





Abstract
In Aspergillus nidulans, the uvsB gene encodes a member of the PI-3K-related kinase family of proteins. We have recently shown that UVSB is required for multiple aspects of the DNA damage response. Since the musN227 mutation is capable of partially suppressing defects caused by uvsB mutations, we sought to understand the mechanism underlying the suppression by cloning the musN gene. Here, we report that musN encodes a RecQ helicase with homology to S. pombe rqh1, S. cerevisiae sgs1, and human BLM and WRN. Phenotypic characterization of musN mutant alleles reveals that MUSN participates in the response to a variety of genotoxic agents. The slow growth and genotoxin sensitivity of a musN null mutant can be partially suppressed by a defect in homologous recombination caused by the uvsC114 mutation. In addition, we present evidence suggesting that MUSN may promote recovery from the DNA damage response. We suggest that a block to recovery caused by the musN227 mutation, coupled with the modest accumulation of recombination intermediates, can suppress defects caused by uvsB mutations. Finally, we report that another RecQ helicase, ORQA, performs a function that partially overlaps that of MUSN.
CELL cycle checkpoints play an integral role in the maintenance of genome integrity. During the past decade, research in yeasts and humans has revealed that cell cycle checkpoints function at the G1/S transition, the G2/M transition, and during S phase to prevent potentially catastrophic attempts to replicate or segregate damaged or incompletely replicated DNA (reviewed by Elledge 1996). Recent observations show that cell cycle checkpoints are directly integrated with the processes of DNA repair and recombination (Rhind and Russell 2000). Despite significant progress in the characterization of several DNA repair mutants in the filamentous fungus Aspergillus nidulans (Kafer and May 1998), the DNA damage response (DDR) remains largely uncharacterized in this organism. We have previously demonstrated that UVSB acts as a central regulator of the A. nidulans DDR, controlling multiple aspects of the response, including cell cycle arrest, inhibition of septation (cytokinesis), and induced mutagenesis (Hofmann and Harris 2000). UVSB is a member of the phosphatidylinositol 3-kinase-related kinase (PIKK) family of proteins. Members of this conserved family include Saccharomyces cerevisiae Mec1p, Schizosaccharomyces pombe Rad3p, and human ATM and ATR, all of which appear to orchestrate the DDR in these organisms. Mutation of the human ATM gene is responsible for ataxia telangiectasia, a neurodegenerative disease associated with a pronounced hypersensitivity to ionizing radiation and a strong predisposition to cancer (Savitsky et al. 1995).
The A. nidulans musN227 and musP234 mutations were isolated in a screen for mutants sensitive to the alkylating agent methyl methanesulfonate (MMS; Kafer and Mayor 1986). It was subsequently shown that both mutations are capable of partially suppressing the MMS sensitivity of the uvsB110 mutant (Kafer and Chae 1994). Given that uvsB encodes a PIKK related to ATM and, thus far, suppressors of the DNA damage sensitivity caused by mutations in these kinases have not been reported, we sought to understand the mechanism behind this suppression. We have previously shown that musN227 is capable of partially suppressing several of the defects caused by mutations in the uvsB gene, including the failure to inhibit septation in the presence of DNA damage (Hofmann and Harris 2000).
Here, we demonstrate that MUSN is a member of the RecQ family of DNA helicases that have been implicated in maintenance of genome integrity. Our results indicate that MUSN is involved in the response to both DNA damage and incomplete replication. Deletion of musN results in more severe genotoxin hypersensitivity compared to musN227, indicating that musN227 is a hypomorphic mutation. The slow growth and genotoxin sensitivity of a musN null mutant can be partially suppressed by a reduction in homologous recombination caused by the uvsC114 mutation. We suggest that MUSN, like the other RecQ helicases, may be involved in preventing promiscuous recombination. Moreover, on the basis of our analysis of septation in musN mutants, we propose that MUSN promotes recovery from the DDR. Accordingly, the suppression of uvsB mutants by musN227 may be related to the involvement of MUSN in promoting recovery from the DDR by regulating recombination. Furthermore, we indicate that, unlike S. cerevisiae and S. pombe, which possess only a single RecQ helicase, A. nidulans possesses a second family member (orqA; Appleyard et al. 2000). We show that expression of orqA can partially compensate for loss of MUSN, suggesting that these two RecQ helicases may have some overlap in function.
MATERIALS AND METHODS
Strains, media, and growth conditions: The following strains were used to carry out this study: AAH13 (musP234; pyrG89 yA2), AAH14 (ΔuvsB; pyrG89 yA2), AAH16 (ΔmusN; pabaA1 yA2), AAH17 (ΔuvsB; musN227; yA2), AAH18 (uvsB110; ΔmusN; yA2), AAH19 (uvsB110; ΔmusN; yA2), AAH22 (pabaA1 yA2 pyrG89; Pyr+ [pRGAMA1]), AAH23 (pabaA1 yA2 pyrG89; Pyr+ [pAH22]), AAH27 (uvsB110; pabaA1 yA2 pyrG89; Pyr+ [pRGAMA1]), AAH28 (uvsB110; pabaA1 yA2 pyrG89; Pyr+ [pAH22]), AAH31 (musP234; pyrG89 yA2; Pyr+ [pRGAMA1]), AAH32 (musP234; pyrG89 yA2; Pyr+ [pAH22]), AAS211 (musN227; pyrG89; chaA1), AAS315 (uvsB110; musN227; pabaA1; acrA1; actA1; riboB2 chaA1), AML8 (pyrG89 pabaA1; argB2; yA2), ASH162 (pyrG89 pabaA1 yA2), ASH201 (uvsB110; chaA1), ASH270 (uvsB110; pyrG89 pabaA1 yA2), ASH383 (musN227; chaA1), ASH581 (ΔmusN; uvsC114; yA2), ASH582, (ΔmusN; uvsC114; wA2), ASH583 (ΔmusN; uvsC114; yA2), ASH587 (ΔmusN; wA2), and ASH588 (ΔmusN; wA2). ASH581–ASH588 may contain additional markers.
