





Abstract
We studied the evolution of high mutation rates and the evolution of fitness in three experimental populations of Escherichia coli adapting to a glucose-limited environment. We identified the mutations responsible for the high mutation rates and show that their rate of substitution in all three populations was too rapid to be accounted for simply by genetic drift. In two of the populations, large gains in fitness relative to the ancestor occurred as the mutator alleles rose to fixation, strongly supporting the conclusion that mutator alleles fixed by hitchhiking with beneficial mutations at other loci. In one population, no significant gain in fitness relative to the ancestor occurred in the population as a whole while the mutator allele rose to fixation, but a substantial and significant gain in fitness occurred in the mutator subpopulation as the mutator neared fixation. The spread of the mutator allele from rarity to fixation took >1000 generations in each population. We show that simultaneous adaptive gains in both the mutator and wild-type subpopulations (clonal interference) retarded the mutator fixation in at least one of the populations. We found little evidence that the evolution of high mutation rates accelerated adaptation in these populations.
MUTATION is ultimately essential for adaptive evolution in all populations. Mutations that affect the phenotype, however, are much more likely to be deleterious than beneficial (Fisher 1958); thus, despite its potential adaptive benefit to populations, mutation is usually harmful to individuals. The prevalence of deleterious mutations over beneficial mutations led A. H. Sturtevant, in an early essay, to conclude that the mutation rate should evolve to the lowest value possible “given the nature of genes” (Sturtevant 1937, p. 466).
Since Sturtevant, theoretical progress in this area has been made by analyzing the indirect effect that selection on deleterious and beneficial mutations has on the frequencies of alleles that modify the mutation rate (Kimura 1967; Leigh 1970, 1973; Painter 1975a,b; Holsinger and Feldman 1983; Liberman and Feldman 1986; Kondrashov 1995; Taddei et al. 1997; Johnson 1999a,b; Tenaillon et al. 1999, 2000; reviewed in Sniegowski et al. 2000). The modifier models have generally upheld Sturtevant’s conclusion that mutation rates should be minimized by natural selection, but they have identified important differences in the evolution of mutation rates between sexual and asexual populations. In sexual populations, indirect selection is weak because recombination erodes linkage disequilibria between mutation rate modifiers and mutations affecting fitness; nonetheless, the prevalence of deleterious mutations ensures that indirect selection continually favors a decreased mutation rate (Leigh 1970, 1973; Liberman and Feldman 1986; Johnson 1999b). In asexual populations, indirect selection is stronger because associations between fitness mutations and mutation rate modifiers persist. Thus, in the absence of beneficial mutations, asexual populations are expected to evolve lower mutation rates than those of sexual populations (Kondrashov 1995). When beneficial mutations are substituting in asexual populations, however, modifier alleles that raise the mutation rate (mutators) can hitchhike to high frequency with them, thereby increasing the average mutation rate in the population (Painter 1975b; Taddei et al. 1997; Tenaillon et al. 1999, 2000).
The capacity for asexual populations to evolve high mutation rates by hitchhiking has been demonstrated by experimental manipulations that enhance the probability of association between mutator alleles and beneficial mutations. When the frequency of a mutator clone is deliberately increased above a certain threshold value in a large experimental population of bacteria, the mutator will spread further in association with new beneficial mutations (Chao and Cox 1983; Tröbner and Piechocki 1984; Giraud et al. 2001); below this threshold, the mutator clone will typically decline in frequency because the wild-type clone is more likely to be associated with new beneficial mutations. When a large bacterial culture is exposed to multiple rounds of lethal (hard) selection, the frequency of clones bearing spontaneous mutator mutations within the culture can be greatly enhanced because these clones have a higher probability of producing mutants that survive the selection (Mao et al. 1997).
Mutator hitchhiking in natural bacterial populations is suggested by the substantial frequencies of mutator strains in some collections of natural isolates (Jyssum 1960; Gross and Siegel 1981; LeClerc et al. 1996; Matic et al. 1997; Oliver et al. 2000). How would hitchhiking proceed without the kinds of experimental inducements described above? Many natural bacterial populations may experience occasional bouts of hard selection, and these could enhance the frequencies of mutator cells. Pathogens, in particular, are likely to encounter hard selection as a consequence of host immune responses. Genetic drift could also occasionally enhance the frequencies of mutator clones to the point where hitchhiking becomes likely. But even without hard selection or drift, chance associations between rare mutator alleles and selectively favored mutations could arise and spread within asexual populations given sufficient time, as predicted by computer simulations (Taddei et al. 1997; Tenaillon et al. 1999, 2000).
In a previous study, we documented the evolution of high mutation rates in three experimental populations of Escherichia coli that were propagated in a constant environment for thousands of generations without any other manipulations (Sniegowski et al. 1997). In these populations, mutator alleles arose by spontaneous mutation rather than by deliberate introduction, and natural selection favored superior competitors rather than the survivors of externally imposed hard selection events. We previously concluded that hitchhiking of mutator alleles was the most likely explanation for the evolution of high mutation rates in these populations, but we acknowledged that two alternative explanations could not be ruled out at that time. These were (i) that mutator alleles had risen to fixation simply as a consequence of genetic drift and (ii) that mutator alleles had risen to fixation as a consequence of direct, rather than indirect, selection.
In this article, we analyze the evolution of high mutation rates in these populations in more depth. We sequence the alleles in the methyl-directed mismatch repair (MMR) pathway that are responsible for the high mutation rates in all three populations, and we show that these alleles spread through the populations too quickly for their fixation to have been caused simply by genetic drift. We test whether MMR mutator alleles are directly beneficial to their bearers in this experimental system by competing a strain carrying one of the three evolved mutator sequences with its isogenic wild-type counterpart. Consistent with previous results (Mao et al. 1997), we find no evidence for direct selection in favor of MMR defects.
