





Abstract
Constitutive stable DNA replication (cSDR) is a mechanism for replisome loading in Escherichia coli K-12. This occurs in a dnaA-independent fashion in an rnhA mutant. cSDR is dependent on recA, priA, and transcription. In this report, it is shown that dnaA rnhA mutants using cSDR for initiation of their DNA replication additionally require priB, but not priC, for viability. Two subtle priA missense mutations either eliminated the ability to grow using cSDR (priA301 C479Y) or resulted in very small colonies (priA300 K230R). DnaC809, a priA suppressor, failed to allow priA or priB mutants to grow using cSDR to initiate DNA replication. Furthermore, unlike dnaC+ strains, dnaC809 strains require priC for cSDR. DnaC809,820, a priC-independent suppressor of priA2::kan phenotypes, allowed priA and priC (but not priB) mutants to grow using cSDR to initiate DNA replication. It is also shown that rep and rnhA mutations are synthetically lethal. DnaC809 and dnaC809,820 mutations suppress this lethality. Rep is further shown to be required for cSDR in a dnaC809 strain. A model whereby these different sets of replication restart proteins interact preferentially with substrates associated with either RecA or SSB during replication restart and cSDR, respectively, is proposed.
IN Escherichia coli, a critical step in DNA replication is the loading of the replicative helicase, DnaB, onto the DNA by DnaC. Loading DnaB at oriC is a sequence-specific, cell-cycle-regulated event regulated and initiated by the DnaA protein (reviewed in Messer and Weigel 1996; Katayama 2001; Messer 2002). Loading DnaB away from oriC at a repaired replication fork occurs by multiple pathways involving the replication restart proteins (RRPs): PriA, PriB, PriC, DnaT, and Rep (Figure 1
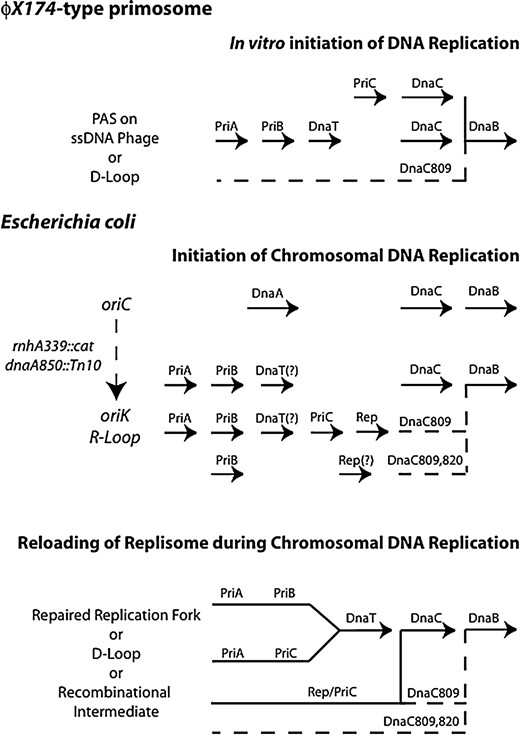
Comparison of different methods studied either in vivo or in vitro for the loading of the DnaB helicase onto DNA and the relationships of the RRPs studied under a particular condition. (Top) Pathways formed by the RRPs using D-loops or ΦX174 single-strand DNA as a substrate. Historically, the RRPs were considered to form a single biochemical pathway. Recently, Marians and colleagues have shown that this reaction is actually the sum of two separate reactions involving PriA, PriB, and DnaT, on the one hand, and PriC on the other hand (K. Marians, personal communication). Both reactions use DnaC to load DnaB. DnaC809 forms a new suppressor pathway denoted by the dashed line. (Middle) The loading of DnaB during initiation of DNA replication as it occurs either at oriC in a DnaA-dependent fashion or using cSDR at oriK in a DnaA-independent fashion. The pathways using the RRPs summarize the new data presented in this report. Each line or reaction shows the gene products required when the strain is dnaC+, dnaC809, or dnaC809,820. DnaT is listed with a question mark since its role has not been explicitly tested. It is hypothesized that since dnaT mutants have the same phenotype as priA mutants (McCool et al. 2004a), the requirement of dnaT will mirror that of priA in these strains. The role of Rep in the dnaC809,820 strain was not tested because the strain grew too poorly and so is listed with a question mark. The dashed lines represent the pathways used by the mutant dnaC proteins. (Bottom) Model showing how the RRPs form multiple pathways to restart replication forks during log-phase growth (Sandler 2000). The dashed lines show pathways available in the presence of particular dnaC alleles.
and Sandler 2000). Two types of priA suppressor mutations have been found and both map to the dnaC gene. The first type is typified by dnaC809 and is dependent on the rep and priC genes (Sandler 2000). The second type of priA suppressor has an additional mutation in dnaC relative to dnaC809 and makes this gene's suppression of priA mutant phenotypes independent of priC and rep. This dnaC allele is called dnaC809,820 (Sandler 2000; Sandler et al. 1999). The multiplicity of replication restart pathways may be of a more general nature in bacteria since Bacillus subtilis also has a similar arrangement of PriA-dependent and PriA-independent pathways (Bruand et al. 2001; Marsin et al. 2001).
Complexes formed during DnaA-dependent initiation of DNA replication at oriC and replication restart differ in their protein components and DNA structure. Both, however, provide a mechanism to unwind the DNA helix and make protein-DNA complexes that are recognized by the DnaC-DnaB complex to load DnaB. Kogoma and colleagues showed that oriC/DnaA-independent initiation of DNA replication could take place in an rnhA mutant strain (Kogoma and von Meyenburg 1983). They called this phenomenon constitutive stable DNA replication (cSDR). Figure 2
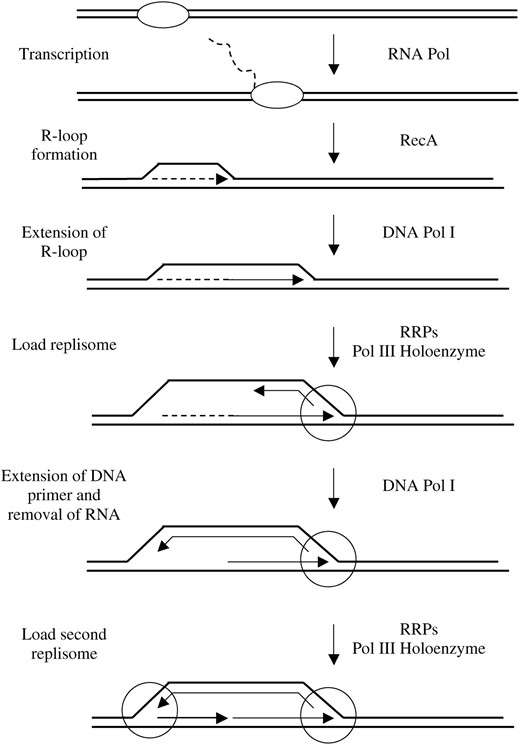
A model for the bidirectional initiation of DNA replication at an oriK using cSDR. This diagram is derived and modified from Kogoma's (1997) model. The oval and the circle represent RNA polymerase and the replisome, respectively. Dashed lines indicate RNA. The wording on the left denotes the process occurring at that step and the wording on the right denotes the enzymes involved.
