





Abstract
The centromere is essential for proper segregation and inheritance of genetic information. Centromeres are generally regulated to occur exactly once per chromosome; failure to do so leads to chromosome loss or damage and loss of linked genetic material. The mechanism for faithful regulation of centromere activity and number is unknown. The presence of ectopic centromeres (neocentromeres) has allowed us to probe the requirements and characteristics of centromere activation, maintenance, and structure. We utilized chromosome derivatives that placed a 290-kilobase “test segment” in three different contexts within the Drosophila melanogaster genome—immediately adjacent to (1) centromeric chromatin, (2) centric heterochromatin, or (3) euchromatin. Using irradiation mutagenesis, we freed this test segment from the source chromosome and genetically assayed whether the liberated “test fragment” exhibited centromere activity. We observed that this test fragment behaved differently with respect to centromere activity when liberated from different chromosomal contexts, despite an apparent sequence identity. Test segments juxtaposed to an active centromere produced fragments with neocentromere activity, whereas test segments far from centromeres did not. Once established, neocentromere activity was stable. The imposition of neocentromere activity on juxtaposed DNA supports the hypothesis that centromere activity and identity is capable of spreading and is regulated epigenetically.
THE metazoan centromere was first identified cytologically as the region of the primary constriction of a metaphase chromosome (Flemming 1880). Later, it was determined that this constriction corresponds to the site of kinetochore formation and polar spindle fiber attachment and is causative in faithful chromosome segregation in mitosis and meiosis (Pluta et al. 1995; Rieder and Salmon 1998; Dobie et al. 1999). Structurally, the genetic activity of these eukaryotic centromeres is located within large arrays of repetitive DNA (Willard 1990; Choo 1997b), and evidence suggests that chromosome inheritance functions may map to numerous redundant elements within each constriction (Zinkowski et al. 1991). This architecture has been referred to as a “regional” centromere (Pluta et al. 1995; Wiens and Sorger 1998). Analysis of the molecular structure of these regional centromeres, which are large and contain hundreds to thousands of kilobases of repetitive DNA, has lagged behind molecular analysis of “point” centromeres (e.g., those of Saccharomyces cerevisiae), whose activities have been mapped to <150 bp (Hyman and Sorger 1995; Pluta et al. 1995; Sunkel and Coelho 1995; Espelin et al. 1997).
The ubiquity of simple repeat DNA at regional centromeres in most metazoa has fostered the belief that these sequences are responsible for determining centromere numbers and sites (centromere identity; Harrington et al. 1997; Losada et al. 1997; Ikeno et al. 1998; Willard 1998; Platero et al. 1999; Abad et al. 2000; Koch 2000). Approaches for de novo artificial mammalian chromosome construction have been based on this interpretation (Harrington et al. 1997; Ikeno et al. 1998; Willard 1998). However, the sufficiency of repetitive DNA for centromere identity and activity has faced two strong criticisms. First, only a subset of an apparently homogenous block of satellite repetitive DNA acts as a centromere (Sun et al. 1997; Vafa and Sullivan 1997; Sullivan and Willard 1998). Corollaries to this are the observations that centromeres may be turned off without apparent alteration in the sequence of the centromeric heterochromatin (Steiner and Clarke 1994; Ekwall et al. 1997; Page and Shaffer 1998; Sullivan and Willard 1998), and arrays of alphoid repeat DNA integrated into human chromosomes are not able to assemble kinetochores (Warburton and Cooke 1997). Second, centromeres may be activated at sites in the genome that are devoid of repetitive DNA, termed neocentromeres (Voullaire et al. 1993; Murphy and Karpen 1995b; Vig et al. 1996; du Sart et al. 1997; Williams et al. 1998; Barry et al. 1999, 2000). Hence, although repeated DNA sequence is found at most centromeres, these DNA sequences are neither sufficient nor necessary for centromere activity.
An alternative model for centromere identity and activity involves chromatin structure and epigenetic regulation (Steiner and Clarke 1994; Murphy and Karpen 1995b, 1998; Choo 1997a, 1998; Ekwall et al. 1997; Karpen and Allshire 1997; Wiens and Sorger 1998). In this model, centromere identity is determined and propagated by a component or modification that is independent of the sequence of the underlying DNA (Karpen and Allshire 1997; Murphy and Karpen 1998). Components that could be responsible for centromere identity and activity include chromosome folding/packaging, association of centromere-specific protein or nucleic acid factors, or chemical modification of either DNA or ubiquitous chromatin-associated factors (Russo et al. 1996).