We show that average population fitness relative to the ancestor increased substantially and significantly while the mutator alleles swept to fixation in two of the populations; this result strongly supports the conclusion that the mutator alleles fixed by hitchhiking in these populations. In the third population, average fitness did not increase significantly while the mutator allele swept to fixation, but a substantial and significant increase in the fitness of the mutator subpopulation occurred. Overall, our evidence indicates that the mutator alleles were fixed in each population by hitchhiking.
Given that their fixation was driven by natural selection, the mutator alleles took a surprisingly long time to progress from rarity to fixation in all three populations—at least 1000 generations in each case and perhaps as long as 2500 generations in one case. We discuss population genetic factors that could explain this observation. Most notably, we show in one case that the wild-type subpopulation underwent significant adaptive evolution even as it was being supplanted by the mutator, as predicted by previous theoretical work (Painter 1975b; Tenaillon et al. 1999). This result provides a clear example of the retarding effect that “clonal interference” (Muller 1932; Gerrish and Lenski 1998; Gerrish 2001) can have on selectively driven allele substitution in an asexual population.
We conclude by considering the general implications of our findings for the causes and consequences of mutation rate evolution in asexual populations.
MATERIALS AND METHODS
Experimental system: We studied asexual experimental populations of E. coli founded and propagated by R. E. Lenski and collaborators (reviewed in Lenski et al. 1998). The common ancestor of these populations was REL606, an Ara- clone of E. coli B (Lederberg 1966). A spontaneous Ara+ revertant was selected from REL606 by Lenski and designated REL607. Six clones each from REL606 (designated Ara-1 through Ara-6) and REL607 (designated Ara+1 through Ara+6) were used to found 12 experimental populations that were propagated with aeration at 37° by daily 100-fold dilutions into 10 ml of fresh Davis minimal (DM) broth (Carlton and Brown 1981) supplemented with glucose at 25 mg/liter (hereafter, DM25). A sample of each population was frozen in 15% glycerol at 100-generation intervals during the first 2000 generations of the experiment and at 500-generation intervals thereafter.
We showed previously that 3 of the 12 Lenski experimental populations—those designated as Ara-2, Ara-4, and Ara+3— evolved ∼100-fold increases in their genomic mutation rates during the first 10,000 generations of propagation (Sniegowski et al. 1997). Genetic experiments showed that each of these populations had become fixed for a defect in the MMR pathway: Low mutation rates were restored in populations Ara-2 and Ara-4 by plasmids expressing wild-type mutL and uvrD alleles and in population Ara+3 by a plasmid expressing a wild-type mutS allele (Sniegowski et al. 1997).
Amplification and sequencing of candidate mutator alleles: Alleles of mutS, mutL, and uvrD were amplified by polymerase chain reaction using primers designed from the E. coli K12 genomic sequence (Blattner et al. 1997). All amplifications were performed using the DyNAzyme EXT system (Finnzymes Oy, Espoo, Finland), which employs a proofreading polymerase.
Nearly full amplifications of mutS were carried out in the ancestor REL606 and in a clonal mutator isolate obtained from population Ara+3 at 10,000 generations, using the primer pair 5′ GAGTGCAATAGAAAATTTCGACG and 5′ TCTTCTGGT ACTGACAGCAAAGAC. The mutS gene in E. coli K12 is 2562 bp in length. The forward primer begins at position 3 and the reverse primer begins at position 2462; amplification of the wild-type gene yielded a product of the predicted size (2359 bp). Amplifications were carried out for 30 cycles in a PTC-100 thermal controller (MJ Research, Watertown, MA) under the following conditions: denaturing step, 30 sec at 95°; annealing step, 1 min at 60°; extension step, 1 min 45 sec at 70°. One unit of polymerase and 25 pmol of genomic DNA were used in a total reaction volume of 50 μl with the following reagent concentrations: 1.5 mm Mg2+, 200 μm of each dNTP, and 0.5 μm of each primer.
Full amplifications of the mutL gene were carried out in the ancestor REL606 and in clonal mutator isolates from populations Ara-2 and Ara-4 at 10,000 generations, using the primer pair 5′ GGCGAGCGACGATTACCAAC and 5′ GCGA CAACCCTTCCAGCAAT. The forward primer starts 150 bp into the 3′ end of the upstream amiB gene; the reverse primer starts 350 bp into the 5′ end of the downstream miaA gene. The mutL gene is 1850 bp long and the primer pair produced an amplicon of size 2350 bp in the ancestor. Amplifications were carried out for 30 cycles under the following conditions: denaturing step, 30 sec at 95°; annealing step, 1 min at 65°; extension step, 1 min 45 sec at 70°. One unit of polymerase and 25 pmol of genomic DNA were used in a total reaction volume of 50 μl with the following reagent concentrations: 2 mm Mg2+, 360 μm of each dNTP, and 0.5 μm of each primer.
A full amplification of the uvrD gene was carried out in a clonal mutator isolate from population Ara-2 at 10,000 generations using the primer pair 5′ TCATGCCAACCTCTCC ACCA and 5′ CGATGTCTTCCAGTTCCGGG. The left primer is 177 bp from the end of the xerC gene, and the right primer is 180 bp into the corA gene. The total amplicon size was 4522 bp, of which the uvrD gene comprised 2163 bp. The conditions for the amplification of the uvrD sequence were the same as those given for mutL, above, except that the reaction mix also contained 9% (v/v) DMSO.
The mutS, mutL, and uvrD amplicons were cloned into the pGEM-T Easy vector (Promega, Madison, WI) and sequenced by primer walks at the University of Pennsylvania Sequencing Center using the Big Dye Taq FS Terminator system (Perkin Elmer, Wellesley, MA) and an ABI 377 Automated Sequencer. Double-stranded sequence was obtained for mutS and mutL; only single-stranded sequence was obtained for uvrD.
