





Abstract
Eyes absent (EYA) proteins are defined by a conserved C-terminal EYA domain (ED) that both contributes to its function as a transcriptional coactivator by mediating protein-protein interactions and possesses intrinsic protein tyrosine phosphatase activity. Mutations in human EYA1 result in an autosomal dominant disorder called branchio-oto-renal (BOR) syndrome as well as congenital cataracts and ocular defects (OD). Both BOR- and OD-associated missense mutations alter residues in the conserved ED as do three missense mutations identified from Drosophila eya alleles. To investigate the molecular mechanisms whereby these mutations disrupt EYA function, we tested their activity in a series of assays that measured in vivo function, phosphatase activity, transcriptional capability, and protein-protein interactions. We find that the OD-associated mutations retain significant in vivo activity whereas those derived from BOR patients show a striking decrease or loss of in vivo functionality. Protein-protein interactions, either with its partner transcription factor Sine oculis or with EYA itself, were not significantly compromised. Finally, the results of the biochemical assays suggest that both loss of protein tyrosine phosphatase activity and reduced transcriptional capability contribute to the impaired EYA function associated with BOR/OD syndrome, thus shedding new light into the molecular mechanisms underlying this disease.
RETINAL specification and development in both vertebrates and invertebrates rely on the concerted actions of a group of evolutionarily conserved transcription factors and cofactors that include twin of eyeless (toy), eyeless (ey), eyes absent (eya), sine oculis (so), and dachshund (dac), with the human homologs referred to as PAX6 (homolog of both toy and ey), EYA, SIX, and DACH, respectively (reviewed in Wawersik and Maas 2000; Pappu and Mardon 2002). PAX6 lies atop the hierarchy and directly activates expression of EYA and SIX family members, which operate synergistically to induce expression of DACH and other downstream genes (reviewed in Pappu and Mardon 2002). Because of their prominent role in eye development, exemplified in Drosophila by the “eyeless” phenotype and visual system defects associated with loss-of-function mutations and the ability to induce formation of ectopic eye tissue upon overexpression, these genes have been referred to collectively as the retinal determination (RD) gene network (Pappu and Mardon 2002). In addition to their roles in the eye, all RD genes, either individually or as a network, contribute to a diverse array of essential developmental processes in Drosophila and in vertebrates. Consequently, null mutations are lethal and exhibit complex defects in a variety of tissues and organs (reviewed in Wawersik and Maas 2000; Silver and Rebay 2005).
EYA family members encode novel nuclear proteins defined by a conserved ∼275-amino-acid C-terminal motif, referred to as the EYA domain (ED), which mediates direct interactions with SO/SIX and DAC/DACH (Bonini et al. 1993; Chen et al. 1997; Pignoni et al. 1997; Zimmerman et al. 1997; Heanue et al. 1999; Ohto et al. 1999). The N terminus of EYA contributes a conserved trans-activation function to an EYA-SO bipartite transcription factor in which the homeodomain protein SO/SIX provides the DNA-binding moiety (Ohto et al. 1999; Silver et al. 2003). The mechanistic implications of EYA-DACH interactions are less clear. DAC/DACH proteins function as both coactivators and corepressors and may also have DNA-binding capability, implying roles in tethering EYA to the DNA and influencing transcription of downstream genes (Ikeda et al. 2002; Kim et al. 2002; Li et al. 2003). Recent work revealed that the ED of EYA also exhibits catalytic activity, functioning as a protein tyrosine phosphatase (Li et al. 2003; Rayapureddi et al. 2003; Tootle et al. 2003b). EYA's phosphatase activity appears critical for switching DACH between corepressor and coactivator states, suggesting an integral contribution to regulating transcriptional output (Li et al. 2003).
The four mammalian EYA paralogs, designated EYA1–4, exhibit distinct but overlapping expression patterns, with EYA1–3 expressed in the developing eye in a PAX6-dependent manner (Xu et al. 1997; Zimmerman et al. 1997; Borsani et al. 1999; Hanson 2001). Emphasizing the high degree of functional conservation among EYA proteins, mammalian EYA1, EYA2, or EYA3 transgenes can rescue the eyeless phenotype of Drosophila eya mutations with comparable efficiency (Bonini et al. 1997; Bui et al. 2000); EYA4 has not yet been tested in such assays. In terms of practical utility, this high degree of structural and functional homology validates the use of Drosophila as an experimentally tractable model system in which to study the function and activity of mammalian EYA proteins in vivo.
Human EYA1 was positionally cloned as the gene responsible for branchio-oto-renal (BOR) syndrome, an autosomal dominant disorder characterized by the association of branchial arch anomalies, otic defects, and a broad spectrum of renal abnormalities (Abdelhak et al. 1997b; Vincent et al. 1997). The clinical features of BOR syndrome manifest early during development with variable penetrance, with hearing loss being the most commonly associated defect (Rodriguez Soriano 2003); for a description of the clinical features of BOR syndrome as well as a list of the associated molecular lesions in EYA1, see http://www.medicine.uiowa.edu/pendredandbor/BOR.htm. Mouse knockouts of Eya1 have a similarly complex phenotype characterized by craniofacial, ear, and kidney defects (Xu et al. 1999).
The fact that BOR patients and murine EYA1 knockout mice generally lack ocular defects has led to the suggestion that in contrast to the fly, where eya plays a pivotal role in eye specification and development, EYA1 function may not be critical for vertebrate eye development. However, sequencing of the EYA1-coding region from a series of human patients with congenital cataracts and ocular segment anomalies, including one who also manifested symptoms of BOR disease, revealed missense mutations in the conserved ED, arguing for involvement of EYA1 in the mammalian eye (Azuma et al. 2000).