presents a model for cSDR. Initiation is thought to begin at a R-loop. An R-loop is a structure of three strands of nucleic acid: two DNA and one RNA. The one strand of RNA is annealed with its complementary strand of DNA, which causes the other strand of DNA to be displaced from the helix. The RNA for the R-loop is transcribed by RNA polymerase and it is thought that RecA is necessary to either form or stabilize the R-loop (Kasahara et al. 2000). The 3′-end of the RNA in the R-loop is then extended by DNA polymerase I to form a structure similar to a D-loop (Kogoma 1997). Kogoma and colleagues showed that cSDR was also dependent on priA (Masai et al. 1994). They suggested that the rest of the replication restart machinery, now known to include PriA, PriB, PriC, DnaT, and Rep, would identify the proper substrate made by RNA polymerase, RecA and DNA polymerase I, and load DnaB and the rest of the replisome. Here we test whether subtle priA missense mutations or mutations in other replication restart genes such as priB, priC, and rep, affect cSDR.
MATERIALS AND METHODS
Bacterial strains:
All bacterial strains used in this work are derivatives of E. coli K-12 and are described in Table 1
Strain list
Strain | priA | priB | priC | dnaA | rnhA | dnaC | Other relevant genotype | Source or derivation |
|---|---|---|---|---|---|---|---|---|
| AQ12551 | + | + | + | + | 339 | + | T. Kogoma | |
| AQ12257 | + | + | + | 850 | 339 | + | T. Kogoma | |
| CAG18431 | + | + | + | + | + | + | ilv-500::Tn10 | E. coli Stock Center |
| CAG18442 | + | + | + | + | + | + | thr-34::Tn10 | E. coli Stock Center |
| JC13509a | + | + | + | + | + | + | Lab stock | |
| JC19008 | + | + | + | + | + | 809 | Sandler et al. (1996) | |
| JC19165 | + | + | 303 | + | + | + | Sandler et al. (1999) | |
| JC19257 | + | + | + | + | + | 809,820 | Sandler et al. (1999) | |
| JJC213 | + | + | + | + | + | + | del(rep)::kan | B. Michel |
| SS135 | + | 302 | + | + | + | + | Sandler et al. (2001) | |
| SS138 | + | 302 | + | + | + | + | thr-34::Tn10 | CAG18442 → SS135b |
| SS140 | + | 302 | + | + | + | 809 | JC19008 → SS138c | |
| SS141 | + | 302 | + | + | + | 809,820 | JC19257 → SS138c | |
| SS145 | + | + | 303 | + | + | + | Sandler et al. (2001) | |
| SS156 | + | + | + | + | + | + | thr-34::Tn10 | CAG18442 → JC13509b |
| SS170 | 2 | + | + | + | + | + | thr-34::Tn10 | JC19008 → SS156c |
| SS171 | 2 | + | + | + | + | 809 | JC19008 → SS170c | |
| SS172 | 2 | + | + | + | + | 809,820 | JC19257 → SS170c | |
| SS494 | 300 | + | + | + | + | + | Sandler et al. (2001) | |
| SS496 | 301 | + | + | + | + | + | Sandler et al. (2001) | |
| SS1091 | + | + | + | + | + | 809 | JC19008 → SS1213c | |
| SS1092 | + | + | + | + | + | 809,820 | JC19257 → SS1213c | |
| SS1099 | + | + | 303 | + | + | 809 | JC19165 → SS1091d | |
| SS1100 | + | + | 303 | + | + | 809,820 | JC19165 → SS1092d | |
| SS1213 | + | + | + | + | + | + | thr-34::Tn10 | CAG18442 → JC13509b |
| SS1531 | + | + | 303 | + | 339 | + | AQ12551 → SS145e | |
| SS1537 | + | 302 | + | + | 339 | 809 | AQ12551 → SS140e | |
| SS1538 | 2 | + | + | + | 339 | 809 | AQ12551 → SS171e | |
| SS1539 | + | + | 303 | + | 339 | 809 | AQ12551 → SS1099e | |
| SS1580 | + | 302 | + | + | 339 | 809,820 | AQ12551 → SS141e | |
| SS1581 | 2 | + | + | + | 339 | 809,820 | AQ12551 → SS172e | |
| SS1582 | + | + | 303 | + | 339 | 809,820 | AQ12551 → SS1100e | |
| SS1627 | 301 | + | + | + | 339 | + | AQ12551 → SS496e | |
| SS1628 | 300 | + | + | + | 339 | + | AQ12551 → SS494e | |
| SS1731 | + | + | + | + | + | + | del(rep)::kan | JJC213 → JC13509d |
| SS1732 | + | + | + | + | 339 | 809 | AQ12551 → SS1091e | |
| SS1733 | + | + | + | + | 339 | 809,820 | AQ12551 → SS1092e | |
| SS1734 | + | 302 | + | + | 339 | + | AQ12551 → SS135e | |
| SS2288 | + | + | + | + | 339 | 809 | del(rep)::kan | JJC213 → SS1732d |
| SS2299 | + | + | + | + | 339 | 809,820 | del(rep)::kan | JJC213 → SS1733d |
| SS3005 | + | + | + | + | + | + | ilv-500::Tn10 del(rep)::kan | CAG18431 → SS1731f |
Strain | priA | priB | priC | dnaA | rnhA | dnaC | Other relevant genotype | Source or derivation |
|---|---|---|---|---|---|---|---|---|
| AQ12551 | + | + | + | + | 339 | + | T. Kogoma | |
| AQ12257 | + | + | + | 850 | 339 | + | T. Kogoma | |
| CAG18431 | + | + | + | + | + | + | ilv-500::Tn10 | E. coli Stock Center |
| CAG18442 | + | + | + | + | + | + | thr-34::Tn10 | E. coli Stock Center |
| JC13509a | + | + | + | + | + | + | Lab stock | |
| JC19008 | + | + | + | + | + | 809 | Sandler et al. (1996) | |
| JC19165 | + | + | 303 | + | + | + | Sandler et al. (1999) | |
| JC19257 | + | + | + | + | + | 809,820 | Sandler et al. (1999) | |
| JJC213 | + | + | + | + | + | + | del(rep)::kan | B. Michel |