Additional amplifications of mutL and mutS were carried out using the above primers on isolates from each population at the beginning and end of the detected sweep of each mutator allele to fixation, using the protocols described above. Because our original sequencing revealed that the mutator mutations were close to the 5′ ends of these genes (see results), we used the forward primers from the amplification reactions for these later sequencing reactions, which were carried out at the Nucleic Acid/Protein Core Research Facility of the Children’s Hospital of Pennsylvania.
Sequences obtained for the wild-type E. coli B mutL and mutS alleles were deposited in GenBank under accession nos. AF440199 (mutL) and AF440200 (mutS).
Estimation of mutator frequencies and isolation of mutator and wild-type clones: We estimated the frequencies of mutator and wild-type cells in the populations at 500-generation intervals, using a variation of a previously described assay (Sniegowski et al. 1997). This assay relies on the fact that cultures with different mutation rates tend to yield divergent numbers of mutants when grown from small inocula (Luria and Delbrück 1943). At each time point of interest, we thawed frozen samples of each population, withdrew 100 μl, and inoculated this into 10 ml of Davis minimal broth containing 1000 mg of glucose per liter (hereafter, DM1000) for overnight growth to stationary density. Growth in this and all subsequent steps was at 37°. We then plated 100 cells from each of these cultures to Luria-Bertani (LB) agar (Miller 1992) and incubated the plates overnight. We next sampled 50 clones at random by lightly touching single, well-isolated colonies with a toothpick and suspending the cells in sterile saline. We then used 100-μl aliquots (containing ∼1000 cells) of each such dilution to inoculate four separate 10-ml DM1000 cultures for each of the sampled clones. These replicate cultures were grown to stationary density. Finally, we sampled 100 μl from each of the four independent cultures representing each clone and deposited these samples separately to an LB agar plate containing 20 mg/liter of nalidixic acid. These plates were incubated for 48 hr, after which the numbers of NalR mutant colonies per spot were recorded.
We generated expected outcomes of this assay by computer simulations using an algorithm that computes the probability of observing a given number of mutants in a population for particular values of the population size and the mutation rate (Ma et al. 1992). In the simulations we used average parameter values that had been measured previously for the appropriate genotypes (Sniegowski et al. 1997): namely, a population size of 1 × 1010 for both types and mutation rates to nalidixic acid resistance of 1.22 × 10-10 for the wild-type and 2.67 × 10-8 for the mutators. We simulated mutation in 1000 replicate mutator cultures and 5000 replicate wild-type cultures. We then simulated spot plating 100 μl (1/100th) of each culture by choosing a random variable from a Poisson distribution with parameter λ= Nm/100, where Nm was the total number of mutants in a given replicate.
We used the results of the simulations to estimate the likelihood of observing any given spot-plate outcome for both phenotypes. The ratio of the likelihoods for a particular outcome was used to provide a basis for classification of individual clones as mutator or wild type. Only clones for which the likelihood ratio was >1000 or <0.001 were included in the frequency estimates; in practice, this included almost all clones isolated.
From each time point studied, we archived multiple mutator and wild-type clones by regrowing them to stationary density in DM1000 and freezing them in 15% glycerol.
Assays of fitness relative to the common ancestor: For each time point studied in all three populations, we competed multiple wild-type and mutator clones individually against the common ancestor using a previously described protocol (Lenski et al. 1991). Pairs of strains to be competed were inoculated from freezer stocks into 10 ml of DM1000, conditioned by growth to stationary density at 37°, and then further conditioned by dilution and growth to stationary density at 37° in 10 ml of DM25. The two competitors were then introduced, each with 200-fold dilution, into a common flask of DM25 and allowed to grow to stationary density with aeration at 37°; competitive fitnesses were thus assayed in an abiotic environment identical to that in which the strains had evolved. Relative fitnesses were calculated as the ratio of the numbers of doublings that the two competitors achieved during overnight growth. All measurements of competitive fitness were replicated at least threefold.
We used the Ara-/Ara+ phenotypic difference to distinguish the competitor strains. Ara- and Ara+ cells produce red and pink colonies, respectively, on tetrazolium-arabinose (TA) indicator agar (Levin et al. 1977; Lenski 1988), and thus it is possible to enumerate the two types in mixed culture by plating suitable dilutions. The Ara-/Ara+ difference is neutral with respect to competitive fitness in DM25 (Lenski 1988; Lenski et al. 1991; Bennett et al. 1992).
Screening of Ara- forward mutants from Ara+3 mutator clones: To facilitate assays of competitive fitness relationships within population Ara+3, we used a variation of a classical enrichment method employing an antibiotic (Lederberg and Zinder 1948) to obtain Ara- forward mutants from Ara+3 mutator isolates. Ara+3 mutator isolates were grown to stationary densities in DM1000, after which each was subcultured 1:10 into 9 ml of DM broth containing 1 g/liter of arabinose as the sole carbon source and incubated for 1 hr with aeration at 37°. Nalidixic acid was then added to a final concentration of 200 μg/liter, and the cultures were incubated for a further 24 hr with aeration at 37°. The cultures were then centrifuged, washed in sterile saline, resuspended in 10 ml of DM1000 (glucose), and incubated overnight at 37° with aeration. Finally, Ara- forward mutants were screened by plating several hundred cells from each culture to TA agar and picking and testing red colonies for the inability to grow on minimal arabinose agar. Only those Ara- forward mutants whose fitnesses were unchanged by the enrichment procedure were utilized in further competition experiments.