The media used in this study were as described previously (Hofmann and Harris 2000). Bleomycin sulfate (Sigma, St. Louis) was resuspended at 5 units/ml and added to the appropriate concentration after autoclaving. Transformations and other genetic manipulations were performed as described previously (Harris et al. 1994). uvsB110ΔmusN double mutants were identified as a class of Pyr+ segregants with increased sensitivity to MMS compared to uvsB110 mutants. Δmus-NuvsC114 double mutants were identified as a class of Pyr+ segregants with decreased sensitivity to hydroxyurea (HU) and MMS compared to the ΔmusN mutant. The genotypes of all double mutants were confirmed by backcrosses to wild type.
Cloning of the musN gene: Strain AAS211 was transformed with a plasmid-based genomic library in the autonomously replicating, pyr-4-containing pRGAMA1 vector (Osherov and May 2000). Transformants capable of growing on minimal vitamin (MNV) media containing 0.01% MMS were recovered at 32°. Transformants were struck for single colonies on 1 mg/ml 5-fluoroorotic acid (5-FOA) to force loss of the pyr-4-containing plasmid. The resulting Pyr− colonies were then tested for their sensitivity to MMS to determine if the MMS resistance phenotype was plasmid dependent. Plasmid DNA was recovered from transformants exhibiting plasmid-dependent MMS resistance by extraction of fungal DNA and subsequent transformation into electro-competent Escherichia coli cells. Plasmid DNA was recovered from the resulting ampicillin-resistant colonies, and restriction digest analysis was performed. The recovered plasmids that contained inserts were retransformed into AAS211 to identify those that complemented the musN227 mutation. A single plasmid containing full complementing activity was identified and named pAH22. Plasmid pAH1 was constructed by cloning a 9-kb KpnI/XbaI fragment from pAH22 into the pBluescript vector (Stratagene, La Jolla, CA). Sequencing was performed by the Molecular Core Facility at the University of Connecticut Health Center. Available databases were searched using the BLAST algorithm at the National Center for Biotechnology Information (NCBI; Altschul et al. 1997). RT-PCR was used to identify intron sequences (Hofmann and Harris 2000).
Localization of the musN227 mutation: Strain AAS211 was transformed with various small, linear fragments of the wild-type musN gene (EcoRV, 4.2 kb; EcoRV, 3 kb; EcoRV, 2.5 kb; EcoRV, 1.8 kb; EcoRV/HindIII, 3 kb; EcoRV/HindIII, 2.5 kb; EcoRV/HindIII, 2 kb; and EcoRV/HindIII, 1.7 kb). Transformants were recovered at 32° on MNV containing 0.01% MMS. The smallest fragment capable of producing MMS-resistant colonies was determined to be the 1.7-kb EcoRV/HindIII fragment. Total RNA was isolated from wild type and musN227 strains as described previously (Hofmann and Harris 2000). RT-PCR of the region of musN containing this fragment was performed using primers designed to the musN cDNA sequence. Sequencing of three independently generated RT-PCR fragments from both wild type and musN227 was performed. All sequences were compared, and only mutations found in all three clones generated from musN227, but not in wild type, were reported.
Construction of the ΔmusN strain: To generate the musN replacement, the pAH49 plasmid was constructed as follows: Plasmid pAH1, which contains the entire musN gene, was digested with ClaI to remove a 2.4-kb fragment of the coding sequence. The remaining ClaI fragment, containing vector sequences and flanking musN sequences, was ligated to a 2-kb ClaI-digested PCR fragment of the Neurospora crassa pyr-4 gene. This pyr-4 fragment was generated using the primers oAH37 (5′ CCATCGATCTCCTTACGCATCTGTGCGG 3′) and oAH38 (5′ CCATCGATGCATCAGAGCAGATTGTACTG 3′) and the pyr-4-containing pRG3 plasmid as template. This construct was transformed into the wild-type strain ASH162. Pyr+ transformants were selected on MNV and subsequently tested for sensitivity to various concentrations of MMS and HU. Genomic DNA from transformants of interest was analyzed by Southern analysis to confirm that the desired double crossover integration had occurred.
Overexpression of musN: pRGAMA1 and pAH22 were transformed into strains ASH162, ASH270, and AAH13. ASH162 and ASH270 transformants were selected on MNV at 32°. Transformants were tested for sensitivity to MMS and HU. Transformants that received the pRGAMA1 plasmid served as a control to which the sensitivities of all other transformants were compared. AAH13 transformants were selected on MNV at 32° and tested for sensitivity to 0.015% MMS. AAH13 transformants were then struck on 5-FOA to force loss of the plasmid. Single FOA-resistant colonies were then retested on 0.015% MMS for sensitivity.
Viability assays: All viability assays were performed as previously described (Hofmann and Harris 2000). The percentage of viability is expressed as the number of surviving colonies on treated plates as compared to untreated control plates. For each treatment, platings were performed in quadruplicate, and the average number of survivors was calculated. All viability assays were repeated twice, and the data shown represent an average of the two independent experiments.
Microscopy: Conidiospores were grown, fixed, and stained on coverslips as previously described (Harris and Kraus 1998; Hofmann and Harris 2000). Calcofluor and Hoechst 33258 were used to stain septa and nuclei, respectively. Nuclear division kinetics, septation, polarization, and chromosome mitotic index (CMI) experiments were performed as previously described (Oakley and Osmani 1993; Hofmann and Harris 2000).
orqA constructs and transformations: Using the published sequence for recQ, primers oAH43 (5′ GATGGCCTGGAGAGCCTATC 3′) and oAH44 (5′ CTGCTGTTAGCTCATCAGGTC 3′) were designed to amplify the gene. PCR was performed on pools of cosmids from each A. nidulans chromosome (Brody et al. 1991). Pools producing the expected 2.6-kb PCR product were subdivided until a single cosmid was identified that produced the desired product. pL17C06 (chromosome II) and pW26D04 (chromosome III) both possess the entire RecQ helicase gene. A 5.5-kb KpnI/SphI fragment from pL17C06 was cloned into a KpnI/SphI-digested pRGAMA1 vector to create pAH54. The 5.5-kb KpnI/SphI fragment from pAH54 was then subcloned into the KpnI/SphI sites on pRG3, an integrating vector containing the N. crassa pyr-4 gene, to create pAH55. Strains AAS211 and AAH13 were then transformed with pAH54 and pAH55. Transformants were selected on MNV at 32° and tested for sensitivity to MMS (0.01% for AAS211 and 0.015% for AAH13).