Previous in vitro analysis of missense mutations in EYA1 identified in human patients with BOR syndrome and/or ocular defects suggested that several of these lesions impaired interactions with SIX family proteins and resulted in defects in transcriptional output from the EYA-SIX transcription factor (Buller et al. 2001; Ozaki et al. 2002). However, not all missense mutations that were examined in these studies impaired function of the EYA-SIX transcription factor, nor were results entirely consistent among the various in vitro assays used in the analyses (Buller et al. 2001; Ozaki et al. 2002). This variability raises the possibility that the molecular basis of BOR disease may be quite complex and emphasizes the importance of establishing in vivo model systems that can provide the critical foundation on which to base subsequent in vitro studies. While such analyses are at best cumbersome in mouse, Drosophila, and in particular the fly eye where the function and regulation of eya have been extensively studied, provides an ideal in vivo system in which to explore the question of how these missense mutations associated with human disease might alter EYA function. The ability of mammalian EYA genes to complement Drosophila eya further validates this approach.
Furthermore, given the recent discovery that EYA, in addition to operating as a transcriptional coactivator in conjunction with SIX proteins (Ohto et al. 1999; Silver et al. 2003), has a second function as a protein phosphatase (reviewed in Li et al. 2003; Rayapureddi et al. 2003; Tootle et al. 2003b; Rebay et al. 2005), we wanted to explore the possibility that some of the missense mutations in the conserved ED might disrupt this new function. Thus, in addition to determining their activity in our Drosophila eye in vivo assays, we have also examined the five missense mutations in EYA1 derived from human patients with BOR syndrome and/or ocular defects, with the three missense mutations identified in Drosophila eya loss-of-function alleles, for defects in phosphatase and/or transcriptional activities. Our work provides the first analysis of human patient-derived EYA mutations in an in vivo developmental context and suggests that defects in both phosphatase and transcription functions likely contribute to the molecular causes of BOR syndrome in humans and to compromised development in flies.
MATERIALS AND METHODS
Molecular biology and transgenic analyses:
Site-directed mutagenesis using Stratagene's quick-change methodology was performed to generate the eight missense mutations in both Drosophila EYA and mouse EYA3. All mutations were confirmed by sequencing. Reverse-complementary primer pairs were used; sequences given correspond to the sense strand primer. Primers used for mutagenesis of Drosophila EYA are:
T497M, 5′ ggatctggacgagatgctcatcatcttcca 3′;
E528K, 5′ tcgccttccgcatgaaggagatggtcttca 3′;
G594S, 5′ ccaccggtgtgaggagcggcgtcgattgga 3′;
T643I, 5′ aatcgaggtggcgatcgacaactgggccac 3′;
S655P, 5′ ggcgctcaagtgcctgcccatgatctcccagcg 3′;
L673R, 5′ aacctccacgcaacgggccccggcgctggc 3′;
R715G, 5′ gtgactcgctttgggggcaagagcacctac 3′;
G723E, 5′ ctacgtggtgattgaggatgggaacgagga 3′.
Primers used for mutagenesis of mouse EYA3 are:
T250M, 5′ tgggacttggacgaaatgatcatcatctttcatt 3′;
E281K, 5′ caggtttaaccatgaaagaaatgatttttg 3′;
G344S, 5′ tcatctgtgggcgttcagtcaggtgtggactgga 3′;
T393I, 5′ agagatcgaggtgctgattgactcctggttaggaa 3′;
L405P, 5′ cgctcaagtccttgcctctcatccagtctc 3′;
L423R, 5′ ctgatcactaccacgcagcgggttccagccctggc 3′;
K465G, 5′ attgtttcgaggtttgggggaaaagtcacatatgt 3′;
G473E, 5′ catatgtagtgattgaagatggacgagatg 3′.
The mutations in mouse Eya3 were introduced into the GST-ED fusion protein (aa 237–510 of mouse EYA3) described in Tootle et al. (2003b). For Drosophila eya, site-directed mutagenesis was performed on a KpnI/SalI fragment of Drosophila eya in Bluescript (Stratagene, La Jolla, CA). A three-piece ligation linked the flag-epitope-tagged N terminus of EYA, obtained as an EcoRI/KpnI fragment from pRmHa3-flag-eya (Silver et al. 2003), with the mutagenized KpnI/SalI EYA C terminus into the EcoRI/SalI sites of pRmHa3 to generate the metallothionein promoter-driven expression constructs used in the transcription and co-immunoprecipitation studies. The mutant constructs were shuttled into the P-element transformation vector pUAST as an EcoRI/SalI fragment. Establishment of transgenic lines and in vivo genetic analysis was performed as previously described (Hsiao et al. 2001; Tootle et al. 2003b). Epitope-tagged versions of wild-type EYA and SO are as described in Silver et al. (2003).
Phosphatase assays:
Phosphatase assays were performed to analyze the ability of GST-mouse EYA3 ED fusion proteins containing the mutations listed in Figure 1

Human and Drosophila-derived missense mutations in the conserved EYA domain. Amino acid sequence alignment of EYA domains (ED) from human (hEYA1), mouse (mEya3), and Drosophila (dEYA) showing the substitution mutations analyzed. Identical amino acid residues that have been mutated are boxed, immediately above each boxed area is the site-directed mutation used in this study, and in superscript is the source of mutation. Residues that are shaded represent the haloacid dehalogenase (HAD) motifs and the putative Sine oculis binding sites are underlined with a solid line. mEya3 protein was used for phosphatase assays and dEYA was used for all other experiments. It should be noted that the S487PBOR mutation affects a residue that is not strictly conserved among EYA proteins. Blast searches reveal multiple variants at this position including the L in mouse EYA3, as well as T, A, and N in various other vertebrate EYA homologs (data not shown). Such variation, when considered in light of the equivalent ability demonstrated by different mammalian EYA paralogs to functionally complement Drosophila eya mutations, suggests that it is the consequences of introducing a P (proline) in this particular position of the protein, rather than the exact identity of the naturally occurring residue, that are important.
to dephosphorylate the peptide I(pY)GEF (CalBiochem, La Jolla, CA) as previously described in Tootle et al. (2003a).
