





Abstract
Microbial cells under growth-limiting stress can generate mutations by mechanisms distinct from those in rapidly growing cells. These mechanisms might be specific stress responses that increase mutation rates, potentially altering rates of evolution, or might reflect non-stress-specific processes in rare growing cells. In an Escherichia coli model system, both frameshift reversion mutations and gene amplifications occur as apparent starvation-induced mutations. Whereas frameshift reversion (“point mutation”) requires recombination proteins, the SOS response, and error-prone DNA polymerase IV (DinB), amplification requires neither SOS nor pol IV. We report that both point mutation and amplification require the stationary-phase and general stress response transcription factor RpoS (σS). Growth-dependent mutation does not. Alternative interpretations are excluded. The results imply, first, that point mutation and amplification are stress responses that occur in differentiated stationary-phase (not rare growing) cells and, second, that transient genetic instability, producing both point mutation and genome rearrangement, may be a previously unrecognized component of the RpoS-dependent general stress response.
MICROBIAL cells exposed to growth-limiting environments can, in some cases, acquire a beneficial or “adaptive” mutation that allows rapid growth. In bacteria and yeast, there are multiple mechanisms of mutation in growth-limiting environments, some demonstrably different from spontaneous mutation in rapidly growing cells (Foster 1999b; Rosenberg 2001). Also called “stationary-phase” mutation mechanisms, these generally occur in populations of cells that are in the stationary phase, defined as displaying no change in population size (Huisman et al. 1996). The mechanisms and regulation of stationary-phase mutation in these systems are of considerable interest because they represent model systems for understanding interactions between the environment and the genome, particularly whether and how stressful conditions influence genetic change. Induction of genetic change in response to stress has broad relevance to mechanisms of evolution and, thus, to the treatment of bacterial infections; the evolution of bacterial pathogens (for example, the generation of antibiotic resistance); the host immune response; and the genetic instability underlying oncogenesis, tumor progression, and resistance to chemotherapeutic drugs (e.g., Loeb 1991; Strauss 1992; Martinez and Baquero 2000).
Since the inception of current interest in stationary-phase mutation mechanisms (Shapiro 1984; Cairns et al. 1988), three broad classes of model have been considered to explain apparent increases in adaptive mutants under some selective conditions (reviewed by Foster 1999b; Rosenberg 2001). In “directed mutation” models (e.g., Cairns et al. 1988), environmental stress was postulated to stimulate formation or retention of mutations specifically in genes whose altered functions were selected. In “cryptic growth” models, mutation rates are specified to be constant (unaffected by environmental conditions) and the emergence of mutants during stress to result from normal growth-dependent mutation mechanisms operating in rare growing cells (e.g., Partridge and Morgan 1988; Lenski et al. 1989; Lenski and Mittler 1993; Galitski and Roth 1995; Andersson et al. 1998; Hendrickson et al. 2002; and see discussion). In “hypermutation” models, mutation rates are suggested to increase globally in response to stress, generating mostly deleterious, but some useful, mutations (e.g., Hall 1990; Ninio 1991; Torkelson et al. 1997; Rosenberg 2001). The ability to increase mutation could itself be selected (for example, as an advantageous by-product of error-prone DNA repair) or not; either way, increased mutation is a stress response. These models can be viewed as representing different views of evolution (and so have been debated vigorously; see Chicurel 2001): the first, somewhat Lamarckian; the second, neoDarwinist in its adherence to constant mutation rates and gradual evolutionary change (e.g., Mayr 1982); and the third, compatible with Darwinism (which allows for changing rates of generation of heritable variations; Darwin 1859). In this article we examine stationary-phase mutation in a well-studied Escherichia coli model system in which directed mutation models were ruled out previously (and hypermutation models supported) by the frequent occurrence of neutral and deleterious mutations along with useful ones (Foster 1997; Torkelson et al. 1997; Rosenberg et al. 1998; Rosche and Foster 1999; Bull et al. 2000, 2001; Godoy et al. 2000). Whereas we have suggested that stationary-phase mutation in this system results from transient genetic instability induced in response to stress, a hypermutation model (e.g., Rosenberg 2001), others favor a cryptic growth model (e.g., Hendrickson et al. 2002). Here, we examine the regulation of mutation in this system and address whether it is part of a stress response, i.e., a response to environmental conditions.
In the Lac system (Cairns and Foster 1991), E. coli cells unable to catabolize lactose (because of a +1 frame-shift mutation in lac genes carried on an F′ conjugative plasmid) are plated on lactose minimal medium, selecting for Lac+ mutants. Lac+ colonies appear after 2 days and continue appearing for >8 days. Most Lac+ colonies present before day 5 carry a compensatory lac frameshift mutation (“Lac+ point mutants”; Foster and Trimarchi 1994; Rosenberg et al. 1994). The remainder (5-10%) are Lac+ because they carry amplified tandem arrays of the leaky lac allele (20-50 copies) that yield sufficient β-galactosidase for growth (“Lac+ amplified clones”; Hastings et al. 2000). The occurrence of amplification in this Lac system (and others previously, see Horiuchi et al. 1963; Tlsty et al. 1984; Whoriskey et al. 1987) was noted previously, but not examined closely (Foster 1994; Foster et al. 1995). Recent work demonstrates that amplified clones make up 40-50% of the Lac+ colonies that appear after day 7 (Hastings et al. 2000). Thus, both permanent genetic changes (point mutation) and reversible genome rearrangements (amplification) occur during starvation, making this system a useful model for both types of apparent environmentally induced genetic instability.
The mechanism of Lac+ stationary-phase point mutation differs from mechanisms of reversion of the same allele in rapidly growing cells in its requirements for homologous recombination and double-strand-break (DSB) repair proteins (Harris et al. 1994, 1996; Foster et al. 1996), the SOS response to DNA damage (Cairns and Foster 1991; McKenzie et al. 2000), the SOS-regulated error-prone DNA polymerase IV (DinB; McKenzie et al. 2001), F′ transfer functions (Foster and Trimarchi 1995a,b; Galitski and Roth 1995), and transiently limiting mismatch repair activity (Harris et al. 1997, 1999; Foster 1999a). We have suggested that stationary-phase mutations are formed during error-prone DNA synthesis (involving pol IV) primed from recombination intermediates created as part of repair of double-strand (DS) ends (Harris et al. 1994; Rosenberg 2001; and see Foster et al. 1996; Rodriguez et al. 2002). Adaptive amplification requires neither SOS nor pol IV (McKenzie et al. 2001).
A key question concerns whether adaptive point mutation and/or amplification are stress responses, as predicted specifically by hypermutation models. In this work we examine the role of RpoS, a “general stress response” regulator, in mutation and amplification in the Lac system. RpoS is a sigma (transcription) factor of RNA polymerase that promotes the increased transcription of >50 genes in response to a variety of environmental stresses (Hengge-Aronis 1996, 2000; Loewen et al. 1998). Inducers of the RpoS regulon include nutrient deprivation (leading to stationary phase), acidic pH, high temperature, oxidative stress, and osmotic stress (Hengge-Aronis 1996). Cross-protection is an important feature of the general stress response (Hengge-Aronis 1993, 2000). For example, when cells enter stationary phase due to carbon starvation, they become protected against oxidative stress and heat shock (Jenkins et al. 1988). RpoS-deficient cells are less viable in long-term culture and are not protected against oxidative, heat, or osmotic stress in stationary phase (Mulvey et al. 1990; Lange and Hengge-Aronis 1991; McCann et al. 1991). Thus, induction of the RpoS regulon leads to the development of a differentiated, protected state.
We present evidence that RpoS is required for stationary-phase mutation and also amplification in the Lac system. This is the first documented genetic requirement for adaptive amplification and the first shared with adaptive mutation, suggesting a possible common early step(s) in both pathways. The results imply that stationary-phase mutation and amplification are stress responses and that both occur in differentiated stationary-phase cells, as hypermutation, but not cryptic-growth, models predict. Further, the data imply that transient genetic instability is a previously unrecognized component of the RpoS-dependent general stress response, at least in response to stationary phase and, perhaps, in response to other stressors as well.
MATERIALS AND METHODS
Bacterial strains and growth: E. coli strains (Table 1) were constructed using standard techniques (Miller 1992). Bacterial growth was at 37°. M9 minimal medium (Miller 1992) had carbon sources added at 0.1% and thiamine (vitamin B1, or B1) at 10 μg/ml, and cultures were incubated for ∼40-48 hr to assure saturation. rpoS M9 glycerol cultures often saturated at cell densities 3- to 5-fold lower than those of rpoS+ and were sometimes concentrated 5-fold before plating. Rarely, all rpoS cultures of an experiment had normal turbidity but viable cell numbers 10- to 100-fold lower than expected; these experiments were unusable and the variable responsible is unknown. Antibiotics and other additives were used at the following concentrations (μg/ml): chloramphenicol, 25; kanamycin, 50; tetracycline, 10; rifampicin, 100; X-gal, 40; threonine, 50; and tryptophan, 50.