| SS135 | + | 302 | + | + | + | + | Sandler et al. (2001) | |
| SS138 | + | 302 | + | + | + | + | thr-34::Tn10 | CAG18442 → SS135b |
| SS140 | + | 302 | + | + | + | 809 | JC19008 → SS138c | |
| SS141 | + | 302 | + | + | + | 809,820 | JC19257 → SS138c | |
| SS145 | + | + | 303 | + | + | + | Sandler et al. (2001) | |
| SS156 | + | + | + | + | + | + | thr-34::Tn10 | CAG18442 → JC13509b |
| SS170 | 2 | + | + | + | + | + | thr-34::Tn10 | JC19008 → SS156c |
| SS171 | 2 | + | + | + | + | 809 | JC19008 → SS170c | |
| SS172 | 2 | + | + | + | + | 809,820 | JC19257 → SS170c | |
| SS494 | 300 | + | + | + | + | + | Sandler et al. (2001) | |
| SS496 | 301 | + | + | + | + | + | Sandler et al. (2001) | |
| SS1091 | + | + | + | + | + | 809 | JC19008 → SS1213c | |
| SS1092 | + | + | + | + | + | 809,820 | JC19257 → SS1213c | |
| SS1099 | + | + | 303 | + | + | 809 | JC19165 → SS1091d | |
| SS1100 | + | + | 303 | + | + | 809,820 | JC19165 → SS1092d | |
| SS1213 | + | + | + | + | + | + | thr-34::Tn10 | CAG18442 → JC13509b |
| SS1531 | + | + | 303 | + | 339 | + | AQ12551 → SS145e | |
| SS1537 | + | 302 | + | + | 339 | 809 | AQ12551 → SS140e | |
| SS1538 | 2 | + | + | + | 339 | 809 | AQ12551 → SS171e | |
| SS1539 | + | + | 303 | + | 339 | 809 | AQ12551 → SS1099e | |
| SS1580 | + | 302 | + | + | 339 | 809,820 | AQ12551 → SS141e | |
| SS1581 | 2 | + | + | + | 339 | 809,820 | AQ12551 → SS172e | |
| SS1582 | + | + | 303 | + | 339 | 809,820 | AQ12551 → SS1100e | |
| SS1627 | 301 | + | + | + | 339 | + | AQ12551 → SS496e | |
| SS1628 | 300 | + | + | + | 339 | + | AQ12551 → SS494e | |
| SS1731 | + | + | + | + | + | + | del(rep)::kan | JJC213 → JC13509d |
| SS1732 | + | + | + | + | 339 | 809 | AQ12551 → SS1091e | |
| SS1733 | + | + | + | + | 339 | 809,820 | AQ12551 → SS1092e | |
| SS1734 | + | 302 | + | + | 339 | + | AQ12551 → SS135e | |
| SS2288 | + | + | + | + | 339 | 809 | del(rep)::kan | JJC213 → SS1732d |
| SS2299 | + | + | + | + | 339 | 809,820 | del(rep)::kan | JJC213 → SS1733d |
| SS3005 | + | + | + | + | + | + | ilv-500::Tn10 del(rep)::kan | CAG18431 → SS1731f |
JC13509 has the following genotype: sulB103− lacMS286 Φ80dIIlacBK1 argE3 his-4 thi-1 xyl-5 mtl-1 SmR T6R. The lacMS286 Φ80dIIlacBK1 code for two nonoverlapping deletions in the lac operon. See Konrad (1977) and Zieg and Kushner (1977).
Selection for tetracycline resistance.
Selection for prototrophic growth and screen for the presence of the dnaC allele by using PCR followed by restriction analysis for the HinFI restriction site polymorphism.
Selection for kanamycin resistance on minimal media.
Selection for chloramphenicol resistance on minimal media.
Selection for tetracycline resistance on Luria media and then screen for kanamycin resistance.
Strain list
Strain | priA | priB | priC | dnaA | rnhA | dnaC | Other relevant genotype | Source or derivation |
|---|---|---|---|---|---|---|---|---|
| AQ12551 | + | + | + | + | 339 | + | T. Kogoma | |
| AQ12257 | + | + | + | 850 | 339 | + | T. Kogoma | |
| CAG18431 | + | + | + | + | + | + | ilv-500::Tn10 | E. coli Stock Center |
| CAG18442 | + | + | + | + | + | + | thr-34::Tn10 | E. coli Stock Center |
| JC13509a | + | + | + | + | + | + | Lab stock | |
| JC19008 | + | + | + | + | + | 809 | Sandler et al. (1996) | |
| JC19165 | + | + | 303 | + | + | + | Sandler et al. (1999) | |
| JC19257 | + | + | + | + | + | 809,820 | Sandler et al. (1999) | |
| JJC213 | + | + | + | + | + | + | del(rep)::kan | B. Michel |
| SS135 | + | 302 | + | + | + | + | Sandler et al. (2001) | |
| SS138 | + | 302 | + | + | + | + | thr-34::Tn10 | CAG18442 → SS135b |
| SS140 | + | 302 | + | + | + | 809 | JC19008 → SS138c | |
| SS141 | + | 302 | + | + | + | 809,820 | JC19257 → SS138c | |
| SS145 | + | + | 303 | + | + | + | Sandler et al. (2001) | |
| SS156 | + | + | + | + | + | + | thr-34::Tn10 | CAG18442 → JC13509b |
| SS170 | 2 | + | + | + | + | + | thr-34::Tn10 | JC19008 → SS156c |
| SS171 | 2 | + | + | + | + | 809 | JC19008 → SS170c | |
| SS172 | 2 | + | + | + | + | 809,820 | JC19257 → SS170c | |
| SS494 | 300 | + | + | + | + | + | Sandler et al. (2001) | |
| SS496 | 301 | + | + | + | + | + | Sandler et al. (2001) | |
| SS1091 | + | + | + | + | + | 809 | JC19008 → SS1213c | |
| SS1092 | + | + | + | + | + | 809,820 | JC19257 → SS1213c | |
| SS1099 | + | + | 303 | + | + | 809 | JC19165 → SS1091d | |
| SS1100 | + | + | 303 | + | + | 809,820 | JC19165 → SS1092d | |
| SS1213 | + | + | + | + | + | + | thr-34::Tn10 | CAG18442 → JC13509b |
| SS1531 | + | + | 303 | + | 339 | + | AQ12551 → SS145e | |
| SS1537 | + | 302 | + | + | 339 | 809 | AQ12551 → SS140e | |
| SS1538 | 2 | + | + | + | 339 | 809 | AQ12551 → SS171e | |
| SS1539 | + | + | 303 | + | 339 | 809 | AQ12551 → SS1099e | |
| SS1580 | + | 302 | + | + | 339 | 809,820 | AQ12551 → SS141e | |