The musN sequence can be found at GenBank, accession no. AF259396. The sequence that we refer to in this article as orqA has been previously described as recQ (Appleyard et al. 2000; GenBank accession no. AJ271844). We have chosen to rename this gene for two reasons. First, a series of rec mutants already exists in A. nidulans (Parag and Parag 1975), and second, according to conventional nomenclature for A. nidulans, the prefix describing a group of genes should be followed by a locus-specific suffix that starts with the letter A and proceeds through the alphabet. Accordingly, we have named the gene orqA, for other RecQ helicase A. In addition, upon further sequencing and RT-PCR of the 3′ end of orqA, we found that the open reading frame extends for an additional 150 bp beyond that originally reported. This amendment to the 3′ end of the sequence has been deposited in GenBank under accession no. AF368289.
RESULTS
musN encodes a RecQ helicase: A screen of a plasmid-based genomic library (Osherov and May 2000), constructed in the autonomously replicating pRGAMA1 vector, yielded a single plasmid capable of complementing the MMS sensitivity caused by the musN227 mutation. Sequencing of the insert and subsequent searches of the available databases revealed an open reading frame with similarity to the RecQ family of helicases, including Rqh1p (S. pombe), QDE-3 (N. crassa), Sgs1p (S. cerevisiae), BLM (Homo sapiens), and WRN (H. Sapiens; Table 1). musN encodes a protein of 1534 amino acids, with a predicted molecular weight of 173 kD. The homology with the members of the RecQ helicase family is confined primarily to the central helicase domain, which possesses the seven conserved helicase motifs, including the characteristic DExH sequence in motif II (Figure 1A). There is a potential nuclear localization signal in the carboxy terminus, similar to WRN and BLM, and acidic patches are present in the amino terminus (Karow et al. 2000b). In addition, there is a putative leucine zipper in the amino terminus (Figure 1A).
To verify that we had cloned the bona fide musN gene, and to determine the nature of the musN227 mutation, we transformed small, linear fragments of the gene into strain AAS211 and assessed their ability to repair the
Similarity of other RecQ helicases to MUSN
| Protein | Organism | BLAST scorea | % identity (% similarity) |
|---|---|---|---|
| QDE-3 | N. crassa | 1558 | 35 (50) |
| Rqh1p | S. pombe | 1367 | 40 (57) |
| Sgs1p | S. cerevisiae | 1309 | 46 (64) |
| BLM | H. sapiens | 1046 | 37 (53) |
| RecQL5 | H. sapiens | 723 | 42 (58) |
| ORQA | A. nidulans | 642 | 35 (51) |
| WRN | H. sapiens | 632 | 32 (50) |
| Protein | Organism | BLAST scorea | % identity (% similarity) |
|---|---|---|---|
| QDE-3 | N. crassa | 1558 | 35 (50) |
| Rqh1p | S. pombe | 1367 | 40 (57) |
| Sgs1p | S. cerevisiae | 1309 | 46 (64) |
| BLM | H. sapiens | 1046 | 37 (53) |
| RecQL5 | H. sapiens | 723 | 42 (58) |
| ORQA | A. nidulans | 642 | 35 (51) |
| WRN | H. sapiens | 632 | 32 (50) |
BLAST scores were calculated by the BLAST algorithm at NCBI using pairwise blastp alignments of each protein with MUSN (Tatusova and Madden 1999).
Similarity of other RecQ helicases to MUSN
| Protein | Organism | BLAST scorea | % identity (% similarity) |
|---|---|---|---|
| QDE-3 | N. crassa | 1558 | 35 (50) |
| Rqh1p | S. pombe | 1367 | 40 (57) |
| Sgs1p | S. cerevisiae | 1309 | 46 (64) |
| BLM | H. sapiens | 1046 | 37 (53) |
| RecQL5 | H. sapiens | 723 | 42 (58) |
| ORQA | A. nidulans | 642 | 35 (51) |
| WRN | H. sapiens | 632 | 32 (50) |
| Protein | Organism | BLAST scorea | % identity (% similarity) |
|---|---|---|---|
| QDE-3 | N. crassa | 1558 | 35 (50) |
| Rqh1p | S. pombe | 1367 | 40 (57) |
| Sgs1p | S. cerevisiae | 1309 | 46 (64) |
| BLM | H. sapiens | 1046 | 37 (53) |
| RecQL5 | H. sapiens | 723 | 42 (58) |
| ORQA | A. nidulans | 642 | 35 (51) |
| WRN | H. sapiens | 632 | 32 (50) |
BLAST scores were calculated by the BLAST algorithm at NCBI using pairwise blastp alignments of each protein with MUSN (Tatusova and Madden 1999).
musN227 mutation. We identified a 1.7-kb EcoRV/HindIII fragment that generated MMS-resistant transformants at low frequency, indicating that this fragment had likely repaired the musN227 mutation via a gene conversion event. Comparison of RT-PCR products generated from this region of wild-type and musN227 strains revealed a missing thymine residue in musN227 just following the helicase domain. Deletion of this thymine residue results in a frameshift that introduces a stop codon after 26 amino acids (Figure 1A). Thus, the musN227 mutation generates a truncated protein that retains the entire helicase domain, but is missing ~27% of the protein at the carboxy terminus.
The musN null mutant has a more severe phenotype than musN227: The phenotype of the musN227 mutant is relatively modest compared to those caused by mutations in other RecQ helicases. Accordingly, to test the possibility that the musN227 mutation does not represent the null phenotype, we created a strain containing a gene replacement. A plasmid was constructed in which ~50% of the musN coding sequence was deleted and replaced with the N. crassa pyr-4 gene as a selectable marker. The region deleted includes all of the conserved helicase domains, with the exception of domain VI (Figure 1A). Transformation of a wild-type strain with this plasmid generated the desired replacement strain as confirmed by Southern analysis and PCR. Phenotypic characterization revealed that ΔmusN has a more severe phenotype than musN227. ΔmusN displays much greater sensitivity to MMS and bleomycin compared to musN227 (Figure 2, A and B). Unlike musN227, the ΔmusN allele also causes sensitivity to HU and UV irradiation (Figure 2, A and B). In addition, ΔmusN displays slower growth and poorer conidiation when compared to either wild type or musN227 (Figure 2B). We have also noted that conidiospore polarization is delayed in ΔmusN mutants compared to wild type (data not shown).