Transcription assays:
The Na, K-ATPase α1 subunit gene (ATP1α1) regulatory element (ARE)-luciferase reporter construct was as described in Silver et al. (2003). The 250-bp lozenge minimal enhancer element (LMEE) was amplified from the LMEE-lacZ plasmid and from versions in which the SO- and GLASS-binding sites were mutated, LMEEso-lacZ and LMEEgl-lacZ, respectively (gifts from U. Banerjee, described in Yan et al. 2003), by PCR using the universal primer and a LMEE-specific primer, 5′ CTGCAGCATTAACAAAATAAAAAAGGGG 3′. The PCR product was digested with KpnI/PstI and ligated into the KpnI/PstI sites upstream of the hsp70 TATA box in BSSK-TATA-luciferase (Silver et al. 2003). Transcription assays were performed in triplicates as previously described (Silver et al. 2003), using 5 μg per assay of the reporter gene and each expression plasmid (expression plasmids previously described in Silver et al. 2003; Tootle et al. 2003b), and normalized using 1 μg of Actin-lacZ per assay.
Co-immunoprecipitation and Western blots:
Transfection and cell lysis [lysis buffer: 300 mm NaCl, 50 mm Tris (pH 7.5), 2 mm EDTA, 2 mm EGTA, 1% NP-40, and one complete mini protease inhibitor cocktail tablet (Roche, Indianapolis) per 10 ml], immunoprecipitation with anti-Flag conjugated beads (Sigma, St. Louis), and Western blotting were done as previously described in Silver et al. (2003). Guinea pig (GP) anti-EYA (1:10,000), GP anti-SO (1:10,000), and mouse anti-MYC (1:10) were used for Western blotting. Anti-SO antibody was raised by injecting guinea pigs with full-length recombinant GST-SO fusion protein.
RESULTS AND DISCUSSION
Missense mutations associated with BOR syndrome and ocular defects compromise EYA function in vivo:
The missense mutations that have been identified either from loss-of-function alleles of Drosophila eya or from human patients suffering from BOR syndrome and/or ocular defects (OD) affect residues that are conserved between vertebrate and invertebrate EYA proteins (Tables 1–4
EYA mutations examined in this study: mutations derived from Drosophila eya loss-of-function alleles
Homologous residue in | ||
|---|---|---|
| Missense mutation in dEYA | hEYA1 | mEya3 |
| T497M | T332 | T250 |
| T643I | T475 | T393 |
| G723E | G555 | G473 |
Homologous residue in | ||
|---|---|---|
| Missense mutation in dEYA | hEYA1 | mEya3 |
| T497M | T332 | T250 |
| T643I | T475 | T393 |
| G723E | G555 | G473 |
EYA mutations examined in this study: mutations derived from Drosophila eya loss-of-function alleles
Homologous residue in | ||
|---|---|---|
| Missense mutation in dEYA | hEYA1 | mEya3 |
| T497M | T332 | T250 |
| T643I | T475 | T393 |
| G723E | G555 | G473 |
Homologous residue in | ||
|---|---|---|
| Missense mutation in dEYA | hEYA1 | mEya3 |
| T497M | T332 | T250 |
| T643I | T475 | T393 |
| G723E | G555 | G473 |
EYA mutations examined in this study: mutations derived from human BOR patients
Homologous residue in | ||
|---|---|---|
| Missense mutation
in hEYA1 | dEYA | mEya3 |
| S487P | S655 | L405 |
| L505R | L673 | L423 |
Homologous residue in | ||
|---|---|---|
| Missense mutation
in hEYA1 | dEYA | mEya3 |
| S487P | S655 | L405 |
| L505R | L673 | L423 |
EYA mutations examined in this study: mutations derived from human BOR patients
Homologous residue in | ||
|---|---|---|
| Missense mutation
in hEYA1 | dEYA | mEya3 |
| S487P | S655 | L405 |
| L505R | L673 | L423 |
Homologous residue in | ||
|---|---|---|
| Missense mutation
in hEYA1 | dEYA | mEya3 |
| S487P | S655 | L405 |
| L505R | L673 | L423 |
EYA mutations examined in this study: mutations derived from human OD patients
Homologous residue in | ||
|---|---|---|
| Missense mutation
in hEYA1 | dEYA | mEya3 |
| E363K | E528 | E281 |
| R547G | R715 | K465 |
Homologous residue in | ||
|---|---|---|
| Missense mutation
in hEYA1 | dEYA | mEya3 |
| E363K | E528 | E281 |
| R547G | R715 | K465 |
EYA mutations examined in this study: mutations derived from human OD patients
Homologous residue in | ||
|---|---|---|
| Missense mutation
in hEYA1 | dEYA | mEya3 |
| E363K | E528 | E281 |
| R547G | R715 | K465 |
Homologous residue in | ||
|---|---|---|
| Missense mutation
in hEYA1 | dEYA | mEya3 |
| E363K | E528 | E281 |
| R547G | R715 | K465 |
EYA mutations examined in this study: mutations derived from human BOR + OD patients
Homologous residue in | ||
|---|---|---|
| Missense mutation
in hEYA1 | dEYA | mEya3 |
| G426S | G594 | G444 |
Homologous residue in | ||
|---|---|---|
| Missense mutation
in hEYA1 | dEYA | mEya3 |
| G426S | G594 | G444 |
EYA mutations examined in this study: mutations derived from human BOR + OD patients
Homologous residue in | ||
|---|---|---|
| Missense mutation
in hEYA1 | dEYA | mEya3 |
| G426S | G594 | G444 |
Homologous residue in | ||
|---|---|---|
| Missense mutation
in hEYA1 | dEYA | mEya3 |
| G426S | G594 | G444 |
and Figure 1; Abdelhak et al. 1997a,b; Vincent et al. 1997; Azuma et al. 2000; Yashima et al. 2003). To understand at a mechanistic level how these mutations might compromise EYA function, we tested their activity in a series of bioassays we have established in the course of our ongoing investigations of EYA function and regulation (Hsiao et al. 2001; Silver et al. 2003; Tootle et al. 2003b). For the analyses described below, site-directed mutagenesis was used to introduce the desired missense mutations into both Drosophila EYA and mouse EYA3 (Figure 1; see materials and methods for details). The Drosophila versions were used for in vivo and cell culture experiments while the murine constructs were used for in vitro biochemical studies. Difficulty with obtaining sufficient amounts of recombinant protein has precluded us, and others previously (Li et al. 2003), from using mammalian EYA1 in the biochemical analyses. However, the ability of both mammalian EYA1 and EYA3 to complement Drosophila eya mutations with comparable efficiency (Bonini et al. 1997; Bui et al. 2000) argues strongly that these are functionally analogous proteins and validates the approach we have taken. For clarity, and to emphasize the origin of the eight mutations examined in this study, we have added the superscripts “FLY,” “BOR,” “OD,” and “BOR+OD” to the mutation name.