| SS1581 | 2 | + | + | + | 339 | 809,820 | AQ12551 → SS172e | |
| SS1582 | + | + | 303 | + | 339 | 809,820 | AQ12551 → SS1100e | |
| SS1627 | 301 | + | + | + | 339 | + | AQ12551 → SS496e | |
| SS1628 | 300 | + | + | + | 339 | + | AQ12551 → SS494e | |
| SS1731 | + | + | + | + | + | + | del(rep)::kan | JJC213 → JC13509d |
| SS1732 | + | + | + | + | 339 | 809 | AQ12551 → SS1091e | |
| SS1733 | + | + | + | + | 339 | 809,820 | AQ12551 → SS1092e | |
| SS1734 | + | 302 | + | + | 339 | + | AQ12551 → SS135e | |
| SS2288 | + | + | + | + | 339 | 809 | del(rep)::kan | JJC213 → SS1732d |
| SS2299 | + | + | + | + | 339 | 809,820 | del(rep)::kan | JJC213 → SS1733d |
| SS3005 | + | + | + | + | + | + | ilv-500::Tn10 del(rep)::kan | CAG18431 → SS1731f |
Strain | priA | priB | priC | dnaA | rnhA | dnaC | Other relevant genotype | Source or derivation |
|---|---|---|---|---|---|---|---|---|
| AQ12551 | + | + | + | + | 339 | + | T. Kogoma | |
| AQ12257 | + | + | + | 850 | 339 | + | T. Kogoma | |
| CAG18431 | + | + | + | + | + | + | ilv-500::Tn10 | E. coli Stock Center |
| CAG18442 | + | + | + | + | + | + | thr-34::Tn10 | E. coli Stock Center |
| JC13509a | + | + | + | + | + | + | Lab stock | |
| JC19008 | + | + | + | + | + | 809 | Sandler et al. (1996) | |
| JC19165 | + | + | 303 | + | + | + | Sandler et al. (1999) | |
| JC19257 | + | + | + | + | + | 809,820 | Sandler et al. (1999) | |
| JJC213 | + | + | + | + | + | + | del(rep)::kan | B. Michel |
| SS135 | + | 302 | + | + | + | + | Sandler et al. (2001) | |
| SS138 | + | 302 | + | + | + | + | thr-34::Tn10 | CAG18442 → SS135b |
| SS140 | + | 302 | + | + | + | 809 | JC19008 → SS138c | |
| SS141 | + | 302 | + | + | + | 809,820 | JC19257 → SS138c | |
| SS145 | + | + | 303 | + | + | + | Sandler et al. (2001) | |
| SS156 | + | + | + | + | + | + | thr-34::Tn10 | CAG18442 → JC13509b |
| SS170 | 2 | + | + | + | + | + | thr-34::Tn10 | JC19008 → SS156c |
| SS171 | 2 | + | + | + | + | 809 | JC19008 → SS170c | |
| SS172 | 2 | + | + | + | + | 809,820 | JC19257 → SS170c | |
| SS494 | 300 | + | + | + | + | + | Sandler et al. (2001) | |
| SS496 | 301 | + | + | + | + | + | Sandler et al. (2001) | |
| SS1091 | + | + | + | + | + | 809 | JC19008 → SS1213c | |
| SS1092 | + | + | + | + | + | 809,820 | JC19257 → SS1213c | |
| SS1099 | + | + | 303 | + | + | 809 | JC19165 → SS1091d | |
| SS1100 | + | + | 303 | + | + | 809,820 | JC19165 → SS1092d | |
| SS1213 | + | + | + | + | + | + | thr-34::Tn10 | CAG18442 → JC13509b |
| SS1531 | + | + | 303 | + | 339 | + | AQ12551 → SS145e | |
| SS1537 | + | 302 | + | + | 339 | 809 | AQ12551 → SS140e | |
| SS1538 | 2 | + | + | + | 339 | 809 | AQ12551 → SS171e | |
| SS1539 | + | + | 303 | + | 339 | 809 | AQ12551 → SS1099e | |
| SS1580 | + | 302 | + | + | 339 | 809,820 | AQ12551 → SS141e | |
| SS1581 | 2 | + | + | + | 339 | 809,820 | AQ12551 → SS172e | |
| SS1582 | + | + | 303 | + | 339 | 809,820 | AQ12551 → SS1100e | |
| SS1627 | 301 | + | + | + | 339 | + | AQ12551 → SS496e | |
| SS1628 | 300 | + | + | + | 339 | + | AQ12551 → SS494e | |
| SS1731 | + | + | + | + | + | + | del(rep)::kan | JJC213 → JC13509d |
| SS1732 | + | + | + | + | 339 | 809 | AQ12551 → SS1091e | |
| SS1733 | + | + | + | + | 339 | 809,820 | AQ12551 → SS1092e | |
| SS1734 | + | 302 | + | + | 339 | + | AQ12551 → SS135e | |
| SS2288 | + | + | + | + | 339 | 809 | del(rep)::kan | JJC213 → SS1732d |
| SS2299 | + | + | + | + | 339 | 809,820 | del(rep)::kan | JJC213 → SS1733d |
| SS3005 | + | + | + | + | + | + | ilv-500::Tn10 del(rep)::kan | CAG18431 → SS1731f |
JC13509 has the following genotype: sulB103− lacMS286 Φ80dIIlacBK1 argE3 his-4 thi-1 xyl-5 mtl-1 SmR T6R. The lacMS286 Φ80dIIlacBK1 code for two nonoverlapping deletions in the lac operon. See Konrad (1977) and Zieg and Kushner (1977).
Selection for tetracycline resistance.
Selection for prototrophic growth and screen for the presence of the dnaC allele by using PCR followed by restriction analysis for the HinFI restriction site polymorphism.
Selection for kanamycin resistance on minimal media.
Selection for chloramphenicol resistance on minimal media.
Selection for tetracycline resistance on Luria media and then screen for kanamycin resistance.
. The protocol for P1 transduction has been described elsewhere (Willetts et al. 1969). All P1 transductions were selected on 2% agar plates made with either Luria broth or 56/2 minimal media (Willetts et al. 1969) supplemented with 0.2% glucose, 0.001% thiamine, specified amino acids, or 0.2% cassamino acids. Selection used the antibiotics kanamycin (25 μg/ml), tetracycline (10 μg/ml), or chloramphemicol (50 μg/ml). All transductants were grown at 37° and purified on the same type of media on which they were selected.