The slow growth of the ΔmusN mutant could conceivably be caused by defects in cell cycle progression. In
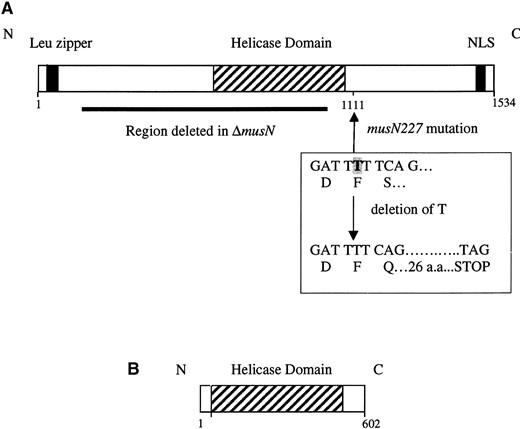
Molecular characterization of MUSN. (A) A schematic representation of MUSN. The location of the musN227 mutation is shown. The single base deletion results in premature truncation of the protein just following the helicase domain. The solid black line represents the region deleted in the ΔmusN strain. Numerical amino acid coordinates are shown. (B) A schematic representation of ORQA.
S. cerevisiae, sgs1srs2 double mutants arrest during mitosis as large, budded cells (McVey et al. 2001; although it is not a member of the RecQ family, Srs2p is a 3′ → 5′ helicase that displays partial overlap in function with Sgs1p; Rong and Klein 1993; Gangloff et al. 2000). To test whether loss of MUSN caused a similar defect, we examined the kinetics of nuclear division in ΔmusN mutants and found that there was a severe delay compared to wild type (Figure 3). In fact, ΔmusN divides with kinetics similar to that of wild type grown in the presence of the genotoxic agent diepoxyoctane (DEO). A comparison of the CMI in wild type vs. ΔmusN revealed a similar percentage of mitotic nuclei in both populations of cells (4%; n = 200). This suggests that mitotic
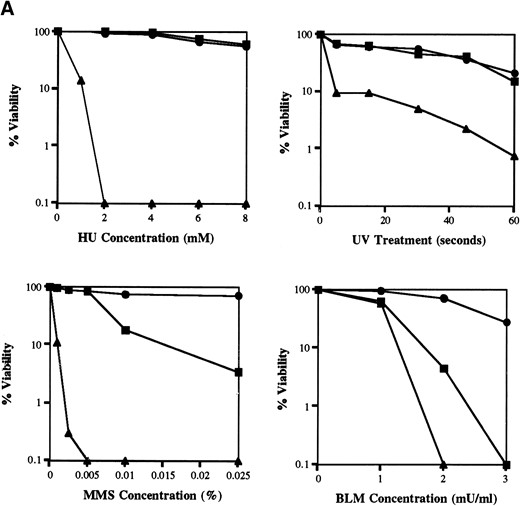
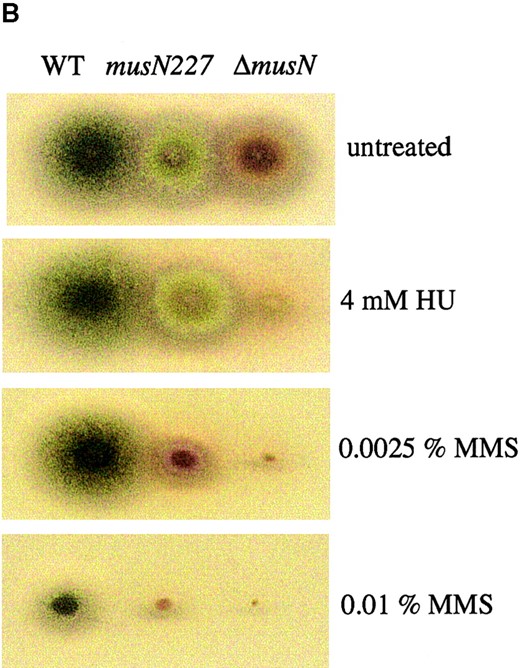
ΔmusN has a more severe phenotype than musN227. (A) Viability assays comparing the HU, UV, MMS, and bleomycin sensitivity of wild type (●; A28), musN227 (▪; ASH383), and ΔmusN (▴; AAH16). (B) A total of 104 conidia from wild type (A28), musN227 (ASH383), and ΔmusN (AAH16) were point inoculated onto MNV media and MNV containing 4 mm HU and 0.0025 or 0.01% MMS and incubated for 4 days at 32°.
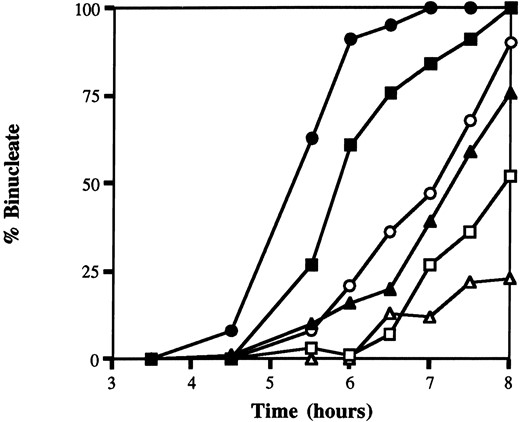
ΔmusN has a significant delay in nuclear division. Nuclear division kinetics of untreated wild type (●; A28), musN227 (▪; ASH383), and ΔmusN (▴; AAH16) strains, and DEO-treated wild type (○; A28), msuN227 (□; ASH383), and ΔmusN (▵; AAH16) strains.
progression is not affected by the ΔmusN mutation. Instead, ΔmusN mutants are presumably delayed in progression through S phase or G2. Since the ΔmusN phenotype is more severe than that caused by the musN227 mutation, we conclude that the musN227 gene product is partially functional.