Escherichia coli K-12 strains
| Strain | Relevant genotype | Reference or source |
|---|---|---|
| CAG12182 | MG1655 cysI3152::Tn10kan | E. coli Genetic Stock Center (Yale); Singer et al. (1989) |
| CAG18425 | MG1655 thrA3092::Tn10kan | E. coli Genetic Stock Center (Yale); Singer et al. (1989) |
| CAG18561 | MG1655 eda-3126::Tn10kan | E. coli Genetic Stock Center (Yale); Singer et al. (1989) |
| CAG18579 | MG1655 trpC3117::Tn10kan | E. coli Genetic Stock Center (Yale); Singer et al. (1989) |
| CAG18604 | MG1655 zgf-3156::Tn10kan | E. coli Genetic Stock Center (Yale); Singer et al. (1989) |
| FC29 | Δ(lac-proB)XIII ara thi[F′Δ(lacI-lacZ)] | Cairns and Foster (1991) |
| FC36 | Δ(lac-proB)XIII ara thi RifR | Cairns and Foster (1991) |
| FC40 | Δ(lac-proB)XIII ara thi RifR[F′ lacI33-lacZ proAB+] | Cairns and Foster (1991) |
| MG1655 | rpoS+ | E. coli Genetic Stock Center (Yale) |
| SMR533 | FC40 malB::Tn9 | FC40 × P1(SMR305) |
| SMR820 | FC40 lexA3 malB::Tn9 | McKenzie et al. (2001) |
| SMR868 | FC40 lexA3 | McKenzie et al. (2000) |
| SMR3462 | Δ(lac-proB)XIII ara thi RifR malB::Tn9 | FC36 × P1(SMR533) |
| SMR3804-3810, SMR3812-3814 | FC40 Lac+ (growth-dependent) | Independent spontaneous growth-dependent mutants; McKenzie et al. (1998) |
| SMR3855-3856, SMR3858-3865 | FC40 Lac+ (day 5) | Independent isolates; McKenzie et al. (1998) |
| SMR4562 | Δ(lac-proB)XIII ara thi RifR[F′ lacI33-lacZ proAB+] | Independent construction of FC40; McKenzie et al. (2000) |
| SMR6413, -6416, -6418, -6429, -6451, -6459, -6464, -6474, -6483 | SMR4562 Lac+ amplified | Independent isolates; Hastings et al. (2000) |
| SMR6499 | SMR4562 cysI3152::Tn10kan | SMR4562 × P1(CAG12182) |
| SMR6541 | SMR4562 rpoS::Tn10 | SMR4562 × P1(ZK1268) |
| SMR6544-6552 | SMR6541 Lac+ | Growth-dependent Lac+ isolates of SMR6541 |
| SMR6553-6560 | SMR4562 rpoS::Tn10 Lac+ | SMR3804-3810, -3812 × P1(ZK1268) |
| SMR6561-6570 | SMR6541 Lac+ | Independent day 5 Lac+ isolates of SMR6541 |
| SMR6577 | SMR4562 rpoS+ (rpoS+ from MG1655) | SMR6499 × P1(MG1655) |
| SMR6578 | SMR6541 lexA3 malB::Tn9 | SMR6541 × P1(SMR820) |
| SMR6579 | SMR6541 lexA3 | Mal+ revertant of SMR6578 |
| SMR6689-6692, SMR6694-6698 | SMR4562 rpoS::Tn10 Lac+ amplified | SMR6413, -6416, -6418, -6429, -6451, -6459, -6464, -6474, -6483 × P1(ZK1268) |
| SMR6993 | SMR4562 thrA3092::Tn10kan | SMR4562 × P1(CAG18425) |
| SMR6994 | SMR6541 thrA3092::Tn10kan | SMR6541 × P1(CAG18425) |
| SMR7037 | SMR4562 trpC3117::Tn10kan | SMR4562 × P1(CAG18579) |
| SMR7038 | SMR6541 trpC3117::Tn10kan | SMR6541 × P1(CAG18579) |
| ZK1268 | rpoS::Tn10 (allele originally named katF13::Tn10) | Loewen and Triggs (1984) |
| Strain | Relevant genotype | Reference or source |
|---|---|---|
| CAG12182 | MG1655 cysI3152::Tn10kan | E. coli Genetic Stock Center (Yale); Singer et al. (1989) |
| CAG18425 | MG1655 thrA3092::Tn10kan | E. coli Genetic Stock Center (Yale); Singer et al. (1989) |
| CAG18561 | MG1655 eda-3126::Tn10kan | E. coli Genetic Stock Center (Yale); Singer et al. (1989) |
| CAG18579 | MG1655 trpC3117::Tn10kan | E. coli Genetic Stock Center (Yale); Singer et al. (1989) |
| CAG18604 | MG1655 zgf-3156::Tn10kan | E. coli Genetic Stock Center (Yale); Singer et al. (1989) |
| FC29 | Δ(lac-proB)XIII ara thi[F′Δ(lacI-lacZ)] | Cairns and Foster (1991) |
| FC36 | Δ(lac-proB)XIII ara thi RifR | Cairns and Foster (1991) |
| FC40 | Δ(lac-proB)XIII ara thi RifR[F′ lacI33-lacZ proAB+] | Cairns and Foster (1991) |
| MG1655 | rpoS+ | E. coli Genetic Stock Center (Yale) |
| SMR533 | FC40 malB::Tn9 | FC40 × P1(SMR305) |
| SMR820 | FC40 lexA3 malB::Tn9 | McKenzie et al. (2001) |
| SMR868 | FC40 lexA3 | McKenzie et al. (2000) |
| SMR3462 | Δ(lac-proB)XIII ara thi RifR malB::Tn9 | FC36 × P1(SMR533) |
| SMR3804-3810, SMR3812-3814 | FC40 Lac+ (growth-dependent) | Independent spontaneous growth-dependent mutants; McKenzie et al. (1998) |
| SMR3855-3856, SMR3858-3865 | FC40 Lac+ (day 5) | Independent isolates; McKenzie et al. (1998) |
| SMR4562 | Δ(lac-proB)XIII ara thi RifR[F′ lacI33-lacZ proAB+] | Independent construction of FC40; McKenzie et al. (2000) |
| SMR6413, -6416, -6418, -6429, -6451, -6459, -6464, -6474, -6483 | SMR4562 Lac+ amplified | Independent isolates; Hastings et al. (2000) |
| SMR6499 | SMR4562 cysI3152::Tn10kan | SMR4562 × P1(CAG12182) |
| SMR6541 | SMR4562 rpoS::Tn10 | SMR4562 × P1(ZK1268) |
| SMR6544-6552 | SMR6541 Lac+ | Growth-dependent Lac+ isolates of SMR6541 |
| SMR6553-6560 | SMR4562 rpoS::Tn10 Lac+ | SMR3804-3810, -3812 × P1(ZK1268) |
| SMR6561-6570 | SMR6541 Lac+ | Independent day 5 Lac+ isolates of SMR6541 |
| SMR6577 | SMR4562 rpoS+ (rpoS+ from MG1655) | SMR6499 × P1(MG1655) |
| SMR6578 | SMR6541 lexA3 malB::Tn9 | SMR6541 × P1(SMR820) |
| SMR6579 | SMR6541 lexA3 | Mal+ revertant of SMR6578 |
| SMR6689-6692, SMR6694-6698 | SMR4562 rpoS::Tn10 Lac+ amplified | SMR6413, -6416, -6418, -6429, -6451, -6459, -6464, -6474, -6483 × P1(ZK1268) |
| SMR6993 | SMR4562 thrA3092::Tn10kan | SMR4562 × P1(CAG18425) |
| SMR6994 | SMR6541 thrA3092::Tn10kan | SMR6541 × P1(CAG18425) |
| SMR7037 | SMR4562 trpC3117::Tn10kan | SMR4562 × P1(CAG18579) |
| SMR7038 | SMR6541 trpC3117::Tn10kan | SMR6541 × P1(CAG18579) |
| ZK1268 | rpoS::Tn10 (allele originally named katF13::Tn10) | Loewen and Triggs (1984) |
Escherichia coli K-12 strains
| Strain | Relevant genotype | Reference or source |
|---|---|---|
| CAG12182 | MG1655 cysI3152::Tn10kan | E. coli Genetic Stock Center (Yale); Singer et al. (1989) |
| CAG18425 | MG1655 thrA3092::Tn10kan | E. coli Genetic Stock Center (Yale); Singer et al. (1989) |
| CAG18561 | MG1655 eda-3126::Tn10kan | E. coli Genetic Stock Center (Yale); Singer et al. (1989) |
| CAG18579 | MG1655 trpC3117::Tn10kan | E. coli Genetic Stock Center (Yale); Singer et al. (1989) |
| CAG18604 | MG1655 zgf-3156::Tn10kan | E. coli Genetic Stock Center (Yale); Singer et al. (1989) |
| FC29 | Δ(lac-proB)XIII ara thi[F′Δ(lacI-lacZ)] | Cairns and Foster (1991) |
| FC36 | Δ(lac-proB)XIII ara thi RifR | Cairns and Foster (1991) |
| FC40 | Δ(lac-proB)XIII ara thi RifR[F′ lacI33-lacZ proAB+] | Cairns and Foster (1991) |
| MG1655 | rpoS+ | E. coli Genetic Stock Center (Yale) |
| SMR533 | FC40 malB::Tn9 | FC40 × P1(SMR305) |
| SMR820 | FC40 lexA3 malB::Tn9 | McKenzie et al. (2001) |
| SMR868 | FC40 lexA3 | McKenzie et al. (2000) |
| SMR3462 | Δ(lac-proB)XIII ara thi RifR malB::Tn9 | FC36 × P1(SMR533) |
| SMR3804-3810, SMR3812-3814 | FC40 Lac+ (growth-dependent) | Independent spontaneous growth-dependent mutants; McKenzie et al. (1998) |
| SMR3855-3856, SMR3858-3865 | FC40 Lac+ (day 5) | Independent isolates; McKenzie et al. (1998) |
| SMR4562 | Δ(lac-proB)XIII ara thi RifR[F′ lacI33-lacZ proAB+] | Independent construction of FC40; McKenzie et al. (2000) |