Epigenetic mechanisms for non-Mendelian inheritance exist in many phyla, including parent-of-origin effects on gene expression or chromosome behavior (Spofford 1976; Golic et al. 1998), limited mating-type switching (Grewal and Klar 1996; Thon and Friis 1997), telomere identity (Biessmann et al. 1990; Donaldson and Karpen 1997; Ahmad and Golic 1999), and paramutation (Hollick et al. 1997). In Drosophila, the observation of variegated position effects demonstrates that gene regulation is capable of maintaining clonality of gene expression or repression, even through numerous generations (Spofford 1976; Dorn et al. 1993; Cavalli and Paro 1998). The common feature of these systems is the differential regulation of identical genetic information, suggesting that chromosomes carry information that is not determined by sequence alone. The epigenetic nature of the centromere was first proposed for Schizosaccharomyces pombe (Steiner and Clarke 1994) and was recently supported by work that has identified centromere-specific factors (Allshire et al. 1994, 1995; Ekwall et al. 1999; Partridge et al. 2000). In these studies, centromeres adopt one of two states (low or high activity) without alteration of the underlying DNA.
One of the strongest arguments in favor of epigenetic determination of regional centromere identity is the existence of neocentromeres, which reflects the ability of normally noncentromeric DNA to acquire and maintain centromere activity (Voullaire et al. 1993; Murphy and Karpen 1995b; Vig et al. 1996; du Sart et al. 1997; Faulkner et al. 1998; Williams et al. 1998; Barry et al. 1999, 2000; Saffery et al. 2000). Sequence information from a human neocentromere and the cognate, centromerically inactive predecessor DNA has irrefutably shown that sequence polymorphism is not sufficient to explain differences in centromere activity (Barry et al. 2000; Maggert and Karpen 2000). Two models have been proposed to account for the formation of neocentromeres in humans and Drosophila. In the first model, centromere activity spreads from extant centromeric regions to neighboring DNA, where it imparts a stable centromere state (Murphy and Karpen 1995b, 1998; Williams et al. 1998). This model predicts that sequences neighboring an active centromere will be more likely to acquire a neocentromere than will sequences elsewhere in the genome. The second model posits that many sites along a chromosome are intrinsically capable of exhibiting centromere activity and nucleating a kinetochore, but are repressed in cis by a more dominant centromere (Choo 1997a, 1998; Platero et al. 1999). This model predicts that sequences with innate centromere activity should manifest centromere activity when removed from repression and that proximity to a functional centromere is not necessary for neocentromere activation.
We have distinguished between these models by comparing three substrate chromosomes in their ability to generate fragments that exhibit neocentromere activity. The structures and stabilities of nearly 100 chromosomal derivatives have been analyzed. Structurally acentric, neocentromeric derivatives were recovered only when the liberated fragment was previously juxtaposed to an active centromere. These data support the hypothesis that centromere identity and activity can spread to an unrelated, linked sequence. The stability of neocentromere activity shows that epigenetic factors play a role in initiating and maintaining centromere activity in Drosophila.
MATERIALS AND METHODS
Fly stocks: Fly stocks, mutations, and chromosome aberrations are as described in Lindsley and Zimm (1992) or at Flybase (http://flybase.bio.indiana.edu/). The mu-2c e ry stock was a gift from Dr. James Mason. The CyO, SUPorP{w+ y+} stock was a gift from Dr. Pamela Geyer. Dp8-23, Dpγ238, and Tγ1337 have been previously described (Le et al. 1995). Y SX.Y L, In(1)EN is abbreviated X^Y. All flies were raised on cornmeal food at 25° in glass bottles supplemented with dry active yeast.