We first investigated the ability of mutant EYA transgenes to function in vivo using the genetically tractable Drosophila system. It has previously been shown that ectopic expression of EYA can induce formation of eye tissue outside of the normal visual field (Bonini et al. 1997; Chen et al. 1997; Pignoni et al. 1997) and that quantification of the efficiency with which a particular transgene induces ectopic eyes can reveal relative activity differences between various EYA mutations (Hsiao et al. 2001; Silver et al. 2003; Tootle et al. 2003b). To control for transgene-specific position effects, multiple independent insertion lines were tested for each mutation and expression of the mutant EYA proteins at levels comparable to that obtained with wild type was confirmed by immunoblot analysis (data not shown).
Interestingly, of the eight EYA mutations tested, only the three OD-derived mutations, R715GOD, E528KOD, and G594SBOR+OD, retained the ability to induce ectopic eyes (Figure 2A)
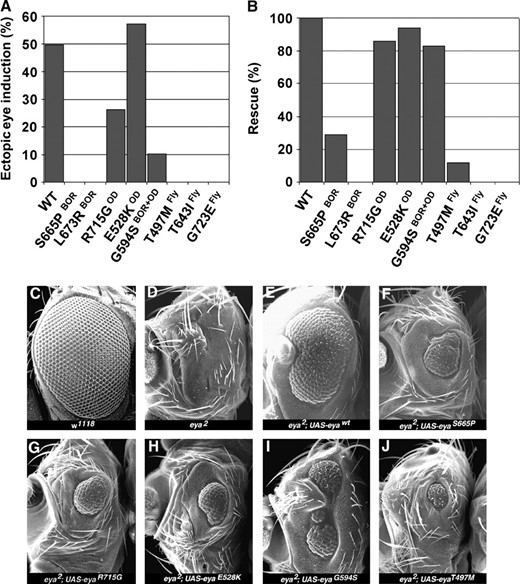
Ectopic eye induction and genetic rescue assays reveal differential in vivo activity among the EYA mutants. (A) The percentage of ectopic eye induction value associated with expression of the different mutations was derived from analysis of multiple independent transgenic lines. The y-axis refers to the penetrance of ectopic eyes and does not account for differences in size of the ectopic eye patch. However, it is important to note that the qualitative nature of the ectopic eye patches correlates tightly with overall penetrance, and hence activity level, of each transgene. (B) The percentage of flies carrying the genotype UAS-eya/dpp-GAL4 that showed rescue of the eya2 eyeless phenotype. Again, the scoring was not weighted to account for differences in size of the patch of rescued eye tissue. (C–J) Scanning electron micrographs of adult eyes showing the rescue of eya2 eyeless phenotype by different transgenes. Genotypes are indicated.
. Of these, E528KOD exhibited activity comparable to that of wild-type EYA transgenes, whereas R715GOD and G594SBOR+OD showed a two- and fivefold respective reduction in activity. The remaining five EYA mutant transgenes, T497MFLY, T643IFLY, S655PBOR, G723EFLY, and L673RBOR, were inactive in this assay. This result indicates a distinct difference in in vivo functionality between OD- and BOR-derived human mutations that may underlie the distinct clinical manifestations.
The second set of in vivo experiments used a genetic rescue assay to assess the function of these eight mutant transgenes in the context of normal, rather than ectopic, eye development. Specifically we tested their ability to complement the eye-specific null allele eya2 that completely lacks eye tissue. Consistent with the results of the ectopic eye induction experiment, transgenic lines carrying mutations with the three OD-derived amino acid substitutions R715GOD, E528KOD, and G594SBOR+OD showed significant rescue of eye tissue. In fact, the percentage of flies showing rescue, defined by the presence of eye tissue within the normal visual field, was almost comparable to that obtained by expressing wild-type EYA transgenes (Figure 2, B, C, and G–I). However, the extent of rescue, judged by comparing the overall size of the recovered eye tissue, reveals that these three mutations have significantly reduced activity, consistent with their association with a pathological condition in human patients (Figure 2).