Transductional assay and controls:
cSDR refers to the type of initiation of DNA replication that occurs in rnhA mutant cells in the absence of DnaA (Kogoma 1997). Experimentally, this can be achieved in a dnaA850::Tn10 rnhA339::cat double-mutant strain. The test for whether a mutation would affect cSDR was performed by first combining the mutations of interests: priA300, priA301, del(priB)302, priC303::kan, dnaC809, and/or dnaC809,820 with rnhA339::cat and then attempting to introduce dnaA850::Tn10 using transduction with a donor P1 lysate grown on AQ12257. If TetR transductants were found at high frequency, then the mutation is judged not to have any major effect on cSDR. If not, then the mutation has an effect. To be sure that the mutations truly caused the effect and that the lack of TetR transductants was not due to other mechanisms, two types of control experiments were performed. The first is that all recipients were tested for their competence for transduction to verify that they are not P1 resistant and that they are recombination proficient by being crossed with a P1 donor (CAG18431) containing the ilv-500::Tn10 mutation. It was found that, for some recipients, it was possible to introduce ilv-500::Tn10 but not dnaA850::Tn10. In these cases, a second control experiment was done. This was to test for the possibility that dnaA850::Tn10 could not be introduced because this insertion mutation may be polar on downstream dnaN and recF expression. To do this, a plasmid expressing dnaA [pAB3 (Gines-Candelaria et al. 1995)] was introduced into each recipient where dnaA850::Tn10 could not be introduced. The transduction introducing dnaA850::Tn10 was then repeated with the modified recipient. If the introduction of dnaA850::Tn10 was now successful, it was concluded that the failure to inherit dnaA850::Tn10 (in the recipient without pAB3) was not due to polar effects. These tests are summarized in Table 2
Ability to transduce rnhA339::cat strains to TetR using P1 grown on strains containing Tn10 inserted into dnaA or ilv
Genotype of recipient strain | Ability to transduce to TetRa | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Strain | priA | priB | priC | dnaC | rep | Donor plasmid: | None | pAB3b | None |
| SS1651 | + | + | + | + | + | + | + | + | |
| SS1628 | 300 | + | + | + | + | + (sm) | + | + | |
| SS1627 | 301 | + | + | + | + | − | + | + | |
| SS1734 | + | 302 | + | + | + | − | + | + | |
| SS1531 | + | + | 303 | + | + | + | ND | + | |
| SS1538 | 2 | + | + | 809 | + | − | + | + | |
| SS1537 | + | 302 | + | 809 | + | − | + | + | |
| SS1539 | + | + | 303 | 809 | + | − | + | + | |
| SS1581 | 2 | + | + | 809,820 | + | + | ND | + | |
| SS1580 | + | 302 | + | 809,820 | + | − | + | + | |
| SS1582 | + | + | 303 | 809,820 | + | + | ND | + | |
| SS1732 | + | + | + | 809 | + | + | ND | ND | |
| SS1733 | + | + | + | 809,820 | + | + | ND | ND | |
| SS2288 | + | + | + | 809 | kan | − | + | +c | |
Genotype of recipient strain | Ability to transduce to TetRa | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Strain | priA | priB | priC | dnaC | rep | Donor plasmid: | None | pAB3b | None |
| SS1651 | + | + | + | + | + | + | + | + | |
| SS1628 | 300 | + | + | + | + | + (sm) | + | + | |
| SS1627 | 301 | + | + | + | + | − | + | + | |
| SS1734 | + | 302 | + | + | + | − | + | + | |
| SS1531 | + | + | 303 | + | + | + | ND | + | |
| SS1538 | 2 | + | + | 809 | + | − | + | + | |
| SS1537 | + | 302 | + | 809 | + | − | + | + | |
| SS1539 | + | + | 303 | 809 | + | − | + | + | |
| SS1581 | 2 | + | + | 809,820 | + | + | ND | + | |
| SS1580 | + | 302 | + | 809,820 | + | − | + | + | |
| SS1582 | + | + | 303 | 809,820 | + | + | ND | + | |
| SS1732 | + | + | + | 809 | + | + | ND | ND | |
| SS1733 | + | + | + | 809,820 | + | + | ND | ND | |
| SS2288 | + | + | + | 809 | kan | − | + | +c | |
(+) refers to the fact that the number of transductants were similar to the rnhA339::cat strain (SS1651) and a 100- to 1000-fold over that seen with a rnhA+ strain (JC13509). P1 transduction of dnaA850::Tn10 into JC13509 typically yielded a few TetR transductants. These strains are presumably dnaA+ and reflect background levels. (−) refers to the observation that the number of transductants was equal or less of that of an rnhA+ strain (JC13509).
pAB3 is a plasmid derived from pACYC177 that has a HindIII-XhoI fragment containing dnaA+ (this plasmid was given to me by Mark Sutton). Its original derivation is in Gines-Candelaria et al. (1995). This plasmid was used to transform the indicated recipient. This new strain was then used as a recipient in a cross with P1 containing dnaA850::Tn10. (+) in this case means that the frequency of transduction was 100- to 500-fold greater than that of the strain without pAB3.
CAG5052 containing btuB3191::Tn10 was used as a donor since it is not tightly linked with the del(rep)::kan mutation.
dnaA refers to dnaA850::Tn10 and ilv refers to ilv-500::Tn10. All transductions reported in this table have been repeated at least twice. sm, a small colony phenotype; ND, not determined.
Ability to transduce rnhA339::cat strains to TetR using P1 grown on strains containing Tn10 inserted into dnaA or ilv
Genotype of recipient strain | Ability to transduce to TetRa | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Strain | priA | priB | priC | dnaC | rep | Donor plasmid: | None | pAB3b | None |
| SS1651 | + | + | + | + | + | + | + | + | |
| SS1628 | 300 | + | + | + | + | + (sm) | + | + | |
| SS1627 | 301 | + | + | + | + | − | + | + | |
| SS1734 | + | 302 | + | + | + | − | + | + | |
| SS1531 | + | + | 303 | + | + | + | ND | + | |
| SS1538 | 2 | + | + | 809 | + | − | + | + | |
| SS1537 | + | 302 | + | 809 | + | − | + | + | |
| SS1539 | + | + | 303 | 809 | + | − | + | + | |
| SS1581 | 2 | + | + | 809,820 | + | + | ND | + | |
| SS1580 | + | 302 | + | 809,820 | + | − | + | + | |
| SS1582 | + | + | 303 | 809,820 | + | + | ND | + | |
| SS1732 | + | + | + | 809 | + | + | ND | ND | |
| SS1733 | + | + | + | 809,820 | + | + | ND | ND | |
| SS2288 | + | + | + | 809 | kan | − | + | +c | |
Genotype of recipient strain | Ability to transduce to TetRa | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Strain | priA | priB | priC | dnaC | rep | Donor plasmid: | None | pAB3b | None |
| SS1651 | + | + | + | + | + | + | + | + | |
| SS1628 | 300 | + | + | + | + | + (sm) | + | + | |
| SS1627 | 301 | + | + | + | + | − | + | + | |
| SS1734 | + | 302 | + | + | + | − | + | + | |
| SS1531 | + | + | 303 | + | + | + | ND | + | |
| SS1538 | 2 | + | + | 809 | + | − | + | + | |
| SS1537 | + | 302 | + | 809 | + | − | + | + | |
| SS1539 | + | + | 303 | 809 | + | − | + | + | |
| SS1581 | 2 | + | + | 809,820 | + | + | ND | + | |
| SS1580 | + | 302 | + | 809,820 | + | − | + | + | |
| SS1582 | + | + | 303 | 809,820 | + | + | ND | + | |
| SS1732 | + | + | + | 809 | + | + | ND | ND | |
| SS1733 | + | + | + | 809,820 | + | + | ND | ND | |
| SS2288 | + | + | + | 809 | kan | − | + | +c | |
(+) refers to the fact that the number of transductants were similar to the rnhA339::cat strain (SS1651) and a 100- to 1000-fold over that seen with a rnhA+ strain (JC13509). P1 transduction of dnaA850::Tn10 into JC13509 typically yielded a few TetR transductants. These strains are presumably dnaA+ and reflect background levels. (−) refers to the observation that the number of transductants was equal or less of that of an rnhA+ strain (JC13509).
pAB3 is a plasmid derived from pACYC177 that has a HindIII-XhoI fragment containing dnaA+ (this plasmid was given to me by Mark Sutton). Its original derivation is in Gines-Candelaria et al. (1995). This plasmid was used to transform the indicated recipient. This new strain was then used as a recipient in a cross with P1 containing dnaA850::Tn10. (+) in this case means that the frequency of transduction was 100- to 500-fold greater than that of the strain without pAB3.
CAG5052 containing btuB3191::Tn10 was used as a donor since it is not tightly linked with the del(rep)::kan mutation.
dnaA refers to dnaA850::Tn10 and ilv refers to ilv-500::Tn10. All transductions reported in this table have been repeated at least twice. sm, a small colony phenotype; ND, not determined.
for all crosses.