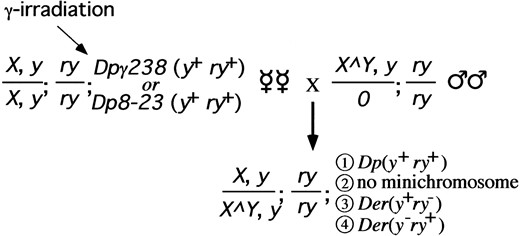
γ-Irradiation: Virgin female flies of the appropriate genotype (see below) were aged for 3–5 days and then subjected to 500 rad of γ-radiation from a 60Co source. Irradiations were done in plastic tubes (13-mm ID, 19-mm OD, 97-mm height). Males of the appropriate genotype (see below) were added, and flies were allowed to mate and lay eggs for 4 days. The flies were transferred to fresh bottles every 3 days and were discarded 16 days after irradiation. Progeny were scored on days 15 and 20.
Screen of Dpγ238 and Dp8-23: Female virgins were y1; mu-2c e ry; Dpγ238, y+ ry+ or y1; mu-2c e ry; Dp8-23, y+ ry+, and males were Y SX.Y L, In(1)EN, y1; ry506 (Figure 3). All male progeny were of genotype X/0 and thus phenotypically sterile (Bridges 1916). All female progeny were Y SX.Y L, In(1)EN, y1/y1 and so carried Y heterochromatin to suppress position effects on the yellow+ and rosy+ genes (Spofford 1976; Murphy and Karpen 1995b) in order to allow all derivatives to be detected. F1 female progeny that were phenotypically yellow+ rosy+ or yellow− rosy− were discarded. Female progeny that were yellow+ rosy− or yellow− rosy+ were outcrossed to y1; ry506 males. F2 and F3 male progeny were outcrossed to y1; ry506 virgins to establish y1; ry506; Dp stocks. mu-2c e ry chromosomes were not selected against and had no effect on the activity of the neocentromere (data not shown).
Screen of Tγ1337: Female virgins were y1; T(2; Dp8-23) γ1337, ry+/CyO, SUPorP{w+ y+}; mu-2c e ry, and males were Y SX.Y L, In(1)EN, y1; ry506 (Figure 4). SUPorP was used to mark the CyO chromosome with yellow+ in order that it may more easily be scored. F1 female progeny that were yellow+ rosy− or yellow− rosy+ were discarded. Female progeny that were yellow− rosy− or yellow+ rosy+ were outcrossed to y1; Sp/SM1; ry506 males. F2 Sternopleural male offspring were outcrossed to y1; Sp/CyO, SUPorP{w+ y+}; ry506 virgins. F3 Sternopleural+ male offspring were collected and outcrossed to y1; Sp/CyO, SUPorP{w+ y+}; ry506 virgins. F4 Sternopleural+ male and female offspring were bred inter se to establish a stock of y1; Df(Tγ1337)/CyO, SUPorP{w+ y+}; ry506. mu-2c e ry chromosomes were not selected against, as mentioned above.
Transmission and brooding tests: Chromosome stability and centromere activity were assayed by crossing single monosomic derivative-bearing males to five y1; ry506 females, or single monosomic derivative-bearing females to three y1; ry506 males. Offspring were scored for presence or absence of the minichromosome on the basis of phenotype (yellow+ rosy− or yellow− rosy+). Brooding (single germ cell division) assays were done by outcrossing individual males with new virgin females every day for 10 days. Transmission was determined for each set of females. Cell division rate was taken as one per 10 hr (Ashburner 1989). Data for each chromosome derivative, including phenotype, size, and transmission, are available elsewhere (Maggert 2000).
Pulsed-field gel electrophoresis, restriction, and Southern blot hybridization: Chromosomes were isolated from embryos, digested, subjected to pulsed-field gel electrophoresis, and hybridized with diagnostic probes as described previously (Le et al. 1995; Karpen 2000).
Cytological preparation and analysis: Chromosome spreads were prepared from salivary glands or from neuroblasts of wandering third instar larvae raised at 18°. Salivary glands were dissected from larvae in 0.7% NaCl and squashed under coverslips in 2% aceto-orcein. Brains were dissected from larvae in 0.7% sodium chloride, incubated in 0.4% sodium citrate for 10 min, and then squashed in methanol:acetic acid:water (at 11:11:2 by volume). Squashed tissues were frozen in liquid nitrogen and then immersed in ethanol and air dried before staining with 1 μg/ml Hoechst 33258 in phosphate-buffered saline. Images were visualized on a Zeiss Axiophot epifluorescence microscope, captured using an ImagePointR chargecoupled device camera (Photometrics, Tucson, AZ), and analyzed on a Macintosh G3 using IP Lab Spectrum (Signal Analytics, Vienna, VA) and Photoshop 3.0 (Adobe) software.