ΔmusN does not suppress uvsB110: Since ΔmusN is a much stronger allele than musN227, we reasoned that it might also be a stronger suppressor of uvsB110. To address this possibility, we generated a uvsB110ΔmusN double mutant. Phenotypic analysis revealed that the double mutant exhibited increased sensitivity to MMS and HU compared to either single mutant (data not shown). Thus, instead of acting as a suppressor, ΔmusN enhances the defects caused by the uvsB110 mutation. Furthermore, this observation implies that the suppression of uvsB110 may be dependent on some function of the truncated musN227 protein. We also noted that the uvsB110ΔmusN double mutant exhibits a phenotype similar to the ΔmusN mutant with respect to its nuclear division kinetics and septation in response to DNA damage (data not shown), which suggests that the defects caused by ΔmusN are not caused by activation of the DNA damage checkpoint.
A defect in homologous recombination partially rescues the genotoxin sensitivity of ΔmusN: The A. nidulans uvsC gene encodes a RecA-like protein homologous to Rad51p from S. cerevisiae (van Heemst et al. 1997). Mutations in uvsC cause a reduction in homologous recombination (Ichioka et al. 2001). In S. cerevisiae, it has been demonstrated that loss of Rad51p rescues the slow growth of Δsrs2Δsgs1 double mutants (Gangloff et al. 2000). Presumably, an early block to homologous recombination prevents the accumulation of lethal recombination intermediates in the double mutant. To test whether homologous recombination is responsible for
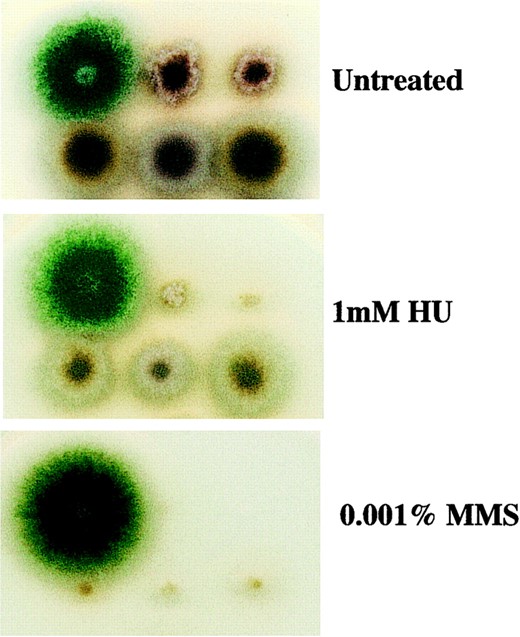
A defect in homologous recombination partially rescues the genotoxin sensitivity of ΔmusN. A total of 104 conidia were spotted onto MNV, MNV + 1 mm HU, and MNV + 0.001% MMS plates and incubated for 3 days at 32°. (Top, from left to right): wild type (A28), ΔmusN (ASH587), and ΔmusN (ASH588). (Bottom, from left to right): ΔmusNuvsC114 (ASH581), ΔmusNuvsC114 (ASH582), and ΔmusNuvsC114 (ASH583). Note that the ΔmusN strains used here (ASH587 and ASH588) possess different genetic backgrounds from the original ΔmusN strain (AAH16).
the slow growth and genotoxin sensitivity of ΔmusN mutants, we constructed a ΔmusNuvsC114 double mutant. Phenotypic analysis revealed that the double mutant was significantly less sensitive to MMS and HU than ΔmusN (Figure 4). Furthermore, whereas 90% of ΔmusN germlings arrested with one nucleus in the presence of 5 mm HU, 82% of ΔmusNuvsC114 double mutants recovered and completed nuclear division (as did 83% of wild-type spores). These observations suggest that the slow growth and genotoxin sensitivity of the ΔmusN mutant are caused by the accumulation of recombination intermediates.
Septation is delayed in musN227 mutants exposed to DNA damage: Since inhibition of septation is one of the effects triggered by activation of the DDR, we have used the percentage of septated hyphae in a population to measure the ability of a strain to recover and resume proliferation. After a prolonged exposure to DNA damage, wild-type hyphae eventually resume nuclear division and form septa (Table 2). In contrast, under identical conditions, fewer musN227 hyphae underwent septum formation (Table 2). Notably, the musN227 hyphae were longer than identically treated wild-type hyphae [average hyphal length (in micrometers): 108 ± 33 for wild type (n = 32) and 213 ± 56 for musN227 (n = 42)] and
musN227 exhibits a defect in recovery of septation following DNA damage
| Strain | Genotype | Septation index (%)a | |||
|---|---|---|---|---|---|
| 12 hr | 16 hr | ||||
| Untreated | + 0.025% DEO | Untreated | + 0.025% DEO | ||
| A28 | Wild type | 97 | 8 | 100 | 53 |
| ASH201 | uvsB110 | 95 | 44 | 100 | 63 |
| ASH383 | musN227 | 63 | 0 | 97 | 15 |
| AAS315 | uvsB110musN227 | 69 | 0 | 95 | 1 |
| Strain | Genotype | Septation index (%)a | |||
|---|---|---|---|---|---|
| 12 hr | 16 hr | ||||
| Untreated | + 0.025% DEO | Untreated | + 0.025% DEO | ||
| A28 | Wild type | 97 | 8 | 100 | 53 |
| ASH201 | uvsB110 | 95 | 44 | 100 | 63 |
| ASH383 | musN227 | 63 | 0 | 97 | 15 |
| AAS315 | uvsB110musN227 | 69 | 0 | 95 | 1 |
The septation index reflects the percentage of germlings possessing at least one septum (n = 200).