The failure to introduce dnaA850::Tn10 to the rnhA mutant does not necessarily mean that the additional mutations interfere with cSDR. It is possible that the dnaA850::Tn10 mutation is synthetically lethal with any one or a combination of the mutations in the recipient strain [priC303::kan was found to be synthetically lethal with certain temperature-sensitive alleles of dnaA at the permissive temperature of 30° (Hinds and Sandler 2004)]. Individual combinations of mutations and dnaA850::Tn10 cannot be tested because dnaA is an essential gene and this dnaA mutant is viable only in the absence of rnhA.
RESULTS
priA300 and priA301 affect cSDR:
PriA has at least four types of activities: ATPase, helicase, the ability to load the replisome, and the ability to interact with other proteins. While it has been shown that the priA1::kan null mutation removes the ability to do cSDR (Masai et al. 1994), it is unclear which activities of PriA are needed for cSDR. Although this issue was partially addressed by the analysis of different mutants on multi-copy plasmid (Tanaka et al. 2003), it has not been addressed by studying subtle priA missense mutants, single copy on the chromosome. By subtle, it is meant that the mutation by itself causes very little if any measurable effect on the cell in terms of growth, DNA repair capacity, and basal levels of SOS expression. Two mutations that have been placed on the chromosome that meet this criteria are priA300 (K230R) and priA301 (C479Y) (Sandler et al. 2001). The former is a mutation in its Walker P-loop motif and is deficient in both ATPase and helicase activities but is still able to load the replisome (Zavitz and Marians 1992). The latter affects the cysteine-rich region of PriA thought to be important for Zn+2 binding, helicase activity, and or protein-protein interactions (Zavitz and Marians 1993; Liu et al. 1996). As a single-copy allele on the chromosome in an otherwise wild-type strain, the only discernible phenotype reported for these mutants thus far is a four- to fivefold lower plating efficiency of Mu phage (Sandler et al. 2001). These mutations, however, are not without consequence. For instance, both mutations when combined with a del(priB)302 mutation yield a cell that is phenotypically like a priA null mutant (Sandler et al. 2001). In combination with other mutations, other phenotypes for priA300 have also been reported (Flores et al. 2002; Jaktaji and Lloyd 2003).
To test whether a chromosomal allele of priA300 or priA301 allows cSDR, a rnhA mutation was first combined with each priA mutation. These double mutants were then the recipients in a cross with a donor lysate grown on a strain containing dnaA850::Tn10. Table 2 shows that for priA300, this triple mutant was viable. The resulting triple mutant, however, produced very small colonies after 2–3 days of growth at 37° (data not shown). When the priA301 rnhA mutant was used as the recipient, no triple-mutant transductants were seen. It is suggested that PriA's ability to load the replisome is not sufficient for cSDR. It is clear from the small colony phenotype of the priA300 mutants that the ATPase and helicase activities of PriA are important for cSDR. There remains, however, at least one other activity of PriA (e.g., protein-protein interactions) that is affected by priA301, and not by priA300, which is essential for cSDR.
PriB, but not PriC, is required for viability in the dnaA850::Tn10 rnhA339::cat strain:
Other work has shown that neither priB nor priC is essential for growth or has any obvious growth or replication phenotype in an otherwise wild-type strain (Sandler et al. 1999). Yet, the priB priC double mutant was inviable (Sandler et al. 1999). Further studies suggested that priB and priC each functioned with priA in redundant pathways for replication restart during normal growth (Sandler 2000). Thus, even though cSDR absolutely required priA, it was possible by the multiple pathways model that cSDR could tolerate either a priB or a priC mutation.
This hypothesis was tested by attempting to introduce the dnaA850::Tn10 mutation into the priB rnhA and priC rnhA double mutants. Table 2 shows that the priC rnhA mutant was able to tolerate the introduction of the dnaA850::Tn10 mutation, but the priB rnhA mutant could not. I conclude that, unlike replication restart in log-phase wild-type cells, cSDR is able to tolerate the absence of priC, but not priB. I hypothesize that there are no redundant mechanisms for loading DnaB at a structure initiated by the formation of an R-loop, as there are for cells during replication restart, and that cSDR proceeds by a mechanism that requires both priA and priB.
DnaC809 does not suppress the absence of priA and priB and creates the additional requirement of priC in cSDR:
Mutations in dnaC (e.g., dnaC809) can indirectly suppress priA2::kan mutant phenotypes. It has been shown that the inviability of priB priC double mutants can be partially rescued by a dnaC809 mutation. These triple-mutant strains, however, still have some defects that can be rescued by a dnaC809,820 mutation (Sandler et al. 1999; Sandler 2000). The molecular basis for this additional suppression is not entirely understood. It has been hypothesized that the dnaC820 mutation makes the dnaC809 mutant priC and rep independent.
To begin to test whether dnaC809 and dnaC809,820 could suppress the absence of priA, priB, or priC in cSDR, I first tested whether dnaC809 and dnaC809,820 mutants were proficient for cSDR. Since dnaC809 and dnaC809,820 have no other obvious phenotypes except the ability to suppress the absence of certain replication restart mutants, it was predicted that these alleles should be proficient for cSDR. This was tested by first combining the dnaC and rnhA mutations into a single strain and then adding dnaA850::Tn10. Table 2 shows that both dnaC809 and dnaC809,820 were viable in the rnhA dnaA strain. The time to form colonies of the dnaC809,820 strain was longer than that of the dnaC809 derivative, which suggests that while both strains are viable, the two dnaC alleles may differentially impart some effects on the double-mutant cell.
The experiments reported above revealed that cSDR was PriC independent. Thus it was first tested whether dnaC809 would suppress the absence of priA in cSDR. Table 2 shows that the dnaA850::Tn10 could not be added to a priA2::kan dnaC809 rnhA strain. It was then hypothesized that dnaC809 also should not suppress the absence of priB since PriB requires PriA for function (Ng and Marians 1996). This was tested directly by constructing the priB dnaC809 rnhA triple mutant and then attempting to introduce dnaA850::Tn10. Table 2 shows that this multiple mutant was not viable. As a control, the priC dnaC809 rnhA triple mutant was also tested for its ability to inherit dnaA850::Tn10. Since priC was not required for cSDR, it was predicted that the triple mutant should allow the introduction of dnaA850::Tn10. Surprisingly, Table 2 shows this is not the case. The dnaC809 strain now required the presence of priC for cSDR, whereas a dnaC does not. From this it is concluded that in the presence of dnaC809, cSDR requires priA, priB, and priC.
DnaC809,820 is able to allow priA and priC mutants to grow using cSDR to initiate DNA replication:
The dnaC809,820 mutation is able to simultaneously suppress the priA-priC and priB-priC synthetic lethalities whereas the dnaC809 mutation cannot (Sandler 2000). It was then tested whether dnaC809,820 could allow growth of priA, priB, and priC mutants using cSDR as their sole method to initiate DNA replication. Once again this was tested by creating the priA, priB, or priC dnaC809,820 rnhA triple-mutant strain and then by attempting to introduce dnaA850::Tn10. Table 1 shows that all triple mutants were viable and Table 2 shows that dnaA850::Tn10 could be added to the strains mutated in priA and priC, but not priB.