Interestingly, the rescue obtained by expressing G594SBOR+OD transgenes was phenotypically distinct from that of all other EYA transgenes, whether wild type or mutant, we have ever tested (Tootle et al. 2003b). Specifically the recovered eye tissue was more dorsally located than usual and was almost always split into multiple independent fields (Figure 2I). Perhaps this distinct in vivo behavior reflects a gain-of-function or neomorphic aspect to this allele that underlies the compound nature of the symptoms, both BOR and OD, exhibited by the human patient from whom the mutation originated.
Much weaker rescue, both quantitatively and qualitatively, was seen with the S655PBOR and T497MFLY transgenes (Figure 2, F and J), while the others lacked activity in this assay. Emphasizing again that these differences reflect activity changes rather than reduced protein levels, comparable protein expression levels and nuclear localization were observed for both mutant and wild-type EYA transgenes (data not shown).
Together, the results of these two in vivo experiments suggest an allelic series in which T643IFLY, G723EFLY, and L673RBOR completely lack activity; S655PBOR and T497MFLY exhibit only slight residual function; and R715GOD, E528KOD, and G594SBOR+OD retain significant activity. Thus, as assayed in the context of in vivo eye development assays in Drosophila, the mutations associated with OD in humans retained high although still lower than normal activity levels, whereas those originating from patients manifesting BOR-specific defects lacked significant function. The loss of activity of the Drosophila-derived mutations is consistent with their having been isolated as loss-of-function alleles (Bui et al. 2000; Rebay et al. 2000).
On the basis of these analyses, it is possible that the molecular mechanisms linking mutations in EYA1 to either BOR or OD are distinct. In the case of the human patient exhibiting both BOR and OD symptoms, it may be informative to sequence the SIX1 coding region to see if a mutation in SIX1 might be responsible for the BOR symptoms, as such mutations have recently been linked to BOR syndrome (Ruf et al. 2004). Future identification and functional analysis of additional missense mutations in human SIX1 and EYA1 will provide important tools with which to explore further the molecular determinants of these disorders.
Loss of phosphatase activity may contribute to BOR syndrome defects:
The collection of eight mutants derived from null Drosophila eya alleles and from human patients suffering from BOR syndrome and/or OD all map to the portion of the conserved ED (Figure 1) that has recently been shown to possess intrinsic protein phosphatase activity (Li et al. 2003; Rayapureddi et al. 2003; Tootle et al. 2003b). Therefore to further our understanding of the physiological relevance of EYA's phosphatase activity, we asked whether any of these nonsense mutations impaired catalytic function.
Specifically, we tested the ability of bacterially expressed and purified GST-ED fusion proteins to dephosphorylate the tyrosyl-phosphorylated peptide I(pY)GEF, which we had previously identified as a good substrate (Table 5
Phosphatase activity of mutant GST-mEYA3 fusion proteins
mEya3 mutations | Km (μm) | Kcat (min−1) |
|---|---|---|
| L405PBOR | NDA | NDA |
| L423RBOR | NDA | NDA |
| K465GOD | Unmeasurable | Unmeasurable |
| E281KOD | NDA | NDA |
| G344SOD+BOR | 871 | 4.7 × 107 |
| T250MFly | 220 | 0.0008 |
| T393IFly | NDA | NDA |
| G473EFly | NDA | NDA |
mEya3 mutations | Km (μm) | Kcat (min−1) |
|---|---|---|
| L405PBOR | NDA | NDA |
| L423RBOR | NDA | NDA |
| K465GOD | Unmeasurable | Unmeasurable |
| E281KOD | NDA | NDA |
| G344SOD+BOR | 871 | 4.7 × 107 |
| T250MFly | 220 | 0.0008 |
| T393IFly | NDA | NDA |
| G473EFly | NDA | NDA |
Km for wild-type mEYA3 ranges from ∼100 to 200 μm. NDA, no detectable activity.
Phosphatase activity of mutant GST-mEYA3 fusion proteins
mEya3 mutations | Km (μm) | Kcat (min−1) |
|---|---|---|
| L405PBOR | NDA | NDA |
| L423RBOR | NDA | NDA |
| K465GOD | Unmeasurable | Unmeasurable |
| E281KOD | NDA | NDA |
| G344SOD+BOR | 871 | 4.7 × 107 |
| T250MFly | 220 | 0.0008 |
| T393IFly | NDA | NDA |
| G473EFly | NDA | NDA |
mEya3 mutations | Km (μm) | Kcat (min−1) |
|---|---|---|
| L405PBOR | NDA | NDA |
| L423RBOR | NDA | NDA |
| K465GOD | Unmeasurable | Unmeasurable |
| E281KOD | NDA | NDA |
| G344SOD+BOR | 871 | 4.7 × 107 |
| T250MFly | 220 | 0.0008 |
| T393IFly | NDA | NDA |
| G473EFly | NDA | NDA |
Km for wild-type mEYA3 ranges from ∼100 to 200 μm. NDA, no detectable activity.
; Tootle et al. 2003b). Phosphatase activity appeared normal in only one of the mutations tested, T250MFLY, a somewhat unexpected result because this mutation affects a conserved residue within motif I of the catalytic domain (Figure 1). The fact that this mutation lacks activity in the in vivo assays (Figure 2) suggests that an essential function other than phosphatase activity has been compromised, such as perhaps interaction with a critical binding partner.
Two additional mutations, G344SBOR+OD and K465GOD, retained measurable phosphatase activity, although in the latter case activity was not sufficiently robust to permit kinetic analyses (Table 5). These same two mutations exhibited robust in vivo activity (Figure 2), consistent with phosphatase activity being important for wild-type function (Tootle et al. 2003b). The remaining five mutations lacked detectable catalytic activity. These included the two BOR patient-derived mutations (L405PBOR and L423RBOR), one OD-derived substitution (E281KOD), and two Drosophila null alleles (G473EFLY and T393IFLY). Of the mutations lacking phosphatase function, only G473EFLY affects a conserved residue within the phosphatase active site (Figure 1). How the other mutations compromise phosphatase activity remains to be determined, although the fact that the ability to interact with at least two of their usual binding partners is retained (Figure 4) makes it unlikely that the mutant EYA proteins are simply misfolded. Therefore these results suggest that loss of phosphatase activity contributes to the BOR phenotype and perhaps to the OD phenotype as well.