musN227 exhibits a defect in recovery of septation following DNA damage
| Strain | Genotype | Septation index (%)a | |||
|---|---|---|---|---|---|
| 12 hr | 16 hr | ||||
| Untreated | + 0.025% DEO | Untreated | + 0.025% DEO | ||
| A28 | Wild type | 97 | 8 | 100 | 53 |
| ASH201 | uvsB110 | 95 | 44 | 100 | 63 |
| ASH383 | musN227 | 63 | 0 | 97 | 15 |
| AAS315 | uvsB110musN227 | 69 | 0 | 95 | 1 |
| Strain | Genotype | Septation index (%)a | |||
|---|---|---|---|---|---|
| 12 hr | 16 hr | ||||
| Untreated | + 0.025% DEO | Untreated | + 0.025% DEO | ||
| A28 | Wild type | 97 | 8 | 100 | 53 |
| ASH201 | uvsB110 | 95 | 44 | 100 | 63 |
| ASH383 | musN227 | 63 | 0 | 97 | 15 |
| AAS315 | uvsB110musN227 | 69 | 0 | 95 | 1 |
The septation index reflects the percentage of germlings possessing at least one septum (n = 200).
had accumulated the same number of nuclei (modal average = 8), suggesting that the effect on septation was most likely not due to a general physiological defect. Furthermore, since uvsB110musN227 double mutants also fail to undergo septation under these conditions, the inability of musN227 mutants to septate following exposure to genotoxic agents is not caused by activation of the DNA damage checkpoint (Harris and Kraus 1998). Instead, we propose that septation is associated with recovery from the DDR and is dependent on proper MUSN function.
Increased expression of musN enhances the genotoxin sensitivity of both wild-type and uvsB110 mutants: If MUSN is involved in promoting recovery from the DDR, increasing its expression may cause sensitivity to DNA-damaging agents by terminating the response before the completion of DNA repair. To determine if presence of additional copies of musN had any affect on MMS sensitivity, a plasmid containing musN on the autonomously replicating pRGAMA1 vector was transformed into appropriately marked wild-type and uvsB110 strains. Both wild-type and uvsB110 transformants containing the musN plasmid exhibited a modest increase in sensitivity to MMS compared to control transformants containing the empty pRGAMA1 vector (Figure 5). Similarly, when tested for their response to HU, both sets of transformants were more sensitive (Figure 5). However, since the transformants were able to restrain septation in the presence of MMS or HU (data not shown), we conclude that neither the DNA damage nor replication checkpoints were abrogated. In contrast, overexpression of rqh1 in fission yeast causes a defect in the replication checkpoint, as cells exhibit premature segregation of chromosomes in the presence of HU (Davey et al. 1998).
The two A. nidulans RecQ helicases may have overlapping functions: A. nidulans possesses a second RecQ helicase that consists essentially of only the helicase domain and is more similar to human RecQL5 (Figure 1B; Table 1; Appleyard et al. 2000). To determine if a functional overlap exists between the two RecQ helicases, we tested the possibility that multicopy expression of orqA could rescue musN227. Transformation of musN227 with orqA on an integrating plasmid partially rescued the MMS sensitivity of musN227 (Figure 6). However, when orqA was supplied on an autonomously replicating vector, the MMS sensitivity of musN227 was not rescued. In fact, these transformants appeared to be sicker than control transformants that received the empty pRGAMA1 vector. This observation may indicate that overexpression of orqA is toxic in the musN227 mutant. In contrast, we were able to recover wild-type strains that had been transformed with multiple copies of orqA. These results suggest that MUSN and ORQA may have overlapping, but not identical, functions in the DDR.
Expression of musN partially complements musP234: Since the musN227 and musP234 mutants display similar
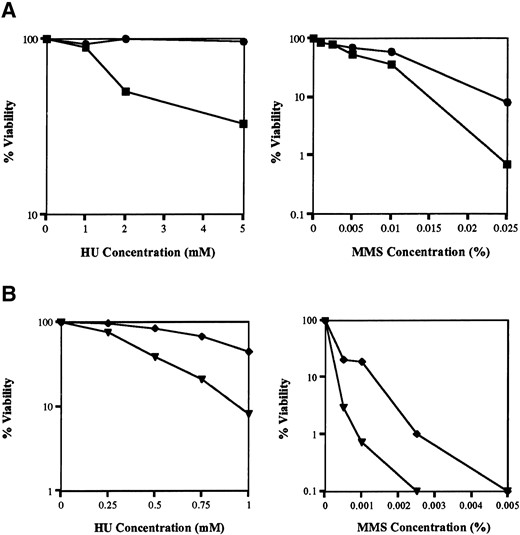
Increased expression of musN increases the MMS and HU sensitivity of wild type. Viability assays compare the MMS and HU sensitivities of (A) wild type + pRGAMA1 vector (●; AAH22) and wild type + pRGAMA1+musN (▪; AAH23) and (B) uvsB110 + pRGAMA1 (♦; AAH27) and uvsB110 + pAH22 (▾; AAH28).
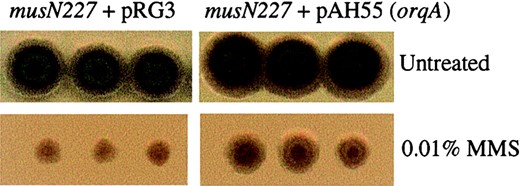
An extra copy of orqA partially rescues the MMS sensitivity of musN227. On the left are patches of the musN227 strain transformed with pRG3 vector alone on CM + Tx plates (top) and CM + Tx + 0.01% MMS (bottom). On the right are patches of the musN227 strain transformed with a copy of orqA on an integrating vector (pAH54).
phenotypes, including the ability to suppress uvsB mutations (Kafer and Chae 1994), we hypothesized that MUSN and MUSP may perform a related function. Therefore, we sought to determine if multiple copies of musN could rescue the musP234 phenotype. An autonomously replicating plasmid containing the entire musN gene was transformed into musP234. All transformants displayed enhanced growth on 0.015% MMS compared to transformants that received the vector alone (Figure 7). The partial suppression was shown to be plasmid dependent, as loss of the plasmid resulted in loss of MMS resistance (Figure 7).
Since the musN and musP genes appear to control related functions, and orqA encodes a RecQ helicase whose function may overlap with that of musN, we tested the possibility that musP234 may be a mutation in orqA. However, transformation of musP234 mutants with orqA on an autonomously replicating plasmid did not result in complementation. In addition, we were able to amplify the orqA gene from a musP234 strain as a single, intact PCR product of the appropriate size. Since the musP234 mutation is presumably caused by a translocation breakpoint (Kafer and Chae 1994), this observation further supports the notion that it is not a mutation in the orqA gene.