Construction and competition of isogenic mutS+ and mutS- strains in the Ara+3 genetic background: We competed nearly isogenic bearers of the evolved mutS- allele and the ancestral mutS+ allele directly against one another on a genetic background derived from the Ara+3 population at 10,000 generations. Because the ancestral and evolved strains bore no selectable genetic differences adjacent to mutS, construction of the mutS-/mutS+ strain pairs required a two-step process. In the first step, we mutagenized and screened a 10,000-generation mutS- isolate for insertions of a miniTn10 conferring cysteine auxotrophy. MiniTn10 mutagenesis was performed according to a method described by Kleckner et al. (1991); the particular transposon construct that we used encoded a kanamycin resistance factor. The cysCNDH operon maps to 61.9 min on the E. coli chromosome, near mutS at 61.5 (Berlyn et al. 1996); thus, some Cys- mutants will map close to mutS. KanR miniTn10 insertion mutants were screened for cysteine auxotrophy by gridding and replica plating them to DM agar (Carlton and Brown 1981) containing 4 g/liter of glucose and to the same medium supplemented with 40 mg/liter of cysteine.
In the second step, we subjected Cys- mutS- KanR miniTn10 insertion mutants to P1 transduction with a lysate prepared from the mutS+ ancestor REL606, using standard P1 methods (Miller 1992). We then selected prototrophic transductants by plating cells to DM agar and verified that these transductants no longer harbored a miniTn10 insertion by confirming that they were kanamycin sensitive. Finally, we identified Cys+ mutS+ cotransductants by screening the prototrophic clones for restoration of the wild-type mutation rate.
We tested the fitnesses of five independent mutS+ transductants in competition with the isogenic mutS- strain. Each fitness assay was conducted with fivefold replication. We employed resistance and sensitivity to bacteriophage T5 to distinguish the isogenic competitors. T5R mutS+ strains were selected by plating cells with excess T5 phage, and selective neutrality of the resistance phenotype itself was verified by competing every T5R mutant against its immediate T5S ancestor. The two phenotypes were enumerated in mixed culture by plating appropriate dilutions on LB agar and then replica plating to LB agar plates spread with ∼109 T5 phage. The numbers of T5S colonies were then obtained by subtraction. All other details of these competition experiments were as described above for competitions between Ara+ and Ara- strains.
RESULTS
Sequence analysis: Sequencing of the candidate mutator loci in populations Ara+3, Ara-2, and Ara-4 identified defective alleles of mutS (Ara+3) and mutL (Ara-2 and Ara-4). In population Ara+3, the mutator phenotype was caused by the insertion of a single guanine nucleotide at position 521. This frameshift was evident at the earliest time point at which the mutS- mutator was detected, immediately after its fixation, and at 10,000 generations. In populations Ara-2 and Ara-4, mutations were present in a single region of mutL (beginning at amino acid 61) in which the six-nucleotide sequence GCGCTG, encoding the amino acid sequence LA, is repeated in tandem three times in the ancestral strain. In population Ara-2 a GCGCTG repeat was added to this array; in population Ara-4 a GCGCTG repeat was deleted from this array. These in-frame mutations are located in a region that is important to the ATPase activity of MutL (Ban and Yang 1998; Ban et al. 1999), and a plausible hypothesis is that they impair this aspect of MutL function; indeed, other in-frame mutations in the ATPase domain of MutL confer a mutator phenotype (Spampinato and Modrich 2000; Tran and Liskay 2000). Three additional lines of evidence connect these mutations to the mutator phenotypes in populations Ara-2 and Ara-4: First, sequencing of mutL in isolates from these populations at the earliest time points at which the mutators were detected and immediately after the mutator substitutions revealed that these same mutant sequences corresponded to the mutator phenotypes. Second, although these mutator phenotypes can be eliminated by wild-type MutL or UvrD proteins expressed from a plasmid (Sniegowski et al. 1997), mutator clones of both populations are no more sensitive to UV exposure than wild-type clones (P. D. Sniegowski and A. Platt, unpublished data); this is contrary to what would be expected with loss of uvrD function (Ogawa et al. 1968; Siegel 1973). Rescue of the wild-type mutation rate by overexpression of uvrD from a plasmid may be a consequence of the fact that MutL and UvrD interact physically during mismatch repair (Hall et al. 1998; Yamaguchi et al. 1998). Finally, sequencing of the complete uvrD gene in a mutator isolate from population Ara-2 revealed only one minor amino acid difference from the published E. coli K12 wild-type sequence of uvrD, a serine-to-threonine substitution at amino acid position 719; it seems unlikely that this difference would cause a loss of uvrD function.
Relative fitnesses of mutS- and mutS+ alleles: Figure 1 illustrates the results of competitions between isogenic mutS+ and mutS- strains derived from a 10,000-generation clonal mutator isolate of population Ara+3. Three of the mutS+ transductants were statistically indistinguishable in fitness from their isogenic mutS- competitor, and no systematic tendency for the mutS+ transductants to be outcompeted by the mutS- strain was observed. Although there was significant heterogeneity in the competitive fitnesses of the mutS+ strains (F4,20 = 14.926, P < 0.0001), this could have been only an artifact of transduction because every transductant was wild type with respect to its mutation rate and hence carried the mutS+ allele.
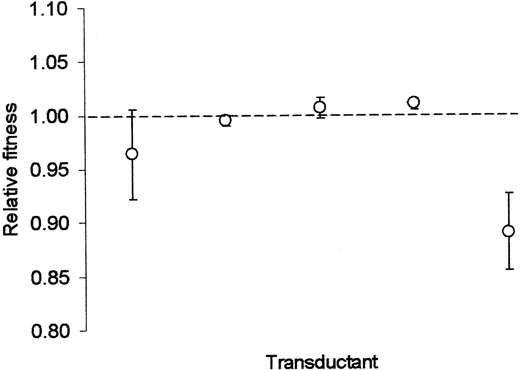
—Fitnesses of five independent mutS+ transductants in direct competition with an isogenic mutS-clone derived from population Ara+3 at 10,000 generations. Each reported fitness is the average of five replicate measurements. Error bars are 95% confidence intervals.