Impaired trans-activation potential of mutant EYA proteins:
To explore the effects of these mutations on EYA's second function as a transcriptional cofactor, we have tested their trans-activation ability in two Drosophila cell-based transcription assays. The first uses a multimerized regulatory element from the mammalian Na+/K+-ATPase α1-subunit fused upstream of the luciferase gene (ARE-luciferase) that we have previously shown to be responsive to the EYA-SO bipartite transcription factor (Figure 3A
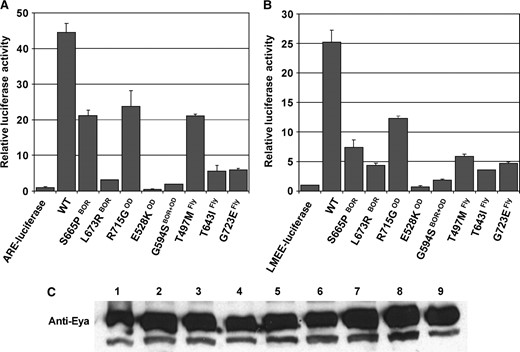
Reduced trans-activation capability of mutant EYA proteins in conjunction with SO. (A) Drosophila S2 cell-based transcriptional assays using ARE-luciferase as the reporter showed that the trans-activational potential of the mutant EYA proteins is lowered compared to that of wild-type protein, and that some of the mutants were transcriptionally inert. (B) Similar trends were seen with the Drosophila lozenge native enhancer LMEE-luciferase reporter. (C) Site-directed mutagenesis of the EYA domain does not affect protein expression levels. Lanes: 1, wild-type EYA; 2, S655PBOR; 3, L673RBOR; 4, R715GOD; 5, E528KOD; 6, G594SBOR+OD; 7, T497MFly; 8, T643IFly; and 9, G723EFly.
; Silver et al. 2003). The second reporter places luciferase downstream of a single copy of a 252-bp minimal enhancer element from the second intron of the Drosophila lozenge (lz) gene (LMEE-luciferase) that has recently been shown to be responsive to SO activity in the fly eye (Yan et al. 2003). In Drosophila S2 cells, LMEE-luciferase is activated to low levels when expressed alone or with either EYA or SO alone, but when coexpressed with both EYA and SO, significant activation is observed (Figure 3B and data not shown). This activation is due to direct binding of SO to the LMEE sequence, as mutation of the SO-binding sites (LMEESO-luciferase; Yan et al. 2003) abolishes activity, while mutation of the binding sites for a different transcription factor (LMEEGL-luciferase; Yan et al. 2003) does not (data not shown).
Using these two independent assays, one relying on an artificial multimerized element (Figure 3A) and the other using a native element from an endogenous Drosophila target gene (Figure 3B), we tested whether the BOR, OD, and Drosophila-derived EYA mutants retain the ability to trans-activate in concert with SO. Although the absolute levels of activation differed between the two reporters, similar trends were observed, with all mutants showing some reduction in activity relative to wild-type EYA. Immunohistochemical and immunoblotting analyses revealed that the mutant proteins all localize appropriately to the nucleus (data not shown) and that expression levels between wild-type and mutant EYA proteins appear comparable (Figure 3C). This confirms that the reduced transcriptional output reflects loss of trans-activation potential rather than abnormal subcellular localization or instability of the mutant proteins.
While all mutants exhibited reduced trans-activation, the degree of impairment varied. R715GOD retained robust trans-activation potential, exhibiting only a twofold activity reduction relative to wild-type EYA in both assays. S655PBOR and T497MFLY also exhibited only mild loss of trans-activation potential, showing two- and fivefold reductions in activity in the ARE and LMEE assays, respectively. More severely compromised were T643IFLY, G723EFLY, and L673RBOR, which exhibited 10- and 5-fold reductions in activity in the ARE and LMEE assays, respectively. Finally, transcriptional output observed with E528KOD and G594SBOR+OD was not significantly above the reporter-alone baseline, indicating a complete loss of trans-activation potential. Together these results suggest that impaired ability to activate transcription in conjunction with SO/SIX appears to be a major consequence of mutations associated with BOR/OD syndrome.