DISCUSSION
A. nidulans possesses multiple RecQ helicases: We have shown here that MUSN is a member of the RecQ family of DNA helicases. It possesses several features characteristic of RecQ helicases (Karow et al. 2000b), including (i) the DExH sequence in helicase motif II, (ii) the helicase-related (HR) domain within the C terminus, and (iii) short acidic stretches within the N terminus. Among the RecQ helicases, MUSN shows greatest homology to S. cerevisiae Sgs1p, S. pombe Rqh1p, and human BLM. Within these family members, there is little homology outside of the helicase motifs and the HR domain. Moreover, other than a canonical nuclear localization sequence at the extreme C terminus and a putative leucine zipper at the N terminus, analysis of
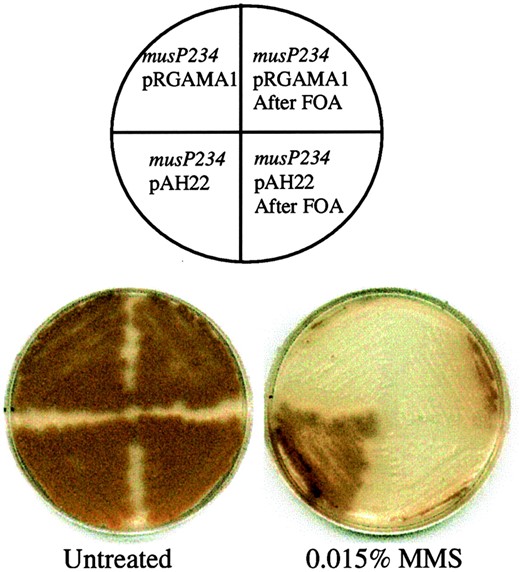
Increased expression of musN rescues the MMS sensitivity of musP234. Comparison of the MMS sensitivity of musP234 transformed with an empty pRGAMA1 vector (AAH31) or pRGAMA1 containing the musN gene (AAH32). Quadrants on the right represent the transformants after being cured of the plasmids using 5-FOA.
the MUSN sequence has yielded little insight into the function of these nonconserved regions.
Surprisingly, A. nidulans possesses a second RecQ helicase, ORQA, that consists of the seven conserved helicase motifs flanked by short N-terminal and C-terminal extensions (Appleyard et al. 2000). Within the RecQ helicase family, ORQA shares greatest homology to human RecQL5. We have found that the musN and orqA genes are both expressed in growing hyphae (data not shown), thus demonstrating that A. nidulans expresses divergent members of the RecQ helicase family in the same cell type. Furthermore, an additional sequence showing homology to RecQ helicases is present on A. nidulans chromosome IV (http://aspergillus-genomics.org/; contig 2000Sep121643_600). Although this sequence shows weak homology to a putative telomere-associated RecQ helicase found in Ustilago maydis (Sanchez-Alonso and Guzman 1998), it is not clear that it is expressed. Nonetheless, unlike the model yeasts S. cerevisiae and S. pombe, which each possess a single RecQ family member, at least two distinct RecQ helicases are found in the A. nidulans proteome. The presence of multiple RecQ helicases may be a common feature of the fungal proteome, since another filamentous fungus, N. crassa, also possesses two distinct family members (Cogoni and Macino 1999).
The yeast RecQ helicases Sgs1p and Rqh1p appear to function in several aspects of chromosomal DNA metabolism (Frei and Gasser 2000; Karow et al. 2000b). In contrast, in those organisms that possess multiple RecQ helicases (i.e., metazoans and plants; Mohaghegh and Hickson 2001), it is not clear if each is endowed with a similar broad range of functions or if each performs a specialized function that is required only under certain circumstances. We suggest that filamentous fungi may be useful models for distinguishing between these possibilities. Although musN and orqA are both expressed in hyphae, and extra copies of orqA can partially suppress the MMS sensitivity caused by the musN227 mutation, we favor the latter possibility for two reasons. First, the larger size of MUSN relative to ORQA, which essentially consists of only the helicase domains, indicates that it may be involved in a broader range of functions. For example, MUSN may be a component of functional complexes that do not contain ORQA. Second, in N. crassa, the larger RecQ helicase (QDE-3) is required exclusively for post-transcriptional gene silencing, whereas the smaller one is most likely involved in the DNA damage response (Cogoni and Macino 1999).
The role of MUSN in the DNA damage response: The sensitivity of the ΔmusN mutant to a diverse range of DNA-damaging agents implies that MUSN performs a critical function during the DNA damage response. We have previously reported that septum formation is an accurate readout for the status of the DNA damage response in A. nidulans (Harris and Kraus 1998). When chromosomal DNA metabolism is perturbed, either by pharmacological means (i.e., HU, MMS, or DEO) or by a mutation (i.e., sepB3, sepJ1, or bimA10), septum formation is blocked (Harris and Kraus 1998; Wolkow et al. 2000). The block depends upon checkpoint signals, since checkpoint mutants such as uvsB110 undergo septation despite the presence of DNA damage (Harris and Kraus 1998; Kraus and Harris 2001). Our results show that the hypomorphic musN227 mutation causes a block to septum formation in hyphae that have been chronically exposed to DNA damage. Although we cannot eliminate the possibility that this is an indirect effect (i.e., caused by abnormal cellular physiology), we note that musN227 hyphae displayed robust growth and continued to undergo nuclear division at a rate comparable to wild type. The observation that the septation block is not alleviated by the uvsB110 mutation raises two important points. First, it demonstrates that MUSN is involved in the regulation of the DNA damage response. If the septation block were due simply to the accumulation of DNA damage caused by compromised MUSN function, it would have been suppressed by the uvsB110 mutation (Harris and Kraus 1998). Second, it implies that MUSN acts downstream of UVSB in the pathway that regulates septum formation in response to DNA damage. We propose that septum formation is associated with recovery from the DNA damage response, and it is blocked in musN227 mutants that have been exposed to genotoxic agents because MUSN is required for recovery.