Inverse PCR and sequencing: DNA was isolated from adult flies by homogenization in 100 mm Tris pH 8.2, 50 mm ethylenediaminetetraacetic acid supplemented with 1% sodium dodecylsulfate and 0.5 μg proteinase K, and allowed to incubate for 1 hr at 65°. The sample was phenol extracted twice, precipitated in ethanol, and resuspended in TE supplemented with 1 μg/ml RNAse A. Restriction with HhaI proceeded in restriction buffer (New England Biolabs, Beverly, MA) for 2 hr before being heat inactivated at 65° for 15 min. The mix was allowed to ligate with 2 units T4 DNA Ligase (New England Biolabs) in 400 μl volume overnight at 4°. The sample was precipitated in ethanol, and 1/75 of the reaction was subjected to polymerase chain reaction [reaction mix contained 2 μm each dNTP, 10 μm each primer, and 2 units Taq polymerase (Perkin-Elmer, Norwalk, CT)]. Reaction proceeded in a Perkin-Elmer 9600 thermocycler at 94° for 2 min, followed by 35 cycles of 94° for 45 sec, 60° for 45 sec, and 72° for 4 min, with a final extension at 72° for 6 min. Bands were run on an agarose gel, excised, and reamplified under the same conditions. The product reaction was sent for automated sequencing on an ABI automated sequencer.
Primer sets for the amplification of the Dpγ238 breakpoint were CGTGTTACACTTGCGAGGCGG and GTTACGTACTATATATCAAATCTAGCAAGC. The sequence of the left arm of the inversion in Dpγ238 is TAATTATTAATCGATGGGTGGATA, AGAATTAATTAAGTGCAGT, where the sequence before the comma is from the yellow locus and the sequence after the comma is from Maupiti [the first A after the comma refers to base 76672 in Maupiti (J. Wahlstrom, X. Sun, H. Le and G. Karpen, unpublished results)]. The right arm of the inverted chromosome has sequence CTACAATATCCTTTATGATACTGTCATCGATCACATTCGATCGATCGATCAATTTGAAACTC, where the sequence before the underline is from the yellow gene, the sequence after the underline is from Maupiti, and the underlined sequence is novel.
Primer sets for the amplification of the Tγ1337 breakpoint were GCAATGTTCCAGGACAAAGGG and TAATCCTCTTCTGTGGACCG. The sequence of the 2P; Dp8-23D element of T(2; Dp8-23)γ1337 is TTTTATTTGTATGCCTTTTCACCATTTTGGTGAAAATCAGCTGTAGCTGATTATGTTGGTATAGGTGT, where the sequence before the underline is from the yellow gene, the sequence after the underline is from 2L (P1 clone no. DS00501), and the underlined sequence is novel. The sequence of the Dp8-23P; 2LD element of T(2; Dp8-23) γ1337 is GATTCAGACCTATAAACTTGGCCGTTGCTATATTCCTTGGCAGAGGAATAAATATGACAATATAT, where the sequence before the underline is from distal 2L, the sequence after the underline is from proximal yellow, and the underlined sequence is novel.
Statistical analyses: Statistical analyses were done using Excel 98 (Microsoft) or Statistica 4.1 (Statsoft) software. Analyses of breakpoint distributions were done on continuous data and nonparametric data grouped into 5-kb bins; in no case did the results differ appreciably. V-square was opted over chi square in the case of small sample size (n ≤ 6). Model I linear regression in Figure 7 was done on independent lntransformed chromosome size and dependent fidelity data (Sokal and Rohlf 1995).
RESULTS
Structure of minichromosome Dp8-23: We have addressed the mechanism of neocentromere formation in Drosophila melanogaster by examining the behavior of a common test segment located in different contexts with respect to centromere proximity. We have exploited a supernumerary minichromosomal derivative of the X chromosome, Dp8-23, and its derivatives. Dp8-23 is fully stable, both meiotically and mitotically, contains genetic markers, and is completely dispensable for viability (Parry and Sandler 1974; Tower et al. 1993).