| SMR6413, -6416, -6418, -6429, -6451, -6459, -6464, -6474, -6483 | SMR4562 Lac+ amplified | Independent isolates; Hastings et al. (2000) |
| SMR6499 | SMR4562 cysI3152::Tn10kan | SMR4562 × P1(CAG12182) |
| SMR6541 | SMR4562 rpoS::Tn10 | SMR4562 × P1(ZK1268) |
| SMR6544-6552 | SMR6541 Lac+ | Growth-dependent Lac+ isolates of SMR6541 |
| SMR6553-6560 | SMR4562 rpoS::Tn10 Lac+ | SMR3804-3810, -3812 × P1(ZK1268) |
| SMR6561-6570 | SMR6541 Lac+ | Independent day 5 Lac+ isolates of SMR6541 |
| SMR6577 | SMR4562 rpoS+ (rpoS+ from MG1655) | SMR6499 × P1(MG1655) |
| SMR6578 | SMR6541 lexA3 malB::Tn9 | SMR6541 × P1(SMR820) |
| SMR6579 | SMR6541 lexA3 | Mal+ revertant of SMR6578 |
| SMR6689-6692, SMR6694-6698 | SMR4562 rpoS::Tn10 Lac+ amplified | SMR6413, -6416, -6418, -6429, -6451, -6459, -6464, -6474, -6483 × P1(ZK1268) |
| SMR6993 | SMR4562 thrA3092::Tn10kan | SMR4562 × P1(CAG18425) |
| SMR6994 | SMR6541 thrA3092::Tn10kan | SMR6541 × P1(CAG18425) |
| SMR7037 | SMR4562 trpC3117::Tn10kan | SMR4562 × P1(CAG18579) |
| SMR7038 | SMR6541 trpC3117::Tn10kan | SMR6541 × P1(CAG18579) |
| ZK1268 | rpoS::Tn10 (allele originally named katF13::Tn10) | Loewen and Triggs (1984) |
| Strain | Relevant genotype | Reference or source |
|---|---|---|
| CAG12182 | MG1655 cysI3152::Tn10kan | E. coli Genetic Stock Center (Yale); Singer et al. (1989) |
| CAG18425 | MG1655 thrA3092::Tn10kan | E. coli Genetic Stock Center (Yale); Singer et al. (1989) |
| CAG18561 | MG1655 eda-3126::Tn10kan | E. coli Genetic Stock Center (Yale); Singer et al. (1989) |
| CAG18579 | MG1655 trpC3117::Tn10kan | E. coli Genetic Stock Center (Yale); Singer et al. (1989) |
| CAG18604 | MG1655 zgf-3156::Tn10kan | E. coli Genetic Stock Center (Yale); Singer et al. (1989) |
| FC29 | Δ(lac-proB)XIII ara thi[F′Δ(lacI-lacZ)] | Cairns and Foster (1991) |
| FC36 | Δ(lac-proB)XIII ara thi RifR | Cairns and Foster (1991) |
| FC40 | Δ(lac-proB)XIII ara thi RifR[F′ lacI33-lacZ proAB+] | Cairns and Foster (1991) |
| MG1655 | rpoS+ | E. coli Genetic Stock Center (Yale) |
| SMR533 | FC40 malB::Tn9 | FC40 × P1(SMR305) |
| SMR820 | FC40 lexA3 malB::Tn9 | McKenzie et al. (2001) |
| SMR868 | FC40 lexA3 | McKenzie et al. (2000) |
| SMR3462 | Δ(lac-proB)XIII ara thi RifR malB::Tn9 | FC36 × P1(SMR533) |
| SMR3804-3810, SMR3812-3814 | FC40 Lac+ (growth-dependent) | Independent spontaneous growth-dependent mutants; McKenzie et al. (1998) |
| SMR3855-3856, SMR3858-3865 | FC40 Lac+ (day 5) | Independent isolates; McKenzie et al. (1998) |
| SMR4562 | Δ(lac-proB)XIII ara thi RifR[F′ lacI33-lacZ proAB+] | Independent construction of FC40; McKenzie et al. (2000) |
| SMR6413, -6416, -6418, -6429, -6451, -6459, -6464, -6474, -6483 | SMR4562 Lac+ amplified | Independent isolates; Hastings et al. (2000) |
| SMR6499 | SMR4562 cysI3152::Tn10kan | SMR4562 × P1(CAG12182) |
| SMR6541 | SMR4562 rpoS::Tn10 | SMR4562 × P1(ZK1268) |
| SMR6544-6552 | SMR6541 Lac+ | Growth-dependent Lac+ isolates of SMR6541 |
| SMR6553-6560 | SMR4562 rpoS::Tn10 Lac+ | SMR3804-3810, -3812 × P1(ZK1268) |
| SMR6561-6570 | SMR6541 Lac+ | Independent day 5 Lac+ isolates of SMR6541 |
| SMR6577 | SMR4562 rpoS+ (rpoS+ from MG1655) | SMR6499 × P1(MG1655) |
| SMR6578 | SMR6541 lexA3 malB::Tn9 | SMR6541 × P1(SMR820) |
| SMR6579 | SMR6541 lexA3 | Mal+ revertant of SMR6578 |
| SMR6689-6692, SMR6694-6698 | SMR4562 rpoS::Tn10 Lac+ amplified | SMR6413, -6416, -6418, -6429, -6451, -6459, -6464, -6474, -6483 × P1(ZK1268) |
| SMR6993 | SMR4562 thrA3092::Tn10kan | SMR4562 × P1(CAG18425) |
| SMR6994 | SMR6541 thrA3092::Tn10kan | SMR6541 × P1(CAG18425) |
| SMR7037 | SMR4562 trpC3117::Tn10kan | SMR4562 × P1(CAG18579) |
| SMR7038 | SMR6541 trpC3117::Tn10kan | SMR6541 × P1(CAG18579) |
| ZK1268 | rpoS::Tn10 (allele originally named katF13::Tn10) | Loewen and Triggs (1984) |
The presence of rpoS::Tn10 in SMR6541 was confirmed by PCR with primers 5′-TCAGCAACCGTAGCAATACGTACA-3′ and 5′-AGGCAATTATCGCGACCGCAG-3′ and also by PCR with those primers (separately) in combination with primer 5′-GACAAGATGTGTATCCACCTTAAC-3′, specific to a sequence present at both ends of Tn10. The presence of rpoS::Tn10 was confirmed similarly in a sampling of 10 day 2 and 10 day 5 rpoS Lac+ colonies, to exclude the possibility that they had lost the Tn10 and were rpoS+. Routinely, rpoS cultures were confirmed to be deficient in catalase activity (poor bubbling when hydrogen peroxide is applied to colonies; Bohannon et al. 1991).
Sequencing: The rpoS gene of SMR4562 including its promoter region (Lange et al. 1995) was sequenced using PCR-generated template DNA (primers flanking rpoS, above) and additional rpoS-specific primers. One strand was sequenced completely. The difference at codon 33 was confirmed by sequencing across that site in independent PCR products. The rpoS allele in SMR6577 was confirmed by sequencing across codon 33, also in PCR-generated DNA. Sequencing was performed by Lone Star Labs (Houston).
Reconstruction experiments: Reconstruction experiments were performed as described (Hastings et al. 2000). Amplified Lac+ isolates were grown in M9 lactose to maintain selection for the amplified array of 20-50 copies of the lac region (Hastings et al. 2000). The number of amplified cells was determined by plating nonselectively to LBH (1% tryptone, 0.5% NaCl, 0.5% yeast extract, 2 μg/ml thymine, pH 7, plate medium solidified with 1.5% agar) with X-gal to allow scoring of amplified cells (which form sectored blue-and-white colonies) or Lac- cells (which form white colonies, having lost the amplified array; Hastings et al. 2000 and references therein).
Mutation, conjugation, and transduction assays: Growth-dependent mutation rates to Lac+ were determined using fluctuation tests, as described (Harris et al. 1997, 1999) and rates were estimated using a modified method of the median (Lea and Coulson 1949; von Borstel 1978). A total of 29 or 30 independent cultures per strain were plated on M9B1 lactose and Lac+ colonies were scored. Rather than scoring only at 48 hr, we controlled for possible differences between genotypes in the time of colony formation and avoided including adaptive mutants (see Harris et al. 1999) by plating four to eight independent Lac+ derivatives of each strain in parallel with the cultures being scored for growth-dependent mutants. Scoring of all plates at ∼1- to 2-hr intervals allows the determination of the median mutant frequency at a time when only 50% of the control Lac+ cells of that genotype have formed visible colonies (t50). The t100 is set at ∼48 hr when ≥95% of the Lac+ controls are visible for all genotypes. The mutation rates were determined from the mutant frequencies at the calculated t50 and multiplied by two (to give the rates at t100).