Trajectories of mutator frequencies during sweeps to fixation: Figure 2 shows that mutator frequencies rose at every successive polymorphic time point during the mutator sweeps. Mutator frequencies may have risen and fallen within these intervals, but these intermediate time points were not available for analysis. It is impossible to say with certainty when each mutator allele began its rise toward fixation (presumably starting from some low quasi-mutation-selection balance frequency; see Johnson 1999a; Sniegowski et al. 2000) and when each neared fixation. Nonetheless, each population was clearly polymorphic for a mutator allele for a considerable length of time: perhaps >1500 generations in the case of population Ara+3, 2000 generations in population Ara-2, and 2500 generations in population Ara-4.
Fitness evolution during the mutator sweeps: Figure 2 shows the fitnesses of mutator and wild-type clones, relative to the common ancestor, estimated at 500-generation intervals during the mutator sweeps. Inspection of these data reveals that two mutator clones—one in population Ara-2 at 3000 generations and one in population Ara+3 at 3000 generations—showed extremely low fitness. In all likelihood, these clones represented mutator lineages that had acquired strongly deleterious mutations and were not destined to contribute further to fitness evolution in these populations. Indeed, the fitnesses of these clones were substantially lower than any other observed fitnesses in the three populations at all time points assayed. The fitnesses of the sets of mutator clones at these time points were heterogeneous with a high level of significance (Ara-2: F2,8 = 41.197; P = 0.0003; Ara+3: F3,16 = 16.698; P < 0.0001). As can be seen from Figure 2, this heterogeneity was entirely due to the presence of the low-fitness clone in each case; fitness was significantly variable among mutator clones at other time points, but not due to the presence of a single low-fitness clone. The relevant F ratios (comparing variance among mutator clones to total variance among mutator replicates for a given time point) were much higher for the two time points with the low-fitness mutator clones than for all other time points with the exception of population Ara-4 at 8500 generations; at that time point, however, the mutator clone with lowest fitness was still well within the observed range of fitnesses for this population. For all of the above reasons, we have treated the low-fitness clones in populations Ara-2 and Ara-4 as outliers and have excluded them from the overall analyses of fitness evolution in these populations.
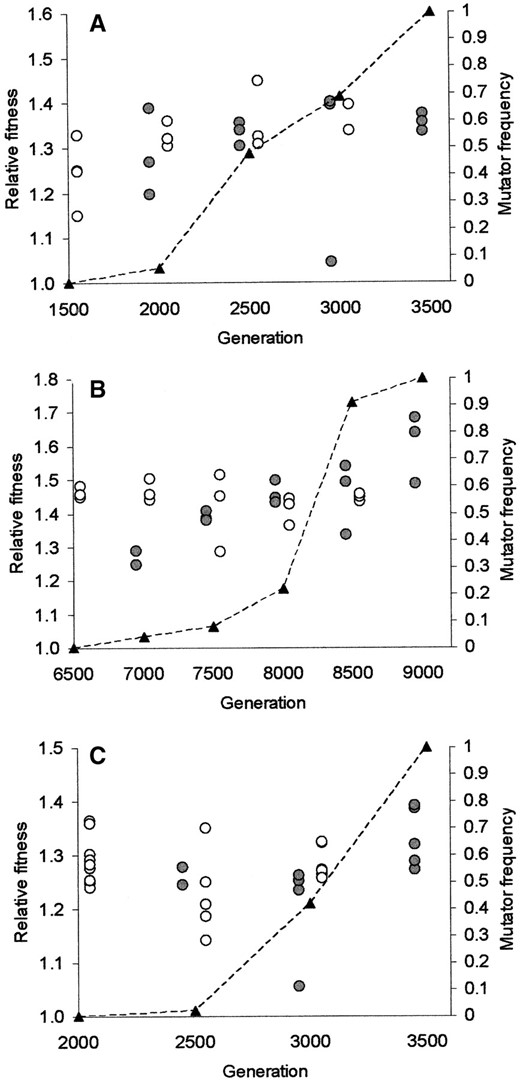
—Fitness measurements from each of the three populations for both wild-type (open) and mutator (shaded) phenotypes (left vertical axis), along with mutator frequency (dashed line, right vertical axis). Measurements from the Ara-2 population (A), the Ara-4 population (B), and the Ara+3 population (C) are shown. Each point represents the fitness of an independently isolated clone. As described in the text, fitness is measured relative to the common ancestor. For the first and last time points sampled in each population, the population was apparently fixed for one phenotype or the other, and thus the fitness of only one phenotype is shown.
Figure 2A plots the fitness estimates obtained for clones from population Ara-2. With the very-low-fitness clone excluded from the analysis, the average relative fitness in this population rose by ∼9% (from 1.24 at generation 1500 to 1.35 at generation 3500) during substitution of the mutator allele; the change is statistically significant (F1,16 = 6.175; P = 0.024). The mutator subpopulation fitness did not rise significantly over time (F3,16 = 1.566; P = 0.236). Notably, however, the wild-type subpopulation fitness rose significantly (from ∼1.24 at generation 1500 to ∼1.38 at generation 3000: F3,16 = 3.811; P = 0.031) even as the wild-type allele was being supplanted by the mutator.
Figure 2B plots the fitness estimates obtained for clones from population Ara-4. The average relative fitness of this population rose significantly (F1,19 = 6.932; P = 0.016) during the mutator sweep, in this case by ∼10% (from ∼1.46 at generation 6500 to ∼1.60 at generation 9000). The fitness of the wild-type subpopulation did not change significantly during the mutator sweep (F4,19 = 0.448; P = 0.773), whereas the mutator subpopulation fitness increased markedly and significantly, from ∼1.27 to ∼1.60 (F4,19 = 8.563; P = 0.0004).