Dp8-23 is composed of one megabase of heterochromatin, including a 260-kb region from the distal heterochromatin (numbered ±0 to +260 kb, Figure 1a), and 740 kb from the base of the X chromosome (numbered +260 to +1000 kb; Karpen and Spradling 1990). These two blocks of heterochromatin were juxtaposed during the ontogeny of Dp8-23, and the interface is referred to as the 1.688/1.672 boundary. Additionally, Dp8-23 contains the terminal 290 kb of the X chromosome (numbered ±0 to −320 kb), including euchromatin containing the yellow locus (y+), and two 14.5-kb telomeric PZ{ry+} transgenes (Karpen and Spradling 1992; Tower et al. 1993). Cytological, molecular, and
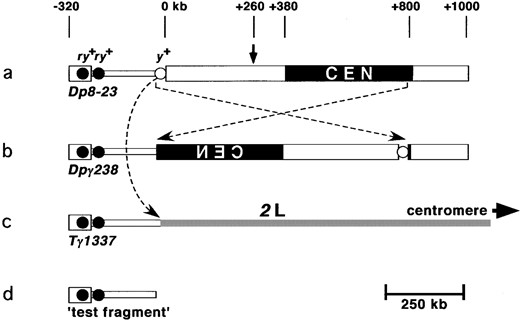
Generation of minichromosome derivatives used in this study. (a) Dp8-23 contains the centromere (CEN) and 1 Mb of pericentric heterochromatin (thick block) of the X chromosome. The arrow indicates where the 1.688 g/cc satellite DNA and 1.672 g/cc satellite DNA are juxtaposed (Le et al. 1995; Williams et al. 1998). Additionally, Dp8-23 contains 320 kb of euchromatin (thin block), the endogenous yellow+ gene (open circle, y+), and subtelomeric DNA (thick block on left), including two introduced PZ{ry+} elements (solid circles, ry+). (b) Pericentric inversion (crossed dotted arrows) rearranged the Dp8-23 chromosome to give Dpγ238. (c) Translocation between Dp8-23 and chromosome 2 (curved dotted arrow) gave rise to Tγ1337. (d) The distalmost 290 kb of all three chromosome are identical and are referred to as the test segment when in situ and test fragment when freed.
genetic experiments show that the centromere of Dp8-23 contained within 420 kb of the heterochromatin (from +380 to +800; Le et al. 1995; Murphy and Karpen 1995b; Sun et al. 1997). The 290-kb segment distal to yellow (−30 to −320 kb) is referred to as the test segment (Figure 1d) and has never been observed to manifest centromere activity in over 75 years of radiation-induced and meiotic recombination-induced structural alteration of the parental X chromosome (Lindsley and Zimm 1992).
Structure of minichromosome Dpγ238: Dpγ238 is a pericentric inversion derivative of Dp8-23 that breaks distal to yellow and very close to the centromere on the right arm (Le et al. 1995; Figure 1b). This inversion placed the yellow gene within centric heterochromatin, which results in variegated effects on yellow expression. The inversion event also juxtaposed the test segment to the active centromere of Dpγ238. The test segment is marked with the PZ{ry+} elements, and all euchromatic DNA distal to the inversion breakpoint are identical to those of Dp8-23 (Williams et al. 1998).
The euchromatic inversion breakpoint of Dpγ238 was known to lie closely distal to yellow (Le et al. 1995), and the heterochromatic breakpoint was thought to lie at the boundary of the centromere and the outlying centric heterochromatin. We exactly mapped the euchromatic and centric breakpoints of Dpγ238 by utilizing inverse polymerase chain reaction (PCR) to regions of the euchromatin near the yellow locus. The sequences of both inversion breakpoints are reported in materials and methods.
The centromere from a related minichromosome, Dpγ1230, has been cloned and partially sequenced (Sun et al. 1997; J. Wahlstrom, X. Sun, H. Le and G. Karpen, unpublished results). When the sequence of the centric breakpoint of Dpγ238 was compared to the sequence of the Dpγ1230 centromeric region, we determined that the heterochromatic Dpγ238 breakpoint was within the centromere, ~3 kb inside the right end of Maupiti. Maupiti is a previously identified cluster of degenerate transposable elements and A+T-rich sequence at one end of the molecularly defined centromere (Sun et al. 1997; J. Wahlstrom, X. Sun, H. Le and G. Karpen, unpublished results). This finding shows that the test segment of Dpγ238 (−30 through −320 kb) is adjacent to a region known to behave as a centromere (Figure 1b).