Stationary-phase mutation assays were performed as described (Harris et al. 1996). All experiments presented had less than twofold net population change during days 1-3 after plating (see results for discussion of the importance of monitoring the Lac- population).
For quantitative conjugation assays, donors were grown to saturation in M9B1 glycerol and then diluted 1:50 into M9 glucose and grown 3-4 hr. Donor and recipient cells (SMR3462, grown to saturation in LBH) were added to prewarmed M9B1 glucose proline and incubated for 30 min at 37°, during which time dilutions of each were plated on LBH to determine viable cells per milliliter. Mating mixes were vortexed and plated on M9B1 glycerol chloramphenicol to select for Pro+ CamR transconjugants. Donor cells were limiting by ≥10-fold to allow calculation of the frequency of conjugation as transconjugants per donor cell.
Transductions were performed as described (Miller 1992) with some modification. Recipient strains were grown to saturation in M9B1 glycerol with threonine or tryptophan added and cells were mixed with phage to give a multiplicity of infection (MOI) of ≤0.01. After 20 min of incubation, transduction mixes were washed once in M9 sodium citrate (20 mm) and plated on M9B1 glycerol 20 mm sodium citrate to select prototrophic transductants. Phage plating efficiencies were similar for both genotypes, indicating that rpoS cells are unimpaired for phage P1 infection and production. The phage titer on the rpoS+ recipient was used in determining the transductant frequency (transductants per phage) for both genotypes.
RESULTS
An rpoS mutation decreases stationary-phase mutation and amplification: An rpoS null allele (Loewen and Triggs 1984; Ivanova et al. 1992) decreases Lac+ colony yield substantially (Figure 1A, “total” values). Multiple experiments give similar results (Figure 1D), with some variability. The rpoS mutation decreases both point mutation and amplification (Figure 1, A and B; a second experiment gives similar results, data not shown). Lac+ point mutant and amplified colonies can be distinguished by streaking to nonselective indicator medium (see materials and methods). The rpoS phenotype in amplification is first detectable on day 5 as a threefold reduction, with greater reductions on later days (five-, six-, and sixfold on days 6, 7, and 8, respectively). RpoS appears to be required for both point mutation and amplification; however, this conclusion depends on control experiments (below) that address other possible explanations for the rpoS phenotype.
rpoS cells are viable during the stationary-phase mutation assay: Mutations in rpoS can decrease survival in stationary-phase cultures, at least in liquid culture (Mulvey et al. 1990; Lange and Hengge-Aronis 1991; McCann et al. 1991), suggesting that death of the rpoS cells might explain the decreases in observed Lac+ point mutants and amplified clones. We monitor viability by counting viable cells in plugs of agar removed from the selective plates each day, beginning the day after plating (day 1). Because Lac+ point mutant cells (the majority of Lac+ before day 7) take 2 days to form a colony (Table 2; McKenzie et al. 1998), any detected growth or death of the population is expected to be reflected in increased or decreased numbers of Lac+ colonies 2 days later. The viability data for the experiment in Figure 1, A and B, are presented in Figure 1C. The data are presented offset by 2 days because the Lac+ point mutant cells that give newly visible colonies on any given day were present in the population 2 days earlier (for example, the day 1 viability is plotted on day 3 because Lac+ cells in the day 1 population grew into the Lac+ colonies observed on day 3). The data demonstrate that the rpoS mutant strain is not poorly viable under these conditions. It is as viable as rpoS+ until 5 days after plating (plotted as day 7 in Figure 1C). This is consistent with previous reports of viability of rpoS strains on solid medium (Bridges and Timms 1998; Bridges et al. 2001). We conclude that net population death does not occur and cannot account for the lack of Lac+ point mutant colonies in the rpoS strain.
The effect of rpoS on formation of Lac+ amplified colonies is also not attributable to altered viability relative to rpoS+. It appears in Figure 1 that lack of growth of the rpoS strain when the rpoS+ begins to grow (late in the experiment) may correlate with the lack of amplified Lac+ colonies in the rpoS strain. However, amplified Lac+ cells take 3-5 days to form a colony (Table 2; Hastings et al. 2000). Thus, changes in the population size 5 days after plating (day 7 in Figure 1, at which point rpoS+ shows growth and the rpoS mutant does not) do not affect amplified colony yield until at least day 8, whereas the difference in amplified colony yield in rpoS is clear even by day 5. If the rpoS Lac+ amplified values are adjusted to reflect the viability 3, 4, or 5 days earlier, the effect of rpoS on amplification is even greater (data not shown). Also, we observe no loss in viability of the rpoS cells, relative to rpoS+, between plating and day 1 (data not shown). Therefore, poor viability of the population does not cause the rpoS phenotype in mutation or amplification.
Previous data imply that a hypermutable cell subpopulation gives rise to point mutants (Torkelson et al. 1997; Rosche and Foster 1999; Godoy et al. 2000), but not amplified Lac+ clones (Hastings et al. 2000). We cannot assess the viability of any subpopulation directly. However, it is unlikely that rpoS reduces point mutation by causing inviability of the hypermutable subpopulation because rpoS also decreases amplified clones, which are not derived from it (Hastings et al. 2000).
![—RpoS in stationary-phase point mutation and amplification. (A-D) RpoS is required for point mutation and amplification. Strains are: rpoS+, SMR4562; rpoS, SMR6541. (A) Values are means ±1 SEM for 10 independent cultures per strain from one experiment. “Total” values are all Lac+ colonies scored. Point mutant and amplified Lac+ values are calculated on the basis of sampling of Lac+ colonies (a total of 100-120 colonies/strain/day, 20-30 from each of six cultures) as described (Hastings et al. 2000): amplified clones form sectored colonies when streaked on LBH rifampicin X-gal, whereas point mutants form solid blue colonies (materials and methods). (B) Data from A, but with the y-axis expanded. (C) Relative viability of the Lac- population on the selection plates was determined as described (Harris et al. 1994), beginning the day after plating (day 1). The relative viability is offset by 2 days (the day 1 viability value is presented on day 3) because the day 1 population contains the Lac+ point mutant cells that will yield the observed day 3 Lac+ colonies. [Lac+ point mutant cells take 2 days to form a colony and thus are present in the population 2 days before they are scored as colonies (McKenzie et al. 1998). Amplified Lac+ cells take longer to form colonies (see text; Table 2; Hastings et al. 2000).] This allows one to track the relationship between the populations that yielded Lac+ on a particular day by looking at the same day on the axis in A-C. Values are means ±1 SEM from five cultures per genotype. Where not visible, error bars are smaller than the symbol. (D) Values are the change in Lac+ colonies per 108 cells plated from day 4 to day 5 (the slope of the line where y is new Lac+ per 108 cells plated and x is days) ±1 SEM. Each experiment included 5-10 independent cultures per strain and displayed no net population change before day 4. Experiment 13 is presented in Figure 1, A-C. (E) The variant rpoS allele in the lac frameshift-bearing strain is not required for stationary-phase mutation (means ±1 SEM, six independent cultures per strain; strains SMR4562 and SMR6577). Relative cell viability of both strains changed less than twofold from day 1 to day 4. A second experiment gave a similar result (data not shown).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/genetics/166/2/10.1093_genetics_166.2.669/3/m_gen1238.f1.jpeg?Expires=1716839424&Signature=TEcB8FhWDPmenwcHi8-jMNLj399N2GZThL1YJWfnxO9qH7lHTicUINfptqQ8nR9n-coihepNnFxLkPOsiV4A5AgLWAYhh5-IQRqGbYhhczz6HGXGiC85l0YX94CuBGFGAAoehiKoqq9rnuHBnY3dd0S-OHklc3OpYxvY8B~vaXgg~FKw6kZbI2qK6MKq3Tvfa~8XPmRzQ5AOG5mOVBpLaBVO2gAYHxOGvHo99KUa78uvxawzm6~P~jWwKo8f5aqpFeq3zFi9buiT5hsuv~tXW6dFDUIj6zOeSh8ruSRPRnfSNj2RgRllFSnVXbo27LQ~zt3seY9ztyVhwbyJGZXvEw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
—RpoS in stationary-phase point mutation and amplification. (A-D) RpoS is required for point mutation and amplification. Strains are: rpoS+, SMR4562; rpoS, SMR6541. (A) Values are means ±1 SEM for 10 independent cultures per strain from one experiment. “Total” values are all Lac+ colonies scored. Point mutant and amplified Lac+ values are calculated on the basis of sampling of Lac+ colonies (a total of 100-120 colonies/strain/day, 20-30 from each of six cultures) as described (Hastings et al. 2000): amplified clones form sectored colonies when streaked on LBH rifampicin X-gal, whereas point mutants form solid blue colonies (materials and methods). (B) Data from A, but with the y-axis expanded. (C) Relative viability of the Lac- population on the selection plates was determined as described (Harris et al. 1994), beginning the day after plating (day 1). The relative viability is offset by 2 days (the day 1 viability value is presented on day 3) because the day 1 population contains the Lac+ point mutant cells that will yield the observed day 3 Lac+ colonies. [Lac+ point mutant cells take 2 days to form a colony and thus are present in the population 2 days before they are scored as colonies (McKenzie et al. 1998). Amplified Lac+ cells take longer to form colonies (see text; Table 2; Hastings et al. 2000).] This allows one to track the relationship between the populations that yielded Lac+ on a particular day by looking at the same day on the axis in A-C. Values are means ±1 SEM from five cultures per genotype. Where not visible, error bars are smaller than the symbol. (D) Values are the change in Lac+ colonies per 108 cells plated from day 4 to day 5 (the slope of the line where y is new Lac+ per 108 cells plated and x is days) ±1 SEM. Each experiment included 5-10 independent cultures per strain and displayed no net population change before day 4. Experiment 13 is presented in Figure 1, A-C. (E) The variant rpoS allele in the lac frameshift-bearing strain is not required for stationary-phase mutation (means ±1 SEM, six independent cultures per strain; strains SMR4562 and SMR6577). Relative cell viability of both strains changed less than twofold from day 1 to day 4. A second experiment gave a similar result (data not shown).