Del(rep)::kan and rnhA339::cat are synthetically lethal:
The rep protein has at least two roles in the cell: it stabilizes the replication fork during DNA synthesis (Uzest et al. 1995; Seigneur et al. 1998) and it functions with PriC in the PriA-independent pathway of replication restart (Sandler 2000). It was therefore of interest to test if the absence of rep would inhibit cSDR. To test this idea, I first attempted to construct the rep rnhA double mutant. Introduction of either the rep mutation into the rnhA strain or the rnhA mutation into the rep strain was unsuccessful, suggesting that the two mutations are synthetically lethal. This was further tested by construction of a ilv-500::Tn10 del(rep)::kan double mutant (SS3005). These two mutations are tightly linked. This strain (SS3005) was then used as donor in a cross with a rnhA+ and rnhA339::cat recipient. Combining the results of two independent sets of transductions, it was seen that when the recipient was rnhA+, 32/48 of TetR transductants were also KanR (thus inheriting del(rep)::kan) and when the recipient was rnhA339::cat, 0/48 were both TetR and KanR. I concluded that del(rep)::kan and rnhA339::cat are synthetically lethal.
DnaC809 and DnaC809,820 suppress the rep-rnhA synthetic lethality:
Several scenarios can be thought of to explain the rep-rnhA synthetic lethality. One idea is that Rep is needed to restart replication forks that have specifically stopped or stalled at R-loops (Kogoma et al. 1993; McCool et al. 2004b). Since dnaC809,820 (and not dnaC809) suppresses priA2::kan in a rep- and priC-independent fashion (Sandler 2000), it is predicted that dnaC809,820 (and not dnaC809) should suppress the rnhA-rep synthetic lethality. Table 1 shows, however, that both dnaC809 and dnaC809,820 suppress the rnhA-rep synthetic lethality. It was also seen that the growth of colonies of the dnaC809 strain is greater than that of the dnaC809,820 strain (data not shown). Large colony variants, probably containing suppressor mutations, were often seen in the dnaC809,820 rnhA rep multiple-mutant strain (data not shown). Since some dnaC mutations can suppress the rnhA-rep synthetic lethality, it is tentatively concluded that the likely cause of the lethality is the failure to restart replication forks that have stopped at R-loops.
Rep is required for cSDR in rnhA dnaC809 strain:
While rep's role in dnaC+ strains could not be tested, it was possible to test if rep was needed during cSDR in a dnaC809 strain. Thus I tested if dnaA850::Tn10 could be introduced into the dnaC809 rnhA rep triple mutant. Table 2 shows that this cross did not yield any viable quadruple-mutant transductants. A control cross was done using P1 grown on a strain carrying btuB3191::Tn10 instead of ilv-500::Tn10, because it is not tightly linked with the del(rep)::kan mutation. This marker was inherited without any problem, so I conclude that rep is required for cSDR in the presence of dnaC809 (like priA, priB, and priC).
DISCUSSION
This report tests whether several mutations known to affect the reloading of the replisome during replication restart, a process that occurs during log-phase growth of a wild-type strain, influence the loading of the replisome during cSDR. This study is important because it shows that the process of loading the replisome at different structures can have different mechanisms even though the same repertoire of proteins are used.
It is interesting that priA and priB are essential for cSDR but not for replication restart in log-phase cells. There are several reasons why this might be true. The first is that there may be only a single pathway for loading a replisome at an R-loop. Evidence of multiple pathways would require the demonstration of genetic redundancy. While it is tempting to speculate that the one proposed pathway for loading during cSDR is very similar to or identical to the PriA-PriB-DnaTΦX174 in vitro system and the PriA-PriB (DnaT) pathway for replication restart, the behavior of the dnaC809 mutant using cSDR suggests that there may be some differences. This will be further addressed below. A second reason why cells using cSDR as their method for initiation of DNA replication require both priA and priB for growth (and cells using DnaA/oriC-dependent initiation of DNA replication do not) is because these cells use priA and priB to both initiate their chromosomal DNA replication and restart repaired replication forks. This may be further compounded if the lack of rnhA creates more situations where replication may stop and then need to be restarted (Kogoma et al. 1993; McCool et al. 2004b). Finally, changes in gene expression created by dnaA850::Tn10 may increase the need for priA and priB in dnaA rnhA mutants. DnaA-binding sites are found at oriC and other sites scattered throughout the chromosome (Roth and Messer 1998; Ogawa et al. 2002). DnaA binding can bend the DNA (Schaper and Messer 1995) and if these sites are within promoter regions, its presence or absence can affect the activity of promoters (Quinones et al. 1997; Glinkowska et al. 2003; Ortenberg et al. 2004). Thus the expression of many genes may be altered in dnaA rnhA mutants and this in turn may create a greater need for priA and priB.
Several differences are seen in the behavior of some of the replication restart mutants during cSDR in dnaA rnhA mutants as compared to normal log-phase growth. The first is between priA300 and priA301. In previous studies, these two mutant alleles in vivo behave similarly in terms of having no phenotype by themselves and creating a priA2::kan-like phenotype in the presence of del(priB)302 (Sandler et al. 2001). These two mutations affect different regions and functions of PriA. PriA300 has been extensively studied biochemically. While this mutant has no ATPase and helicase activities, it maintains the ability to load replisomes (Zavitz and Marians 1992; Kogoma et al. 1996; Sandler et al. 1996). PriA301 has not been characterized biochemically. However, other PriA proteins mutant in the zinc-binding/protein interactions/helicase region have been (Zavitz and Marians 1993; Liu et al. 1996; Tanaka et al. 2003). Many of these are proficient in ATP hydrolysis and deficient in helicase activity. Thus one can speculate that the difference between these two mutants during cSDR is due to the ability to interact with other proteins.
A second difference between the behavior of mutations during replication restart and cSDR lies with the priB and priC mutations. PriB and PriC form redundant, PriA-dependent pathways for reloading replication forks. PriC also participates in a PriA-independent pathway. Initially, few differences were documented between the two PriA-dependent pathways. More recently, it has been shown that the helicase activity of PriA may be important for the PriA-PriC pathway (Sandler et al. 2001) and that dnaC1331 cannot function with the PriA-PriB pathway (Harinarayanan and Gowrishankar 2004). An interpretation of these new results is that the substrate (R-loop) used during cSDR is very specific in nature and requires PriA and PriB (and not PriA and PriC). Although this pathway may be similar to the PriA-PriB pathway used during normal growth and the PriA-PriB-DnaT biochemical pathway for ΦX174 DNA replication in that both do not require PriC, a greater specificity for dnaC+ is also apparent in that dnaC809 will not substitute.