Structure of translocation chromosome Tγ1337: In previous studies, γ-irradiation of Dp8-23 produced a number of internal deficiencies and translocations (Le et al. 1995). Tγ1337 was characterized as a translocation involving exchange of material between Dp8-23 and chromosome 2. Salivary gland chromosomes from individuals heterozygous for T(2;Dp8-23)γ1337 were used to determine the location of the chromosome 2 breakpoint. The tip of 2L was consistently bifurcated and showed a different banding pattern in both halves (Figure 2a). Consensus from numerous spreads suggests that the breakpoint lies within salivary gland region 21B. T(Dp8-23P;2D)γ1337 chromosomes were not visible in these preparations, possibly due to their small size and inclusion in the chromocenter. Two small chromosomes were visible in neuroblast squashes from homozygous individuals (Figure 2b, arrows), indicating that the translocation was reciprocal (Figure 2c) and that the tip of 2L is carried by the Dp8-23 centromere in Tγ1337 animals.
Inverse PCR was used to clone and sequence the translocation breakpoint of Tγ1337. The 2L breakpoint of Tγ1337 lies within P1 clone DS00501 (see materials and methods). This region of the chromosome is near the telomere, appears to be euchromatic, and is ~20 Mb from centric or centromeric heterochromatin (Adams et al. 2000).
The Dp8-23-linked breakpoint of the Tγ1337 translocation occurs in the intron of yellow, consistent with its yellow− rosy+ phenotype. Restriction digests of the Dp8-23D material distal to the breakpoint suggests that the 2P; Dp8-23D is unaltered in structure. That is, the structure of Tγ1337 places the rosy+-containing test segment in a euchromatic context without any other alterations in sequence from that of Dp8-23 (Figures 1c and 2c).
Rationale for the γ-irradiation screen: We used the above chromosome derivatives to address the role of epigenetic regulation and to determine if proximity to a functional centromere is required for neocentromere
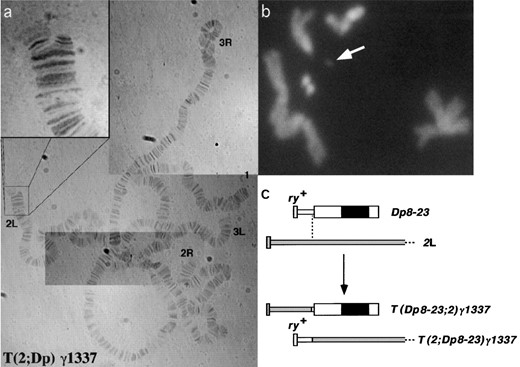
Structure of Tγ1337. (a) Aceto-orcein-stained salivary gland squash from a Tγ1337/+ individual. Inset shows the bifurcation of 2L, indicating a translocation with distal 2L. (b) Mitotic neuroblast of Tγ1337/Tγ1337, stained with 4′,6-diamidino-2-phenylindole, showing a small chromosome that is presumably the Dp8-23 centromere carrying the tip of 2L (white arrow). (c) Schematic depicting the translocation between 2L and Dp8-23 (thick and thin bars represent heterochromatin and euchromatin, respectively; solid rectangle represents the centromere; open blocks are Dp8-23 derived and shaded blocks are chromosome 2 derived).
activation. Previous work demonstrated that the test segment is capable of manifesting neocentromere activity when present in Dpγ238 (Murphy and Karpen 1995b). If neocentromere activation was sequence dependent and involved derepression of latent centromeres, the test segment should behave identically in all three substrate chromosomes. Alternatively, context-dependent recovery of neocentromere activity would suggest that neocentromere function is acquired by spreading of centromere identity from neighboring DNA.
We irradiated Dp8-23, Dpγ238, and Tγ1337 in parallel. The irradiation was done in females homozygous for the mutagen-sensitive mutation, mutator-2 (mu-2c; Mason et al. 1984). This mutation confers sensitivity to low doses of ionizing radiation and allows the recovery of terminal deficiencies despite the absence of telomeric repeats (Mason et al. 1984, 1986, 1997). This approach has been shown to be fruitful in previous studies of the Dp8-23 centromere (Murphy and Karpen 1995b). Mason et al. (1984) and Murphy and Karpen (Murphy and Karpen 1995b; Murphy and Karpen 1998) have shown that the preponderance of 500-rad-induced break events are one-break terminal deficiencies. The irradiation and subsequent crosses are shown in Figures 3 and 4. All female offspring were scored for the loss of one diagnostic marker (i.e., yellow+ rosy− or yellow− rosy+), indicating a breakage event within the test segment-containing chromosome.