rpoS phenotype cannot be attributed to poor colony formation or loss of amplification: Perhaps Lac+ mutations and amplified arrays form normally in rpoS cells, but cells carrying them form colonies slowly, giving the appearance of decreased mutation and amplification. We performed reconstruction experiments (as previously, McKenzie et al. 1998; Hastings et al. 2000) with multiple day 2 (presumed growth-dependent) and day 5 (stationary-phase) Lac+ isolates to determine how efficiently and rapidly rpoS Lac+ cells form colonies under the exact conditions of the stationary-phase mutation assay. Both day 2 and day 5 rpoS Lac+ point mutant cells form Lac+ colonies with unit efficiency (∼100% of viable cells plated yield Lac+ colonies) and as rapidly as the rpoS+ controls (Table 2A). To control for the possibility that rpoS Lac+ cells had acquired mutations that facilitate colony formation under selective conditions, we performed similar experiments with rpoS+ day 2 Lac+ point mutants that were transduced with rpoS:: Tn10. These gave similar results (data not shown).
To determine whether rpoS slows colony formation of Lac+ amplified cells, we introduced rpoS::Tn10 into 9 independent amplified Lac+ strains with different amplified arrays that confer varying growth rates on lactose medium (Hastings et al. 2000). The rpoS derivatives retain the sectoring phenotype (Figure 1 legend) that is the hallmark of lac amplification, and the time required for colony formation is similar (Table 2B). Thus, rpoS does not decrease numbers of mutant and amplified colonies by slowing colony formation. Rather, rpoS cells appear to be impaired in forming mutations and amplified arrays.
rpoS:: Tn 10 does not affect Lac+ colony formation
| Average days to Lac+ colony formation ± SDa | |||
|---|---|---|---|
| A. Point mutant Lac+ | rpoS+ | rpoS::Tn10 | |
| Day 2 | 2.1 ± 0.2 | 2.1 ± 0.03 | |
| Day 5 | 2.1 ± 0.04 | 2.1 ± 0.05 | |
| Average days to Lac+ colony formation (single experiment)b | |||
| B. Amplified Lac+ | rpoS+ | rpoS::Tn10 | Average of rpoS+/rpoS ± SD (three experiments)c |
| Isolate 1 | 3.5 | 3.3 | 1.0 ± 0.12 |
| Isolate 2 | 4.5 | 4.7 | 0.92 ± 0.06 |
| Isolate 3 | 4.0 | 3.8 | 1.2 ± 0.15 |
| Isolate 4 | 5.0 | 3.8 | 1.2 ± 0.10 |
| Isolate 5 | 5.0 | 4.2 | 1.2 ± 0.04 |
| Isolate 6 | 4.6 | 4.7 | 0.93 ± 0.07 |
| Isolate 7 | 4.5 | 3.7 | 1.2 ± 0.03 |
| Isolate 8 | 5.8 | 4.6 | 1.0, 1.3c |
| Isolate 9 | 3.3 | 2.6 | 1.4 ± 0.19 |
| Average days to Lac+ colony formation ± SDa | |||
|---|---|---|---|
| A. Point mutant Lac+ | rpoS+ | rpoS::Tn10 | |
| Day 2 | 2.1 ± 0.2 | 2.1 ± 0.03 | |
| Day 5 | 2.1 ± 0.04 | 2.1 ± 0.05 | |
| Average days to Lac+ colony formation (single experiment)b | |||
| B. Amplified Lac+ | rpoS+ | rpoS::Tn10 | Average of rpoS+/rpoS ± SD (three experiments)c |
| Isolate 1 | 3.5 | 3.3 | 1.0 ± 0.12 |
| Isolate 2 | 4.5 | 4.7 | 0.92 ± 0.06 |
| Isolate 3 | 4.0 | 3.8 | 1.2 ± 0.15 |
| Isolate 4 | 5.0 | 3.8 | 1.2 ± 0.10 |
| Isolate 5 | 5.0 | 4.2 | 1.2 ± 0.04 |
| Isolate 6 | 4.6 | 4.7 | 0.93 ± 0.07 |
| Isolate 7 | 4.5 | 3.7 | 1.2 ± 0.03 |
| Isolate 8 | 5.8 | 4.6 | 1.0, 1.3c |
| Isolate 9 | 3.3 | 2.6 | 1.4 ± 0.19 |
Values are the averaged “average days to colony formation” (determined in reconstruction experiments as described; Hastings et al. 2000) of 10 independent Lac+ isolates for rpoS+ and 9 for rpoS::Tn10. Lac+ colonies were scored each day for 5 days. Point mutant isolates are: day 2 rpoS+, SMR3804-3810, SMR3812-3814; day 5 rpoS+, SMR3855-3856, SMR3858-3865; day 2 rpoS, SMR6544-6552; day 5 rpoS, SMR6561-6570. Two additional experiments gave similar results.
Values are for a single culture of each genotype from one experiment. Lac+ colonies were scored each day for 9 days. Amplified isolates (in order 1-9) are: rpoS+, SMR6413, SMR6416, SMR6418, SMR6429, SMR6451, SMR6459, SMR6464, SMR6474, SMR6483; rpoS, SMR6689-6692, SMR6694-6698.
Average of the ratio (rpoS+/rpoS) of the “average days to colony formation” from three experiments with one culture of each genotype, except for isolate 8 (ratios for two experiments are given).
rpoS:: Tn 10 does not affect Lac+ colony formation
| Average days to Lac+ colony formation ± SDa | |||
|---|---|---|---|
| A. Point mutant Lac+ | rpoS+ | rpoS::Tn10 | |
| Day 2 | 2.1 ± 0.2 | 2.1 ± 0.03 | |
| Day 5 | 2.1 ± 0.04 | 2.1 ± 0.05 | |
| Average days to Lac+ colony formation (single experiment)b | |||
| B. Amplified Lac+ | rpoS+ | rpoS::Tn10 | Average of rpoS+/rpoS ± SD (three experiments)c |
| Isolate 1 | 3.5 | 3.3 | 1.0 ± 0.12 |
| Isolate 2 | 4.5 | 4.7 | 0.92 ± 0.06 |
| Isolate 3 | 4.0 | 3.8 | 1.2 ± 0.15 |
| Isolate 4 | 5.0 | 3.8 | 1.2 ± 0.10 |
| Isolate 5 | 5.0 | 4.2 | 1.2 ± 0.04 |
| Isolate 6 | 4.6 | 4.7 | 0.93 ± 0.07 |
| Isolate 7 | 4.5 | 3.7 | 1.2 ± 0.03 |
| Isolate 8 | 5.8 | 4.6 | 1.0, 1.3c |
| Isolate 9 | 3.3 | 2.6 | 1.4 ± 0.19 |
| Average days to Lac+ colony formation ± SDa | |||
|---|---|---|---|
| A. Point mutant Lac+ | rpoS+ | rpoS::Tn10 | |
| Day 2 | 2.1 ± 0.2 | 2.1 ± 0.03 | |
| Day 5 | 2.1 ± 0.04 | 2.1 ± 0.05 | |
| Average days to Lac+ colony formation (single experiment)b | |||
| B. Amplified Lac+ | rpoS+ | rpoS::Tn10 | Average of rpoS+/rpoS ± SD (three experiments)c |
| Isolate 1 | 3.5 | 3.3 | 1.0 ± 0.12 |
| Isolate 2 | 4.5 | 4.7 | 0.92 ± 0.06 |
| Isolate 3 | 4.0 | 3.8 | 1.2 ± 0.15 |
| Isolate 4 | 5.0 | 3.8 | 1.2 ± 0.10 |
| Isolate 5 | 5.0 | 4.2 | 1.2 ± 0.04 |
| Isolate 6 | 4.6 | 4.7 | 0.93 ± 0.07 |
| Isolate 7 | 4.5 | 3.7 | 1.2 ± 0.03 |
| Isolate 8 | 5.8 | 4.6 | 1.0, 1.3c |
| Isolate 9 | 3.3 | 2.6 | 1.4 ± 0.19 |
Values are the averaged “average days to colony formation” (determined in reconstruction experiments as described; Hastings et al. 2000) of 10 independent Lac+ isolates for rpoS+ and 9 for rpoS::Tn10. Lac+ colonies were scored each day for 5 days. Point mutant isolates are: day 2 rpoS+, SMR3804-3810, SMR3812-3814; day 5 rpoS+, SMR3855-3856, SMR3858-3865; day 2 rpoS, SMR6544-6552; day 5 rpoS, SMR6561-6570. Two additional experiments gave similar results.