One possible explanation for reduced transcriptional output is that the specific missense mutations compromise the ability of EYA to bind to SO, the DNA-binding component of the EYA-SO transcription factor. Indeed, previous in vitro analyses using yeast two-hybrid and GST pull-down assays suggested that this might be the case (Bui et al. 2000; Buller et al. 2001; Ozaki et al. 2002). However, the artificial context of the assay systems, combined with the fact that full-length proteins were not tested, raised the possibility that the reduced binding capacity might not accurately reflect the situation in vivo. To investigate this possibility, co-immunoprecipitation studies were performed from Drosophila cells cotransfected with full-length epitope-tagged expression constructs. All mutant EYA proteins complexed efficiently with SO although S655PBOR-SO interactions were reduced by almost threefold relative to wild-type EYA-SO and G723EFLY also showed a slight reduction in binding to SO (Figure 4A)
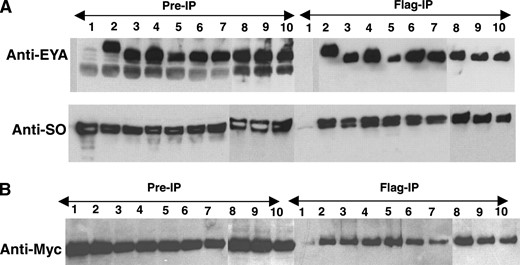
EYA domain mutations retain the ability to complex with SO and EYA. (A) Co-immmunoprecipitation and Western blot of Drosophila S2 cell extracts transfected with wild-type and mutant Flag-EYA and Myc-SO, immunoprecipitated by anti-Flag-conjugated beads (Sigma) and detected by anti-EYA and anti-SO antibodies. Lanes: 1, Myc-So; 2, Myc-So + Flag-Eya; 3, Myc-So + Flag-S655PBOR; 4, Myc-So + Flag-L673RBOR; 5, Myc-So + Flag-R715GOD; 6, Myc-So + Flag-E528KOD; 7, Myc-So + Flag-G594SBOR+OD; 8, Myc-So + Flag-T497MFly; 9, Myc-So + Flag-T643IFly; and 10, Myc-So + Flag-G723EFly. (B) Extracts from S2 cells transfected with Myc-EYA and Flag-EYA, immunoprecipitated by anti-Flag-conjugated beads, Western blotted, and probed with anti-Myc. Lanes: 1, Myc-Eya; 2, Myc-Eya + Flag-Eya; 3, Myc-Eya + Flag-S655PBOR; 4, Myc-Eya + Flag-L673RBOR; 5, Myc-Eya + Flag-R715GOD; 6, Myc-Eya + Flag-E528KOD; 7, Myc-Eya + Flag-G594SBOR+OD; 8, Myc-Eya + Flag-T497MFly; 9, Myc-Eya + Flag-T643IFly; and 10, Myc-Eya + Flag-G723EFly.
. Thus, our co-immunoprecipitation studies argue strongly that the various EYA mutants do not significantly compromise interactions with SO when placed in the more physiologically relevant context of a Drosophila cell and that therefore reduced binding efficiency is unlikely to be a primary contributor to their impaired trans-activation ability.
With respect to the extent of impairment of transcriptional output, our results agree well with previous studies, although several differences raise the interesting possibility that the effects of these mutations on transcriptional output may differ from target gene to target gene. For example, in our two assays, the E528KOD mutation is inactive whereas in myogenin promoter-based transcriptional assay activity it was equivalent to wild type (Ozaki et al. 2002). Conversely, in our systems S655PBOR retained significant, albeit reduced, transcriptional activity whereas activity was not detected in the myogenin promoter-based assay (Ozaki et al. 2002). Given the extensive conservation that has been demonstrated among EYA homologs, the most plausible explanation is that such differences reflect the physiological complexity of transcriptional output mediated by EYA. Specifically, the mutant EYA proteins might recruit different sets of interacting proteins to the target promoters, leading to abnormal transcriptional output. If correct, then to fully understand the molecular basis of BOR syndrome and optical defects in humans, a better understanding of the full spectrum of target genes regulated by the EYA-SIX transcription factor and how the transcriptional profile is altered in the various EYA mutant backgrounds will be required. Furthermore, our finding that phosphatase activity is impaired in many of the associated mutations suggests that the contribution of EYA's phosphatase activity to transcriptional output at different target genes may also be critical.
EYA mutations do not abolish interactions with wild-type EYA:
BOR and OD syndrome are autosomal dominant disorders in which a mutation in one copy of the EYA1 gene is sufficient to cause the disease (Abdelhak et al. 1997b). Because EYA has recently been shown to self-associate (Silver et al. 2003), we wondered if any of the BOR and OD mutations abolished interactions with wild-type EYA. To test this, we performed co-immunoprecipitation studies from S2 cells cotransfected with Myc epitope-tagged wild-type EYA and different Flag epitope-tagged EYA mutants. The results indicated that all EYA mutants co-immunoprecipitate with wild-type EYA with an efficiency comparable to that of wild-type EYA-EYA interactions (Figure 4B). The apparently normal interaction of the EYA mutants with wild-type EYA suggests that the autosomal dominant nature of BOR/OD syndrome results not from an inability of mutant EYA to complex with wild-type EYA, but from impaired function of a complex containing both wild-type and mutant EYA proteins. In this light, it is interesting to note that mutations in EYA4 result in late-onset familial deafness (Wayne et al. 2001). Very speculatively, perhaps impaired function of a complex containing both EYA1 and EYA4 in the ear might contribute to the otic defects in BOR syndrome patients and/or to hearing loss in EYA4 mutant individuals. Future proteomic comparisons of EYA-containing complexes found in normal vs. BOR/OD model tissues should shed new light on the molecular basis of BOR/OD diseases in humans.
In conclusion, our work provides the first functional analysis of human patient-derived EYA mutations in an in vivo developmental context and suggests that defects in both phosphatase and transcription functions likely contribute to the molecular causes of BOR syndrome in humans and to compromised development in flies (Table 6)
Summary of relative activities of mutant EYA proteins
mEya3 | PTPase | dEYA | Rescue | Transcription activity |
|---|---|---|---|---|
| L405PBOR | − | S655PBOR | + | ++ |
| L423RBOR | − | L673RBOR | − | +/− |
| K465GOD | + | R715GOD | ++ | ++ |
| E281KOD | − | E528KOD | ++ | − |
| G344SOD+BOR | ++ | G594SBOR+OD | ++ | − |
| T250MFly | +++ | T497MFly | + | ++ |
| T393IFly | − | T643IFly | − | + |
| G473EFly | − | G723EFly | − | + |
mEya3 | PTPase | dEYA | Rescue | Transcription activity |
|---|---|---|---|---|
| L405PBOR | − | S655PBOR | + | ++ |
| L423RBOR | − | L673RBOR | − | +/− |
| K465GOD | + | R715GOD | ++ | ++ |
| E281KOD | − | E528KOD | ++ | − |
| G344SOD+BOR | ++ | G594SBOR+OD | ++ | − |
| T250MFly | +++ | T497MFly | + | ++ |
| T393IFly | − | T643IFly | − | + |
| G473EFly | − | G723EFly | − | + |
+++, wild-type activity; ++, almost wild-type activity; +, moderate activity; +/− slight activity; −, no activity.