Figure 2C plots the fitness estimates obtained for clones from population Ara+3. With the very-low-fitness clone excluded from the analysis, average relative fitness in this population did not change significantly during the mutator sweep between generations 2000 and 3500 (F1,22 = 1.967; P = 0.175). The wild-type subpopulation showed no significant increase in fitness during the mutator sweep (F2,22 = 2.757; P = 0.0854), but the mutator subpopulation showed a marginally nonsignificant gain in fitness (F2,22 = 3.176; P = 0.0614). Between generations 3000 and 3500, the mutator subpopulation gained substantially (∼11%) and significantly (two-tailed P < 0.05) in fitness as determined by multiple unplanned comparisons using Studentized distributions.
To investigate fitness evolution in population Ara+3 in more depth, we studied single clones previously isolated from this population by Lenski et al. (1991) at generations 1600, 1700, 1800, 1900, 2100, 2200, 2300, and 2400. Each clone’s fitness relative to the ancestor was measured with threefold replication. We found that the fitnesses of these clones were approximately equal to the value that we had obtained at the 2000-generation time point (data not shown). Overall, our extensive series of fitness measurements in population Ara+3 provided no evidence that its average fitness relative to the ancestor increased between generations 1600 and 3000; a regression of the average values for all of our fitness estimates in population Ara+3 against time during this interval was markedly nonsignificant (R2 = 0.008399; slope =-0.0000116; F1,9 = 0.0762; P = 0.7887).
The frequency of the mutS- allele increased from 0 to 42% between generations 2000 and 3000 in population Ara+3 despite the absence of fitness increase during this time. We therefore hypothesized that adaptive evolution in this population might have occurred without changes in fitness relative to the ancestor during this interval. To test this hypothesis, we measured the fitnesses of each of the isolated mutator clones (made Ara- by the enrichment procedure described above) in direct competition with each of the isolated wild-type clones in all of the possible pairwise combinations at 2500 generations and at 3000 generations. We then compared these observed fitnesses to expected values on the basis of the ratio of the mutator and wild-type fitnesses measured relative to the common ancestor. Although we observed some higher- and lower-than-expected fitnesses in clones of both phenotypes, none of these was significant when a correction for multiple tests (Ury 1976) was applied.
DISCUSSION
Theoretical studies have predicted that rare mutator mutations can hitchhike to fixation in asexual populations under a regime of soft selection (Painter 1975b; Taddei et al. 1997; Tenaillon et al. 1999). The dynamics and adaptive consequences of this process, however, have received limited experimental attention (Boe et al. 2000; Notley-McRobb and Ferenci 2000; Notley-McRobb et al. 2002). Here, we have described the first experimental study to examine the relationship between the spread of rare mutator mutations and the evolution of fitness. Our study builds on previous work in which we showed that 3 of 12 experimental populations of E. coli evolved 100-fold elevated mutation rates during 10,000 generations of evolution (Sniegowski et al. 1997).
Our main results are as follows:
A frameshift in mutS caused the mutator phenotype in population Ara+3, and alterations of repeat number in mutL apparently caused the mutator phenotypes in populations Ara-2 and Ara-4.
Each population was polymorphic for its mutator allele for well over 1000 generations.
Competitions between isogenic strains bearing the evolved mutS- allele and the ancestral mutS+ allele provided no evidence that MMR defects were favored by direct selection.
Populations Ara-2 and Ara-4 increased substantially and significantly in average fitness relative to the common ancestor while substituting their mutator allele; population Ara+3 did not, but its mutator subpopulation underwent a substantial and significant increase in average fitness during part of this time.
In population Ara-2, the wild-type subpopulation increased significantly in fitness as the mutator allele was sweeping.
Causes of mutator allele fixation: The amount of time required for the mutator sweeps (Figure 2) rules out simple genetic drift as their cause in all three populations. With no selection on linked loci, drift of a neutral allele to fixation is expected to take a number of generations roughly equal to the effective population size (Kimura and Ohta 1969). The effective size of our experimental populations is ∼3.3 × 107 (Lenski et al. 1991), yet the mutator alleles rose from rarity to fixation in each population in ≤3000 generations; natural selection must have driven the fixation of these alleles.
Fixation of the mutator alleles by direct selection was also not supported by the data. Competitions between isogenic mutS- and mutS+ strains (Figure 1) provided no evidence that MMR mutator alleles were intrinsically fitter; most of the mutS+ transductants tested showed no competitive inferiority to the isogenic mutS- strain (Figure 1). This result was perhaps not surprising; no evidence for direct positive effects of mutator alleles on fitness has been observed in previous studies (e.g., Chao and Cox 1983; Mao et al. 1997). A lack of selectable genetic markers near mutL prevented us from performing the transductions required to test experimentally for direct positive effects of the evolved mutL- alleles in populations Ara-2 and Ara-4. However, it appears that these mutant alleles were still being expressed (as indicated by the ability of both wild-type mutL and uvrD alleles to restore low mutation rates in these populations; Sniegowski et al. 1997), and this argues against any direct advantage to the cell due to energy or time savings. Indeed, early in the mutator sweep in population Ara-4 the isolated mutator clones had lower fitness than that of the contemporaneous wild-type clones (Figure 2B); this clearly indicates that the mutator allele itself did not confer a sufficient selective advantage at that time point.
The ∼10% fitness increases observed in populations Ara-2 and Ara-4 during their mutator sweeps are incommensurate with any measured and inferred direct effects of the mutator alleles on fitness described above, and they provide positive evidence that the mutators hitchhiked to fixation in these populations. Indeed, only indirect selective effects can explain the rise to fixation of the mutator allele in population Ara-4 given the clear inferiority of the sampled mutator strains at generation 7000.