Values are for a single culture of each genotype from one experiment. Lac+ colonies were scored each day for 9 days. Amplified isolates (in order 1-9) are: rpoS+, SMR6413, SMR6416, SMR6418, SMR6429, SMR6451, SMR6459, SMR6464, SMR6474, SMR6483; rpoS, SMR6689-6692, SMR6694-6698.
Average of the ratio (rpoS+/rpoS) of the “average days to colony formation” from three experiments with one culture of each genotype, except for isolate 8 (ratios for two experiments are given).
The retention of the amplified (sectored colony) phenotype when rpoS::Tn10 is introduced by transduction also indicates that rpoS does not cause loss of amplified arrays, which presumably occurs by homologous recombination and segregation. This contrasts with recA mutations, which stabilize preformed lac amplification in E. coli (Tlsty et al. 1984) and Salmonella (Andersson et al. 1998). Apparently rpoS mutation does not promote loss of amplified arrays, but instead inhibits their formation.
rpoS and growth-dependent Lac+ mutation: Several genes required for Lac+ stationary-phase mutation (e.g., recA) are not required for reversion of the same allele in rapidly growing cells (growth-dependent mutation), providing clear distinctions between the stationary-phase and growth-dependent mutation mechanisms (Cairns and Foster 1991; Harris et al. 1994, 1996; Foster et al. 1996; McKenzie et al. 2001). We find that growth-dependent Lac+ mutation is higher in rpoS cells than in rpoS+ (Table 3; paired Student’s t-test on the three sets of data, t =-4.33, d.f. = 2, P = 0.050), which is the opposite of its effect on stationary-phase mutation (Figure 1). This is the first direct evidence we are aware of that RpoS can affect mutation in growing cells. We conclude that RpoS is required specifically for Lac+ mutation and amplification in stationary phase and that it may suppress mutation in growing cells.
The rpoS genotype of the scavenger cells does not affect Lac+ colony yield: In adaptive mutation experiments, the lac frameshift-bearing cells are plated with an excess of nonrevertible (Δlac) “scavenger” cells to remove nonlactose carbon sources (Cairns and Foster 1991). rpoS cells might appear deficient in mutation because the rpoS+ scavenger cells might, for example, prevent growth of rpoS Lac+ mutant colonies. If so, rpoS::Tn10 scavengers might have given a different result. However, we find that the yield of Lac+ colonies in rpoS+ and rpoS::Tn10 strains is unaffected by the rpoS allele of the scavengers (data not shown). This excludes a key role for the scavengers in the rpoS mutation/amplification phenotype.
DNA recombination and F transfer are not defective in rpoS cells: The results presented above indicate that RpoS is required specifically for formation of stationary-phase mutations and amplified arrays and exclude several trivial reasons for the rpoS phenotype. The experiments in this and the following section address several possible mechanisms by which RpoS (and the genes it controls) might promote stationary-phase mutation and amplification.
RpoS is not required for Lac+ mutation in rapidly growing cells
| Relevant genotypea | Experiment | Mutation rate (×10-10; mutations per cell per generation)b | Average mutation rate (×10-10; ±SEM) |
|---|---|---|---|
| rpoS+ | 1 | 1.5 | 3.1 ± 1.0 |
| 2 | 2.8 | ||
| 3 | 5.0 | ||
| rpoS::Tn10 | 1 | 8.4 | 12 ± 1.9 |
| 2 | 15 | ||
| 3 | 11 |
| Relevant genotypea | Experiment | Mutation rate (×10-10; mutations per cell per generation)b | Average mutation rate (×10-10; ±SEM) |
|---|---|---|---|
| rpoS+ | 1 | 1.5 | 3.1 ± 1.0 |
| 2 | 2.8 | ||
| 3 | 5.0 | ||
| rpoS::Tn10 | 1 | 8.4 | 12 ± 1.9 |
| 2 | 15 | ||
| 3 | 11 |
Strains are: rpoS+, SMR4562; rpoS::Tn10, SMR6541.
Mutant frequencies were determined in 29 or 30 tube fluctuation tests and mutation rates were calculated using control Lac+ strains: rpoS+, SMR3804-3810, SMR3812; rpoS, SMR6544-6551 (see materials and methods). In experiment 1, four rpoS+ Lac+ and eight rpoS Lac+ control strains were used, and in experiments 2 and 3, eight Lac+ control strains of each genotype were used. Lac+ controls were independent isolates confirmed as mutant, not amplified, by colony appearance on LBH rifampicin X-gal plates (Figure 1 legend).
RpoS is not required for Lac+ mutation in rapidly growing cells
| Relevant genotypea | Experiment | Mutation rate (×10-10; mutations per cell per generation)b | Average mutation rate (×10-10; ±SEM) |
|---|---|---|---|
| rpoS+ | 1 | 1.5 | 3.1 ± 1.0 |
| 2 | 2.8 | ||
| 3 | 5.0 | ||
| rpoS::Tn10 | 1 | 8.4 | 12 ± 1.9 |
| 2 | 15 | ||
| 3 | 11 |
| Relevant genotypea | Experiment | Mutation rate (×10-10; mutations per cell per generation)b | Average mutation rate (×10-10; ±SEM) |
|---|---|---|---|
| rpoS+ | 1 | 1.5 | 3.1 ± 1.0 |
| 2 | 2.8 | ||
| 3 | 5.0 | ||
| rpoS::Tn10 | 1 | 8.4 | 12 ± 1.9 |
| 2 | 15 | ||
| 3 | 11 |
Strains are: rpoS+, SMR4562; rpoS::Tn10, SMR6541.
Mutant frequencies were determined in 29 or 30 tube fluctuation tests and mutation rates were calculated using control Lac+ strains: rpoS+, SMR3804-3810, SMR3812; rpoS, SMR6544-6551 (see materials and methods). In experiment 1, four rpoS+ Lac+ and eight rpoS Lac+ control strains were used, and in experiments 2 and 3, eight Lac+ control strains of each genotype were used. Lac+ controls were independent isolates confirmed as mutant, not amplified, by colony appearance on LBH rifampicin X-gal plates (Figure 1 legend).
Stationary-phase mutation of the F′-borne lac allele requires F transfer functions (Foster and Trimarchi 1995a,b; Galitski and Roth 1995) and RecBC-dependent homologous recombination proteins (Harris et al. 1994, 1996; Foster et al. 1996). We find that neither F transfer nor RecBC-dependent recombination is deficient in rpoS cells. rpoS+ and rpoS stationary-phase cells are similarly active as donors in F transfer assays (consistent with previous results; Frost and Manchak 1998). The average numbers of transconjugants per donor cell are (experiment 1 and experiment 2; values are the means of three cultures): rpoS+ (strain SMR4562), 7.7 × 10-3, 4.0 × 10-3; rpoS (SMR6541), 14 × 10-3, 3.8 × 10-3. Also, the frequency of transduction (a RecBC-dependent process; Lloyd and Low 1996) is not different for rpoS+ and rpoS minimal B1 glycerol stationary-phase cultures (Table 4; also true for minimal B1 glucose and rich medium cultures, data not shown). These are the only experiments we are aware of that address a role for RpoS in homologous recombination. Because rpoS mutation affects neither F transfer nor homologous recombination, the rpoS requirements in stationary-phase mutation and amplification are unlikely to be to due to defects in either of these processes.
rpoS:: Tn10 does not reduce transductional recombination
| Transductant frequency (×10-4)b | Fold decrease | Mean fold decrease (±SEM) | |||
|---|---|---|---|---|---|
| Selectiona | Experiment | rpoS+ | rpoS::Tn10 | ||
| Thr+ | 1 | 5.8 | 3.4 | 1.7 | 0.94 ± 0.4 |
| 2 | 1.2 | 2.0 | 0.60 | ||
| 3 | 1.2 | 2.3 | 0.52 | ||
| Trp+ | 1 | 0.86 | 2.7 | 0.32 | 1.2 ± 0.8 |
| 2 | 0.28 | 0.10 | 2.8 | ||
| 3 | 6.1 | 13 | 0.47 | ||
| Transductant frequency (×10-4)b | Fold decrease | Mean fold decrease (±SEM) | |||
|---|---|---|---|---|---|
| Selectiona | Experiment | rpoS+ | rpoS::Tn10 | ||
| Thr+ | 1 | 5.8 | 3.4 | 1.7 | 0.94 ± 0.4 |
| 2 | 1.2 | 2.0 | 0.60 | ||
| 3 | 1.2 | 2.3 | 0.52 | ||
| Trp+ | 1 | 0.86 | 2.7 | 0.32 | 1.2 ± 0.8 |
| 2 | 0.28 | 0.10 | 2.8 | ||
| 3 | 6.1 | 13 | 0.47 | ||
For threonine or tryptophan prototrophy (materials and methods).