Summary of relative activities of mutant EYA proteins
mEya3 | PTPase | dEYA | Rescue | Transcription activity |
|---|---|---|---|---|
| L405PBOR | − | S655PBOR | + | ++ |
| L423RBOR | − | L673RBOR | − | +/− |
| K465GOD | + | R715GOD | ++ | ++ |
| E281KOD | − | E528KOD | ++ | − |
| G344SOD+BOR | ++ | G594SBOR+OD | ++ | − |
| T250MFly | +++ | T497MFly | + | ++ |
| T393IFly | − | T643IFly | − | + |
| G473EFly | − | G723EFly | − | + |
mEya3 | PTPase | dEYA | Rescue | Transcription activity |
|---|---|---|---|---|
| L405PBOR | − | S655PBOR | + | ++ |
| L423RBOR | − | L673RBOR | − | +/− |
| K465GOD | + | R715GOD | ++ | ++ |
| E281KOD | − | E528KOD | ++ | − |
| G344SOD+BOR | ++ | G594SBOR+OD | ++ | − |
| T250MFly | +++ | T497MFly | + | ++ |
| T393IFly | − | T643IFly | − | + |
| G473EFly | − | G723EFly | − | + |
+++, wild-type activity; ++, almost wild-type activity; +, moderate activity; +/− slight activity; −, no activity.
. Intriguingly, OD patient-derived mutations retain significant in vivo functionality relative to that of BOR patient-derived mutations, suggesting that distinct molecular determinants may underly the different phenotypic manifestations of these two classes of human EYA mutations. Continued exploitation of the powerful Drosophila in vivo model systems we have established should provide additional insight into how EYA's two functions as transcription factor and phosphatase are coordinated and coregulated during normal development and by extension how misregulation of one or both functions contributes to human disease.
Footnotes
Communicating editor: R. S. Hawley
Acknowledgement
Utpal Banerjee generously provided the LMEE reagents prior to publication. We thank members of the Rebay lab for helpful discussions throughout the course of this work. S.J.S. was supported by a Howard Hughes Medical Institute predoctoral fellowship, T.L.T. was supported by a Ludwig Foundation predoctoral fellowship, and this work was supported in part by National Institutes of Health grant R01 EY 12549-06 to I.R.
References
Abdelhak, S., V. Kalatzis, R. Heilig, S. Compain, D. Samson et al.,
Abdelhak, S., V. Kalatzis, R. Heilig, S. Compain, D. Samson et al.,
Azuma, N., A. Hirakiyama, T. Inoue, A. Asaka and M. Yamada,
Bonini, N. M., W. M. Leiserson and S. Benzer,
Bonini, N. M., Q. T. Bui, G. L. Gray-Board and J. M. Warrick,
Borsani, G., A. DeGrandi, A. Ballabio, A. Bulfone, L. Bernard et al.,
Bui, Q. T., J. E. Zimmerman, H. Liu and N. M. Bonini,
Buller, C., X. Xu, V. Marquis, R. Schwanke and P. X. Xu,
Chen, R., M. Amoui, Z. Zhang and G. Mardon,
Hanson, I. M.,
Heanue, T. A., R. Reshef, R. J. Davis, G. Mardon, G. Oliver et al.,
Hsiao, F. C., A. Williams, E. L. Davies and I. Rebay,
Ikeda, K., Y. Watanabe, H. Ohto and K. Kawakami,
Kim, S. S., R. Zhang, S. E. Braunstein, A. Joachimiak, A. Cvekl et al.,
Li, X., K. A. Oghi, J. Zhang, A. Krones, K. T. Bush et al.,
Ohto, H., S. Kamada, K. Tago, S. I. Tominaga, H. Ozaki et al.,
Ozaki, H., Y. Watanabe, K. Ikeda and K. Kawakami,
Pappu, K., and G. Mardon,
Pignoni, F., B. Hu, K. H. Zavitz, J. Xiao, P. A. Garrity et al.,
Rayapureddi, J. P., C. Kattamuri, B. D. Steinmetz, B. J. Frankfort, E. J. Ostrin et al.,
Rebay, I., F. Chen, F. Hsiao, P. A. Kolodziej, B. H. Kuang et al.,
Rebay, I., S. Silver and T. L. Tootle,
Ruf, R. G., P. X. Xu, D. Silvius, E. A. Otto, F. Beekmann et al.,
Silver, S., and I. Rebay,
Silver, S. J., E. L. Davies, L. Doyon and I. Rebay,
Tootle, T. L., P. S. Lee and I. Rebay,
Tootle, T. L., S. J. Silver, E. L. Davies, V. Newman, R. R. Latek et al.,
Vincent, C., V. Kalatzis, S. Abdelhak, H. Chaib, S. Compain et al.,
Wawersik, S., and R. L. Maas,
Wayne, S., N. G. Robertson, F. DeClau, N. Chen, K. Verhoeven et al.,
Xu, P. X., I. Woo, H. Her, D. R. Beier and R. L. Maas,
Xu, P. X., J. Adams, H. Peters, M. C. Brown, S. Heaney et al.,
Yan, H., J. Canon and U. Banerjee,
Yashima, T., Y. Noguchi, K. Ishikawa, H. Mizusawa and K. Kitamura,
Zimmerman, J. E., Q. T. Bui, E. Steingrimsson, D. L. Nagle, W. Fu et al.,

{kind=link}
{kind=link}
{kind=link}
{kind=link}