The sensitivity of the ΔmusN mutant to several DNA-damaging agents is suppressed by a mutation in uvsC, which encodes a Rad51 ortholog required for homologous recombination (van Heemst et al. 1997; Ichioka et al. 2001). The ability of Rad51 mutations to suppress defects caused by the functional inactivation of RecQ helicases has also been noted in other organisms (Gangloff et al. 2000; McVey et al. 2001). These observations suggest that the DNA damage sensitivity caused by musN mutations is caused by the accumulation of lethal recombination intermediates. Indeed, RecQ helicases appear to play a general role in preventing promiscuous recombination during the processing of damaged DNA (Stewart et al. 1997; Harmon and Kowalczykowski 1998; Gangloff et al. 2000; Myung et al. 2001). They may do so by removing Holliday junctions via reverse branch migration (Harmon and Kowalczykowski 1998; Karow et al. 2000a). Since the elimination of Holliday junctions is likely to be a prerequisite for recovery from the DNA damage response, this could account for the apparent role of MUSN in recovery. Moreover, it could explain the inability of musN227 mutants to septate once they have been exposed to DNA-damaging agents, since they would effectively be trapped in the DNA damage response.
Homologous recombination is a preferred mechanism for the repair of double-strand breaks (DSBs; Paques and Haber 1999). In support of this notion, Rad55p, which plays an important role in the strand exchange reaction (Sung 1997), appears to be activated in a Mec1p-dependent manner early in the S. cerevisiae DNA damage response (Bashkirov et al. 2000). However, mitotic recombination is typically not associated with crossing over (Paques and Haber 1999), which could lead to deleterious chromosomal rearrangements. For example, although mutations in PIKKs cause increased recombination, the majority of the events appear to be nonreciprocal (Bashkirov et al. 2000). Accordingly, the presence of Holliday junctions during recombination-mediated repair of DSBs may be dangerous. Thus, in addition to promoting recovery, the removal of Holliday junctions by RecQ helicases such as MUSN may ensure that recombination intermediates are funneled into pathways that do not lead to crossing over (i.e., synthesis-dependent strand annealing; Paques and Haber 1999).
In addition to its role in the DNA damage response, the HU sensitivity of the ΔmusN mutant suggests that MUSN is required when DNA replication is perturbed. In S. cerevisiae, Sgs1p appears to perform a critical signaling function in the response to stalled replication forks (Frei and Gasser 2000). Although we cannot rule out a similar function for MUSN, the observation that the ΔmusN mutant does not septate in the presence of low concentrations of HU (data not shown) suggests that the S phase checkpoint is intact. In contrast, checkpoint mutants such as uvsB, uvsD, and sntA form septa under similar conditions (Harris and Kraus 1998; Kraus and Harris 2001). Instead, as suggested for other RecQ helicases (Stewart et al. 1997; Harmon and Kowalczykowski 1998; Gangloff et al. 2000; Myung et al. 2001), we propose that MUSN protects stalled replication forks from undergoing promiscuous recombination events that could trigger genome instability.
Suppression of uvsB defects by the musN227 mutation: The ability of the musN227 mutation to suppress the genotoxin sensitivity of uvsB and uvsD mutants initially suggested that suppression was caused by inactivation of a pathway that normally antagonizes the functions(s) of UVSB and UVSD (Hofmann and Harris 2000). However, we still cannot eliminate the possibility of a direct interaction between MUSN and UVSB. Moreover, it has recently been demonstrated that human homologs of MUSN and UVSB (BLM and ATM, respectively) coexist within a large supercomplex of proteins that respond to DNA damage (Wang et al. 2000). Such a complex may also exist in A. nidulans, and musN227 may suppress uvsB mutations by affecting its assembly or function.
Understanding the basis of the genetic interaction between uvsB and musN is further complicated by the observation that the ΔmusN mutation does not suppress, but instead enhances, the sensitivity of uvsB mutants to MMS and HU. Accordingly, since musN227 appears to be a hypomorphic mutation, we suggest that MUSN function can be compromised only to a certain extent to allow suppression of uvsB defects. For example, a modest decrease in MUSN function (i.e., musN227) may suppress uvsB mutations by causing a small increase in the number of Holliday junctions, but a more severe decrease in function (i.e., ΔmusN) may trigger a potentially lethal increase in promiscuous recombination.
We propose two related models to explain the suppression of uvsB defects by the musN227 mutation. In the first model, suppression is an indirect effect of a block to recovery. By prolonging the DNA damage response, the musN227 mutation could allow more time for the repair of potentially lethal genotoxic damage. Indeed, since the level of homologous recombination associated with crossing over is elevated in musN227 mutants (Zhao and Kafer 1992), the accumulation of Holliday junctions could facilitate the repair of such damage. The second model is based on the premise that MUSN may be a substrate for phosphorylation by UVSB (note that, by analogy to the model yeasts, A. nidulans is likely to possess a second ATM-related PIKK capable of modifying MUSN; Karow et al. 2000b). In support of this idea, MUSN possesses two clusters of consensus PIKK phosphorylation sites (Kim et al. 1999), one in the extreme N terminus and another in the extreme C terminus. Perhaps UVSB sets the stage for recovery from the DNA damage response by modifying MUSN and thus influencing its activity. Since the potential cluster of PIKK phosphorylation sites located at the C terminus would be missing from the musN227 gene product, it may be less efficiently modified by PIKKs, thus delaying recovery.
Our observation that a mutation in a RecQ helicase partially suppresses the growth defects and genotoxin sensitivity of an ATM homolog may have clinical relevance. For example, mutations in human RecQ helicases such as BLM, WRN, or RecQL5 could serve as phenotypic modifiers (Hartman et al. 2001) of the disease ataxia telangiectasia. This could account for some of the clinical heterogeneity associated with this disorder. In addition, the apparent susceptibility of atm+/− heterozygotes to cancer (Swift et al. 1987, 1990; see also Fitz-Gerald et al. 1997) could be influenced by their genotypes at BLM, WRN, or RecQL5.
Acknowledgement
We thank Greg May for providing the pRG3-AMA genomic library. We also thank Peter Kraus and Tom Wolkow for providing helpful comments that improved this manuscript. This work was supported by awards from the A-T Children's Project and the American Cancer Society (RPG-99-214-01-MBC).
Footnotes
Communicating editor: J. J. Loros
LITERATURE CITED

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}