Recipient and donor strains are: rpoS+ thr, SMR6993; rpoS::Tn10 thr, SMR6994; rpoS+ trp, SMR7037; rpoS::Tn10 trp, SMR7038; P1 phage donor, SMR4562. Transductant frequencies are means from two to four transductions of one rpoS+ or rpoS recipient culture at varying MOIs (all <0.01 phage per cell).
rpoS:: Tn10 does not reduce transductional recombination
| Transductant frequency (×10-4)b | Fold decrease | Mean fold decrease (±SEM) | |||
|---|---|---|---|---|---|
| Selectiona | Experiment | rpoS+ | rpoS::Tn10 | ||
| Thr+ | 1 | 5.8 | 3.4 | 1.7 | 0.94 ± 0.4 |
| 2 | 1.2 | 2.0 | 0.60 | ||
| 3 | 1.2 | 2.3 | 0.52 | ||
| Trp+ | 1 | 0.86 | 2.7 | 0.32 | 1.2 ± 0.8 |
| 2 | 0.28 | 0.10 | 2.8 | ||
| 3 | 6.1 | 13 | 0.47 | ||
| Transductant frequency (×10-4)b | Fold decrease | Mean fold decrease (±SEM) | |||
|---|---|---|---|---|---|
| Selectiona | Experiment | rpoS+ | rpoS::Tn10 | ||
| Thr+ | 1 | 5.8 | 3.4 | 1.7 | 0.94 ± 0.4 |
| 2 | 1.2 | 2.0 | 0.60 | ||
| 3 | 1.2 | 2.3 | 0.52 | ||
| Trp+ | 1 | 0.86 | 2.7 | 0.32 | 1.2 ± 0.8 |
| 2 | 0.28 | 0.10 | 2.8 | ||
| 3 | 6.1 | 13 | 0.47 | ||
For threonine or tryptophan prototrophy (materials and methods).
Recipient and donor strains are: rpoS+ thr, SMR6993; rpoS::Tn10 thr, SMR6994; rpoS+ trp, SMR7037; rpoS::Tn10 trp, SMR7038; P1 phage donor, SMR4562. Transductant frequencies are means from two to four transductions of one rpoS+ or rpoS recipient culture at varying MOIs (all <0.01 phage per cell).
rpoS and the SOS response: Additional evidence supporting the involvement of RpoS in the SOS-dependent point mutation pathway (Cairns and Foster 1991; McKenzie et al. 2000) comes from epistasis analysis of lexA3 and rpoS mutations. lexA3 encodes a noncleavable LexA repressor; a lexA3 strain is therefore unable to induce the Lex-dependent genes of the SOS response (Walker et al. 2000). lexA3 prevents most stationary-phase mutation (Cairns and Foster 1991; McKenzie et al. 2000), but has no effect on amplification (McKenzie et al. 2001). If RpoS and the SOS response act in separate pathways leading to point mutation, then the rpoS lexA3 double mutant should have a more severe decrease in Lac+ yield than either single mutant. We find, in contrast, that the double mutant is similar to the rpoS strain (Figure 2), indicating that the RpoS and the SOS regulons act in the same pathway. That amplification also requires RpoS (Figure 1; Table 2), but not SOS (McKenzie et al. 2001), suggests that RpoS might act upstream of SOS before a common pathway branches into SOS-dependent point mutation and SOS-independent amplification routes. However, it is not yet clear whether RpoS plays the same role in amplification as it does in point mutation.
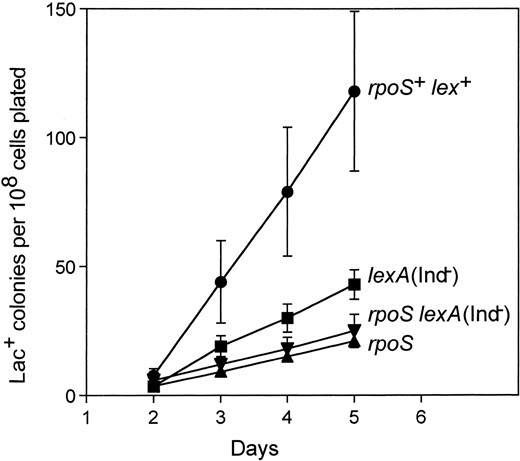
—rpoS::Tn10 is epistatic to lexA3(Ind-). Values are means ±1 SEM of Lac+ colony counts per 108 cells plated from three experiments of six cultures each. Viability in each of the experiments varied less than twofold from day 1 to day 4 for all strains (data not shown).
Mutation and amplification do not require a special rpoS allele: Many E. coli laboratory strains carry rpoS mutations that attenuate RpoS function (Ivanova et al. 1992; Visick and Clarke 1997; Atlung et al. 2002), and the acquisition of an rpoSatt allele is a key feature of the growth advantage in stationary phase (GASP) phenotype (Zambrano et al. 1993; Zambrano and Kolter 1996). In GASP, cells in long-term culture acquire advantageous mutations, the first being rpoSatt, and then outcompete their parental genotypes in successive waves, each requiring additional mutations (Zambrano and Kolter 1996). Might Lac+ stationary-phase mutation require a special allele of rpoS?
We find that the rpoS allele of the lac frameshift strain SMR4562 [an independent construction of FC40 (Cairns and Foster 1991) from the same parental strains] has a mutation relative to the sequence in the E. coli K12 “wild-type” strain MG1655 (Blattner et al. 1997). rpoS codon 33 in SMR4562 is TTG, encoding leucine, instead of the CAG, encoding glutamine, in MG1655. This codon varies in laboratory strains (glutamine or an amber codon are common; Ivanova et al. 1992; Visick and Clarke 1997; Atlung et al. 2002). However, this variant allele is not important for stationary-phase mutation and amplification because neither is altered in an SMR4562 derivative carrying a nonattenuated rpoS+ allele (Atlung et al. 2002) from wild-type MG1655 (Figure 1E and additional data not shown). Therefore, the FC40 variant rpoS allele does not specifically promote mutation and amplification, and further, attenuated rpoS function conferred by mutation, which underlies GASP, does not underlie these stationary-phase mutations.
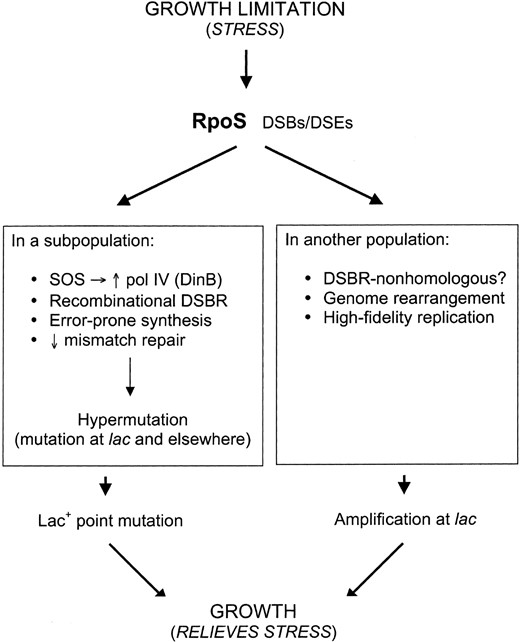
—A branched pathway model for adaptive point mutation and amplification as RpoS-dependent stress responses (modified from Hastings et al. 2000; McKenzie et al. 2001; Rosenberg 2001; Hastings and Rosenberg 2002). Amplification and point mutation are proposed to be alternative outcomes of a branched pathway, in which the only common genetic requirement known currently, RpoS, acts early, before the pathways diverge. Both processes are proposed to be stress responses that result in genetic change, some of which can promote growth in the growth-limiting environment. In point mutation, double-strand-break repair (DSBR) becomes error prone in stationary phase via use of pol IV. DSBs, double-strand breaks; DSEs, double-strand ends.
DISCUSSION
Summary: We found that RpoS, which controls a large regulatory network induced in response to various environmental stresses, including the onset of stationary phase (Loewen and Hengge-Aronis 1994), is required for most stationary-phase point mutation and amplification in the Lac system (Figure 1; Table 2; results reported in the text). RpoS is required specifically for mutation in stationary phase and not in rapidly growing cells (Table 3). RpoS and the SOS regulon act in the same pathway leading to stationary-phase point mutation (Figure 2), with RpoS possibly acting upstream of SOS at a point before the amplification route branches from the point mutation route (e.g., Figure 3). These results support the idea that transient genetic instability in this system is a stress response, specifically one induced in stationary phase.
These conclusions are possible only because other explanations for the rpoS phenotype (poor viability, poor colony formation, and loss of amplified arrays) were excluded experimentally. A previous study reported data indicating that an rpoS mutation reduced late Lac+ colonies, but the authors dismissed the phenotype because it correlated with altered growth relative to rpoS+ before day 4 (Bridges et al. 2001). Their experimental conditions differed sufficiently as to allow growth during the early days of the experiment, which confounds interpretation of the data. For example, if the population grows, some unknown number of Lac+ colonies must be attributed to non-stationary-phase (rec- and din-independent) mutation mechanisms. Here, we controlled carefully for effects on growth and viability and other alternative explanations, including RpoS effects on mutation in growing cells, and conclude that RpoS is required specifically for most stationary-phase mutation and amplification.