The low fitness of the sampled mutator strains at generation 7000 in population Ara-4 nonetheless raises the question of how the mutator subpopulation reached its estimated frequency of 4% by this time. Genetic drift is an unlikely explanation: The selection coefficients indicated by the fitness data would overwhelm drift given the large effective size of these asexual populations. It is possible that the mutator subpopulation’s frequency and fitness were higher at some earlier time point but declined by generation 7000; this would be consistent with the dynamics sometimes observed in computer simulations of mutators in asexual populations (Taddei et al. 1997; Tenaillon et al. 1999). Unfortunately, there is no way to test this possibility because the populations were stored only at 500-generation intervals during this part of Lenski’s experiment. Whatever the explanation, we reiterate that the low fitness of the mutator isolates at 7000 generations in population Ara-4 is logically consistent only with subsequent fixation of the mutator allele by hitchhiking rather than by drift or direct selection.
The substantial (∼11%) and significant gain in the average fitness of the mutator subpopulation in population Ara+3 between generations 3000 and 3500 was consistent with hitchhiking. A perplexing result in population Ara+3, however, was the lack of any detectable fitness evolution between generations 1600 and 3000, a period in which the mutator rose in estimated frequency from 0 to 42%. The hypothesis that adaptive evolution was intransitive during this time—that it occurred without gains in fitness relative to the ancestor—was not supported by our competitions between contemporaneous mutator and wild-type clones from generations 2500 and 3000. It remains possible that both the mutator and wild-type populations increased in competitive ability relative to each other early in the mutator sweep without increasing their fitness relative to the ancestor; that is, that simultaneous intransitive fitness evolution took place in both subpopulations. If so, then fitness inequities need not have been revealed by competitions between contemporaneous mutator and wild-type clones in this population. One might still predict, however, that clones from later time points during the mutator sweep would be superior in direct competition against clones from earlier time points during the sweep. We are currently testing this prediction.
Dynamics of mutator substitution: As noted previously, populations Ara-2 and Ara-4 underwent ∼10% increases in average fitness (relative to the ancestor) during the 1000 generations or more required to effect their mutator sweeps. If the mutator alleles had simply hitchhiked with a unique beneficial mutation of 10% effect in each population, then their fixation would have been at least 10 times faster than that observed: Standard selection theory (Crow and Kimura 1970) indicates that a clone with a 10% fitness advantage will rise in frequency from 1 to 99% in ∼92 generations in a haploid population. Three factors could explain the slowness with which the mutators swept through the three populations: (i) clonal interference (Muller 1932; Gerrish and Lenski 1998; Gerrish 2001) caused by the acquisition of beneficial mutations in both mutator and wild-type subpopulations during the mutator sweeps, (ii) limitation in the rate and magnititude of beneficial mutations, and (iii) the effect of increased deleterious mutation on the mutator subpopulations.
Clonal interference is strongly supported by the fitness evolution observed in population Ara-2 (Figure 2A). In this population, the wild-type subpopulation underwent a substantial and significant fitness gain even as it was being supplanted by the mutator. This could have occurred only if the wild-type subpopulation had also been acquiring beneficial mutations during the mutator substitution event, consequently closing the fitness gap on the mutator and slowing its spread through the population as a whole. This sort of adaptive race between mutator and wild-type subpopulations was originally modeled analytically by Painter (1975b) and has been studied more extensively in computer simulation by Tenaillon et al. (1999). Our results provide the first experimental demonstration of its occurrence, and they provide an illustration of the retarding effect that clonal interference can have on the selectively driven substitution of an allele in an asexual population.
Limitation in the rate and/or magnitude of beneficial mutations is suggested by the fitness evolution observed in population Ara-4. In this case, the fitness of the wild-type subpopulation remained steady while the mutator was substituting whereas the average fitness of the mutator subpopulation increased substantially (Figure 2B). It is noteworthy that this population substituted its mutator allele more slowly and thousands of generations later in the Lenski experiment than did the other two populations. Previous studies have shown that the rate of fitness increase relative to the ancestor in all 12 Lenski experimental populations between 5000 and 10,000 generations was less than one-thirtieth of its value between 0 and 5000 generations (Lenski et al. 1991; Lenski and Travisano 1994; Elena and Lenski 1997). It is thus conceivable that the rarity and/or weakness of beneficial mutations slowed the sweep of the mutator allele and prevented the wild-type subpopulation, with its 100-fold lower mutation rate, from increasing its fitness.
The presence of two very-low-fitness clones in the samples from the mutator subpopulations suggests that deleterious mutations could also have affected the kinetics of mutator substitution. The fitness advantage of a beneficial allele in an asexual population is eroded by newly arising deleterious mutations (Peck 1994; Rice and Chippendale 2001), and one might well expect this same effect to slow the hitchhiking of a mutator. In the absence of beneficial mutations, the equilibrium fitness disadvantage of a mutator that raises the genomic mutation rate U by a factor m ⪢ 1 relative to the wild-type rate is ∼mU (reviewed in Sniegowski et al. 2000). The total genomic rate of deleterious mutation in E. coli B in a glucose-limited environment has previously been estimated as at least 0.0002 per generation by a mutation-accumulation experiment (Kibota and Lynch 1996), and thus the equilibrium fitness disadvantage of an E. coli mutator that raises the mutation rate 100-fold will be at least 2%. Johnson (1999a), however, has shown that this equilibrium disadvantage is approached very slowly in a large population; thus, where beneficial mutations accumulate in the mutator subpopulation, the effect of deleterious mutations on the kinetics of mutator hitchhiking may be weak.