RnhA mutations are synthetically lethal with mutations in a number of other genes: polA (Kogoma and Maldonado 1997), recB (Itaya and Crouch 1991), and recG (Hong et al. 1995). In this report, rep is added to the list. It is important to note that these synthetic lethalities are in cells that initiate DNA replication using DnaA and oriC. In some cases (recG), the synthetic lethality is thought to occur because the gene products have the ability to remove R-loops on the chromosome and that their combined absence leads to too many R-loops (Hong et al. 1995). In other cases (polA and recB), it is thought that the ability to process DNA damage created by the encounter of replication forks with R-loops is essential (Kogoma et al. 1993). Rep has at least two roles in the cell: stabilization of replication forks and PriA-independent replication restart (Uzest et al. 1995; Seigneur et al. 1998; Sandler 2000). While other models are also possible, the observation that dnaC809 can rescue the rep-rnhA synthetic lethality suggests that Rep's essential role in rnhA mutants may be to help restart forks that were inhibited at R-loops (Kogoma et al. 1993; McCool et al. 2004b).
The behavior of the dnaC mutations was surprising on several levels. First, dnaC809 did not suppress the absence of priA in cSDR. All previous experiments show that this allele will suppress all of the phenotypes of priA2::kan that have been tested. It was also quite surprising that the presence of dnaC809 increased the requirement for priC during cSDR. It was less surprising that dnaC809,820 should have a different effect because it is known that the two dnaC mutations can also behave quite differently with respect to priA-rep, priA-priC, and priB-priC synthetic lethalities (Sandler 2000). Although the pattern of suppression of dnaC809,820 was similar in replication restart and cSDR (it suppressed the absence of priA and priC), the pattern of suppression of dnaC809 was different. In normal cells during replication restart, dnaC809 suppresses the absence of priA in a way that requires priC and rep. In cSDR, dnaC809 did not suppress the absence of priA and it did suppress the rnhA-rep synthetic lethality.
It is proposed that the differences in behavior of replication restart mutations during normal log-phase growth and during cSDR are due to the differences in the substrates to which these proteins load. While the models of replication restart during log-phase growth and cSDR that have been postulated so far (Kogoma 1997; Cox et al. 2000) have some similarities, there are some differences. The way in which the substrates are formed and the proteins that may be associated with them may explain some of the different requirements. For instance, during replication restart, D-loops are formed by processes involving RecBCD and RecFOR helping RecA onto the DNA (Cox et al. 2000; Kowalczykowski 2000). These proteins can make structures in vitro that are substrates for the RRPs (Xu and Marians 2003). The way in which the R-loop is formed is different. It has been proposed that transcription by RNA polymerase creates the RNA. The R-loop is formed and stabilized by RecA. This then creates a primer that can be extended by DNA polymerase I (Figure 2) (Kogoma 1997; Kogoma and Maldonado 1997). The proteins that are left behind after the helix unwinding or repair event that may interact with the RRPs are also different. Since recombination is thought to be a highly used pathway for fixing replication forks, it is possible that the repaired fork may still have RecA associated with it at the time of reloading. In cSDR, DNA polymerase I is thought to extend an RNA primer at the R-loop as a prelude to loading the replisome. It is reasonable to hypothesize that the displaced strand that occurs during this DNA synthesis is coated with single-strand DNA binding protein (SSB) to protect it from nucleases. SSB has been shown to bind this type of “bubble” substrate in vitro (Xu and Marians 2000). Thus it is possible that the ability of these different sets of proteins to load the replisome at these two types of substrates is their ability to interact with DNA structures associated with either RecA or SSB. It has been shown recently that PriA can interact specifically with SSB bound to certain substrates in vitro (Cadman and McGlynn 2004; Chen et al. 2004). A second idea is that RecA bound to a D-loop is different enough from RecA bound to an R-loop to be seen by the RRPs. A drawback to this model is that it has no role for DNA polymerase I, which is known to be required for cSDR.
Footnotes
Communicating editor: S. Lovett
Acknowledgement
I thank Ken Marians, Martin Marinus, and Benedicte Michel for reading the manuscript before publication and helpful suggestions. This work was supported by grant RPG-99-194-04-GMC from the American Cancer Society.
References
Bruand, C., M. Farache, S. McGovern, S. D. Ehrlich and P. Polard,
Cadman, C. J., and P. McGlynn,
Chen, H. W., S. H. North and H. Nakai,
Cox, M. M., M. F. Goodman, K. N. Kreuzer, D. J. Sherratt, S. J. Sandler et al.,
Flores, M. J., S. D. Ehrlich and B. Michel,
Gines-Candelaria, E., A. Blinkova and J. R. Walker,
Glinkowska, M., J. Majka, W. Messer and G. Wegrzyn,
Harinarayanan, R., and J. Gowrishankar,
Hinds, T., and S. J. Sandler,
Hong, X., G. W. Cadwell and T. Kogoma,
Itaya, M., and R. J. Crouch,
Jaktaji, R. P., and R. G. Lloyd,
Kasahara, M., J. A. Clikeman, D. B. Bates and T. Kogoma,
Katayama, T.,
Kogoma, T.,
Kogoma, T., and R. R. Maldonado,
Kogoma, T., and K. von Meyenburg,
Kogoma, T., X. Hong, G. W. Cadwell, K. G. Barnard and T. Asai,
Kogoma, T., G. W. Cadwell, K. G. Barnard and T. Asai,
Konrad, E. B.,
Kowalczykowski, S. C.,
Liu, J., P. Nurse and K. J. Marians,
Marsin, S., S. McGovern, S. D. Ehrlich, C. Bruand and P. Polard,
Masai, H., T. Asai, Y. Kubuta, K. Arai and T. Kogoma,
McCool, J. D., C. C. Ford and S. J. Sandler,
McCool, J. D., E. Long, J. F. Petrosino, H. A. Sandler, S. M. Rosenberg et al.,
Messer, W.,
Messer, W., and C. Weigel, 1996 Initiation of chromsome replication, pp. 1597–1601 in Escherichia coli and Salmonella: Cellular and Molecular Biology, edited by F. C. Neidhardt. American Society for Microbiology, Washington, DC.
Ng, J. Y., and K. J. Marians,
Ogawa, T., Y. Yamada, T. Kuroda, T. Kishi and S. Moriya,
Ortenberg, R., S. Gon, A. Porat and J. Beckwith,
Quinones, A., G. Wandt, S. Kleinstauber and W. Messer,
Roth, A., and W. Messer,
Sandler, S. J.,
Sandler, S. J., H. S. Samra and A. J. Clark,
Sandler, S. J., K. J. Marians, K. H. Zavitz, J. Coutu, M. A. Parent et al.,
Sandler, S. J., J. D. McCool, T. T. Do and R. U. Johansen,
Schaper, S., and W. Messer,
Seigneur, M., V. Bidnenko, S. D. Ehrlich and B. Michel,
Tanaka, T., C. Taniyama, K. Arai and H. Masai,
Uzest, M., S. D. Ehrlich and B. Michel,
Willetts, N. S., A. J. Clark and B. Low,
Xu, L., and K. J. Marians,
Xu, L., and K. J. Marians,
Zavitz, K. H., and K. J. Marians,
Zavitz, K. H., and K. J. Marians,
Zieg, J., and S. R. Kushner,

{kind=link}
{kind=link}