Mutation and amplification are stress responses—implications for hypermutation and cryptic growth models: Three broad classes of model, two of which persist, were proposed as possible explanations for adaptive mutation in this and other assay systems (reviewed by Foster 1999b; Rosenberg 2001). One class, postulating mutations directed to genes under selection (directed mutation models, e.g., Cairns et al. 1988), was rejected for the Lac system (see Introduction; Rosenberg 2001). The two extant classes of model evoke either a global increase in mutation rates in a stress response to the environment (hypermutation models) or “normal” mutation rates and mechanisms acting on multiple DNA copies in rare growing cells (cryptic growth models, an example is discussed below). In the Lac system, hypermutation models are supported not solely by detection of mutation at non-lac sites (see Introduction; reviewed by Rosenberg 2001), but also by requirements for components well known to contribute to increased mutation in other contexts and/or assay systems (Friedberg et al. 1995; Kim et al. 1997; Wagner and Nohmi 2000; Walker et al. 2000): the mutagenic SOS response (Cairns and Foster 1991; McKenzie et al. 2000), the low-fidelity DNA pol IV (McKenzie et al. 2001), and limiting mismatch repair (Harris et al. 1997, 1999).
A current cryptic growth model (Andersson et al. 1998; Hendrickson et al. 2002) proposes preexisting spontaneous amplification of the lac region as a mechanism to achieve cryptic growth and mutation in some rare cells: multiple lac copies (more β-galactosidase) allow growth; mutation rates are normal, and the increased Lac+ mutants in stationary phase result mainly from growth and extra lac copies. Recent versions of this model account for the extensively documented genome-wide hypermutation in Lac+ mutants (Torkelson et al. 1997; Rosche and Foster 1999; Bull et al. 2000, 2001; Godoy et al. 2000) and for the requirement for SOS-inducible DNA pol IV (McKenzie et al. 2001) by invoking induction of the SOS response by amplification (Hendrickson et al. 2002) or by amplification of the gene encoding pol IV (Kofoid et al. 2003). This cryptic growth model thus makes hypermutation a by-product of, rather than central to, formation of Lac+ adaptive point mutants. Lac+ mutations are proposed to result mainly from growth and increased replication of multiple gene copies, occurring more robustly with increasing growth rate, not as part of a stress response or in differentiated stationary-phase cells. The requirement for an RpoS-dependent stress response (Figure 1; Table 2) and the implication that the mutating cells are in stationary phase are incompatible with such cryptic growth models that allow no special circumstances (other than growth) and instead support hypermutation models, which feature increased mutation rates as part of a stress response in growth-limited cells (e.g., Figure 3).
RpoS and models for transient genetic instability: In Figure 3, we suggest that RpoS acts upstream in a common stress response pathway that branches to yield either point mutation or amplification, rather than downstream in promoting survival of cells carrying mutation or amplification. This order of action is supported by the well-characterized role of RpoS in establishing the differentiated state of stationary phase (Hengge-Aronis 2000) and by the epistasis of rpoS to a lexA(Ind-) mutation (Figure 2). Although it is economical to hypothesize a single role for RpoS in the two genetic instability routes in the common part of a branched pathway, it is also possible that RpoS plays different roles in point mutation and amplification.
The two branches (to point mutation or to amplification; Figure 3) are clearly delineated: the SOS response and DNA pol IV are required only for mutation, not for amplification (McKenzie et al. 2001), and amplified isolates do not display hypermutation as point mutants do (Hastings et al. 2000). The point mutation pathway is better defined, requiring DSB-repair proteins (Harris et al. 1994, 1996; Foster et al. 1996), F transfer proteins (Foster and Trimarchi 1995a,b; Galitski and Roth 1995), an SOS response (Cairns and Foster 1991; McKenzie et al. 2000), SOS-induced DNA pol IV (DinB; McKenzie et al. 2001), and limiting mismatch repair (Harris et al. 1997, 1999), and is associated with transient genome-wide hypermutation (Torkelson et al. 1997; Rosche and Foster 1999; Bull et al. 2000, 2001; Godoy et al. 2000). In contrast, amplification requires neither SOS nor pol IV (McKenzie et al. 2001) and is not associated with genome-wide hypermutation (Hastings et al. 2000). We suggest that point mutation occurs in a transiently hypermutable cell subpopulation in which the SOS response is induced, leading to increased production of error-prone pol IV. The mutations could result from error-prone synthesis (using pol IV) primed during homologous recombinational DSB repair (Harris et al. 1994). Amplification might result when DS ends (possible sources reviewed by Rosenberg 2001) are repaired beginning with a nonhomologous end-joining event, creating either a duplicated segment or a rolling circle intermediate, using high-fidelity DNA synthesis in non-SOS induced cells (Hastings and Rosenberg 2002).
In this model (Figure 3), acquisition of either a Lac+ point mutation or amplified lac DNA allows growth, curtails the RpoS-dependent stress response, and restores genetic stability. That amplification is an endpoint, rather than an intermediate en route to point mutation (as in single-pathway models, e.g., Hendrickson et al. 2002), is supported by previous findings that amplified clones are neither hypermutated nor particularly mutable and do not lead readily to formation of adaptive point mutants under selection (Hastings et al. 2000).
RpoS in other stationary-phase mutation mechanisms: RpoS is required for stress-induced mutation in another assay system in E. coli (Bjedov et al. 2003). In that system mutation in aged colonies also requires RecA, but in contrast to mutation in the Lac system does not have strong dependence on either RecB or the LexA regulon. Thus this appears to be a different mechanism, but one that also requires the RpoS regulon, supporting a general role for RpoS in multiple stationary-phase mutation mechanisms (although not in all stationary-phase mutation systems; see Bridges and Timms 1998). RpoS is required for mutation-dependent adaptation to long-term culture conditions that can give some cells a growth advantage in stationary phase (see results; Zambrano and Kolter 1996), as are pol IV and two other SOS-inducible polymerases (Yeiser et al. 2002). Also, apparent stationary-phase mutations in Pseudomonas seem to require RpoS (Saumaa et al. 2002) as do stationary-phase genome rearrangements associated with transposable elements in E. coli (Gomez-Gomez et al. 1997; Lamrani et al. 1999) and other bacteria (Ilves et al. 2001; Saumaa et al. 2002).
Candidate genes in the RpoS regulon: RpoS-regulated gene products (Loewen et al. 1998) that might be involved at the DNA level in mutation and amplification include proteins involved in base excision repair (Exo-nuclease III), methylation damage repair (AidB), chromosome segregation/cell division (FtsQ, FtsZ), and prevention of oxidative damage (catalase HPII, Dps). In addition, RpoS downregulates methyl-directed mismatch repair in stationary phase, apparently by decreasing the levels of MutS and MutH (Feng et al. 1996; Tsui et al. 1997). An apparent insufficiency of functional MutL facilitates stationary-phase mutation in this system (Harris et al. 1997, 1999; and perhaps amplification, because the work was done prior to the discovery of amplification as a major component on later days).
Transient genetic instability and the general stress response: The data presented here imply that transient genetic instability in the Lac system occurs in cells that are differentiated by RpoS-dependent gene expression. The well-characterized role of the RpoS regulon (Hengge-Aronis 2000) in protecting cells (particularly at the DNA level) from environmental stresses may seem at odds with a role in supporting genetic instability. However, the evidence that only a subpopulation of cells experiences genetic instability in this system (reviewed in Rosenberg 2001) means that the population as a whole is not threatened. Only a subpopulation is subject to the potentially damaging effects of increased point mutation rates. Recent work demonstrates that the majority of wild E. coli experience stress-inducible mutation, supporting the relevance of increased mutation rates to bacterial survival (Bjedov et al. 2003; Rosenberg and Hastings 2003). The role of RpoS as the regulator of the general stress response (Hengge-Aronis 2000) suggests that stationary phase might not be the only condition that stimulates mutation and genetic rearrangements such as amplification. Other inducers of the general stress response might also cause transient genetic instability and so promote bacterial adaptation to stressful environments.
Note: A prepublication report released during the review of this manuscript shows that RpoS positively regulates the DinB error-prone DNA polymerase (pol IV; Layton and Foster 2003). Because DinB is required for adaptive point mutation (McKenzie et al. 2001), this could account for some or all of the role of RpoS in adaptive point mutation. Because DinB is not required for adaptive amplification (McKenzie et al. 2001), RpoS must play some other role in that process.
Acknowledgement
For providing strains, we thank Mary Berlyn and the E. coli Genetic Stock Center, Roberto Kolter and Steve Finkel, Joe Peters, and P. J. Hastings. For comments on the manuscript, we thank Bryn Bridges, Harold Bull, Susan Gottesman, Steve Finkel, P. J. Hastings, Megan Hersh, Greg McKenzie, Rebecca Ponder, Miroslav Radman, Peg Riley, Andrew Slack, and Shirley Yang. This work was supported by National Institutes of Health grants F32-GM19909 (M.-J.L.) and R01-GM53158.
Footnotes
Communicating editor: R. S. Hawley
LITERATURE CITED
Author notes
Present address: Department of Microbiology and Immunology, University of Maryland School of Medicine, 685 W. Baltimore St., Baltimore, MD 21201.
Present address: University of Pecs, Pharmaceutical Technological Institute, 7624 Pecs, Rokus utca 2, Hungary.

{kind=link}
{kind=link}
{kind=link}