Adaptive consequences of mutator substitution: Because asexual populations cannot generate genetic variation by recombination, their rate of adaptive evolution can be limited by the rate at which beneficial mutations arise. However, the adaptive significance of the tendency for high mutation rates to evolve in asexual populations is unclear (Sniegowski et al. 2000). Computer simulations have predicted that if a small number of beneficial mutations is to be substituted in an asexual population during a bout of adaptation, then a population that substitutes a mutator allele early in the bout of adaptation will sometimes reach this goal sooner (Taddei et al. 1997; Tenaillon et al. 1999). Studies of laboratory E. coli strains colonizing the guts of germ-free mice have provided some support for this prediction (Giraud et al. 2001). On the other hand, analytical models of adaptive evolution in asexual populations (Gerrish and Lenski 1998; Gerrish 2001) predict a diminishing increase in the rate of adaptation with increasing mutation rates. At higher rates of beneficial mutation, the overall speed of adaptation in an asexual population becomes limited more by intrapopulation competition between clones bearing different beneficial mutations than by the rate at which new beneficial mutations arise. Experimental work (de Visser et al. 1999) has shown that this clonal interference constrains the adaptive usefulness of a high mutation rate to situations in which beneficial mutations are rare. As de Visser et al. (1999, p. 405) have emphasized, “Mutators need not—and often will not—substantially accelerate adaptive evolution.”
In a population in which a mutator has just hitchhiked to fixation, the mutator subpopulation must have outcompeted the wild-type subpopulation. But does this necessarily mean that during the hitchhiking event the population adapted faster than equivalent populations in which mutator hitchhiking did not occur? With the data in this study and those previously reported by Lenski and Travisano (1994), it is possible to address this question in our experimental system; we can compare the increases in fitness relative to the ancestor attained by the Ara-2, Ara-4, and Ara+3 populations during their mutator sweeps to contemporaneous increases in the nine populations that never substituted mutators. Interestingly, populations Ara-2 and Ara-4 had the highest fitness gains during the relevant time interval (data not shown). This result supports the idea that adaptation is accelerated during mutator sweeps. The results from population Ara+3, however, do not support this idea: Eight of the nonmutator populations gained more fitness than population Ara+3 as its mutator was sweeping.
The above comparisons of fitness gains should be regarded with caution for several reasons. First, the fitness estimates of Lenski and Travisano (1994) that we used to make these comparisons were replicated only threefold for each time point in each population and thus are subject to substantial experimental uncertainty. Second, our estimates of the times required for the mutator sweeps are also subject to uncertainty in that we were constrained to working with population samples archived at 500-generation intervals. Third, our measures of fitness gain do not take into account the possibility of intransitive fitness evolution; this may have been important in population Ara+3, as discussed above. Finally, as de Visser et al. (1999) have pointed out, mutator substitution is more likely in populations that are adapting rapidly precisely because there are more opportunities for mutator alleles to hitchhike with beneficial alleles that are increasing in frequency. Thus, even if the data we used to make these comparisons had been highly reliable, the question of whether hitchhiking caused the increased rate of adaptation or vice versa would remain.
There is no evidence that evolving a high mutation rate increased the subsequent rate of adaptive evolution in these populations. Mutability and fitness relative to the ancestor do not correlate significantly in the first 10,000 generations of the Lenski long-term experiment, the time during which all of the mutators substituted (Sniegowski et al. 1997). The MMR mutator alleles and phenotypes are retained in the Ara-2, Ara-4, and Ara+3 populations to the 20,000-generation time point (A. C. Shaver and P. D. Sniegowski, unpublished data), but these populations are not significantly fitter than the nine wild-type populations at this time point (V. S. Cooper, unpublished data).
Persistence of mutator alleles and mutator phenotypes: Whatever their beneficial effects might be in the short term, mutator phenotypes must usually be disfavored over the long term; otherwise, all asexual populations would have high mutation rates, which is clearly not the case. Why, then, have high mutation rates been maintained in the Lenski experimental populations? The possibility that the mutator alleles have persisted simply because they cannot revert (LeClerc et al. 1996; Radman et al. 1999; Notley-McRobb et al. 2002) is ruled out by the sequence data we have presented here. Indeed, the fact that mutations in the same repeat region of mutL apparently caused the mutator phenotypes in populations Ara-2 and Ara-4 suggests that this region is a mutational hotspot that could revert rapidly.
We can suggest three evolutionary explanations for the persistence of high mutation rates in these populations. First, many revertants or modifiers that decrease the mutation rate will have no initial selective advantage, as they will arise on a genetic background that is just as loaded with deleterious mutations as the average mutator background. Because the effective size of these populations is very large, the buildup of mutational load in the majority mutator subpopulation will be very slow; hence, the selective advantage of such revertants or modifiers will grow very slowly. Second, adaptive improvement relative to the ancestor is still taking place in these populations after 20,000 generations, albeit slowly (Cooper and Lenski 2000), and it may mask the effect of increased deleterious mutation. Finally, one might speculate that intransitive fitness evolution is occurring continually in this system, producing a kind of Red Queen dynamic that favors mutator clones. Sorting out these possibilities is a matter for future research.
Acknowledgement
We thank R. E. Lenski for generous access to his strains and V. S. Cooper for the 20,000-generation fitness data from the Lenski long-term experiment. We are grateful to E. Fingerman, K. Lasnowski, A. Platt, and A. Wilmot for technical assistance; W. J. Ewens and P. Petraitis for statistical advice; and P. J. Gerrish, R. E. Lenski, and especially T. Johnson for insightful discussions. The manuscript was improved by thoughtful comments from P. D. Keightley and two anonymous reviewers. A.C.S. is supported by a predoctoral fellowship from the Howard Hughes Medical Institute. This research was supported by grants to P.D.S. from the Alfred P. Sloan Foundation (98-4-3-ME) and the National Science Foundation (DEB 9981518).
Footnotes
Communicating editor: P. D. Keightley
LITERATURE CITED

{kind=link}
{kind=link}