nauche and W. G. Hol,
Zanella, A., M. Mariani, M. B. Colombo, C. Borgna-Pignatti, P. De Stefano et al.,

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}