Irradiation of Dpγ238: We irradiated and screened 24,978 Dpγ238 chromosomes for new breakage events (Figure 3). Twenty-eight new yellow+ rosy− derivatives and 49 new yellow− rosy+ derivatives were recovered. Transmission tests confirmed that all yellow+ rosy− derivatives were transmitted at 100% fidelity, indicating that they retained a fully functional centromere and other components necessary for normal transmission. Pulsed-field gel electrophoresis and Southern hybridization analyses showed that all of these derivatives were at least 1 Mb in size and contained all of the heterochromatin. In agreement with previous irradiation of Dpγ238, the yellow− rosy+ derivatives fell into three categories based on transmission—“eucentric” (45–55% transmission through females), “midcentric” (20–40% transmission through females), and “structurally acentric” (2–15% transmission through females; see Figure 5, Dpγ238 column). Transmission correlated with size and, in general,
Irradiation screen for Dp8-23 and Dpγ238. Females were irradiated and outcrossed, and F1 females were screened for presence or absence of the minichromosome or minichromosome derivatives. Class 1 were unbroken minichromosomes and were discarded. Class 2 were null for the supernumerary minichromosome. Class 3 were characterized in yellow+ rosy− individuals. Class 4 were characterized in yellow− rosy+ individuals. Potential neocentromere activation events were a subset of Class 4. Genetic nomenclature is explained in materials and methods.
![Irradiation screen for Tγ1337. Females were irradiated and outcrossed, and F1 females were screened for the loss of the telomeric rosy+ marker or activation of a neocentromere. Class 1 were unbroken Tγ1337 chromosomes and were discarded. Class 2 contained the homologous CyO, y+ chromosome. Class 3 were terminal deficiencies of Tγ1337 that lost the ry+ markers [Der(ry−)]. Class 4 were potential activation of neocentromeres on the test fragment, followed by segregation with the CyO, y+ homologue [Dp(ry+)]. Genetic nomenclature is explained in materials and methods.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/genetics/158/4/10.1093_genetics_158.4.1615/2/m_gen9818.f4.jpeg?Expires=1716916415&Signature=wWDnC4UAjLObA23wLz5gzvBDtLf3NFimNX~JWs2Lw0LodVm72VjN34PJbWfu3gEUim5hM0y5cgND2by8ZhNOOmN1mlCFIHy61YRrMeSoSdziP7f-gnkKrHlAutYwa5w02tvZsXBt4oIr1rBC8TWAMQdw2DwERrNr6c2X3jWquUHsiXt1DIU~YgiVtK23AXyCIM9H1DpTlpm6WhbZ-x17QjG8CB5b-~I1QQeS8eF1uA8u9eWN9Dxo6oUD~NqcPnEKFKMRjkOd03h-NHFRCWb7WY6GFsAUk8OJEcfCuMHa-I2w2EUu6CPj5iaMKsPIsgzisPrVAObq~Awejtk5fznoWg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Irradiation screen for Tγ1337. Females were irradiated and outcrossed, and F1 females were screened for the loss of the telomeric rosy+ marker or activation of a neocentromere. Class 1 were unbroken Tγ1337 chromosomes and were discarded. Class 2 contained the homologous CyO, y+ chromosome. Class 3 were terminal deficiencies of Tγ1337 that lost the ry+ markers [Der(ry−)]. Class 4 were potential activation of neocentromeres on the test fragment, followed by segregation with the CyO, y+ homologue [Dp(ry+)]. Genetic nomenclature is explained in materials and methods.
a chromosome derivative was more stable if it contained more centromeric chromatin (discussed below).
Structural analysis of Dpγ238-derived chromosomes: We performed structural analyses on the derivatives of Dpγ238 to assess the distribution of breakpoints and to determine which derivatives were neocentromeric. The yellow+ rosy− derivatives have breakpoints that are dispersed randomly between the proximal PZ{ry+} transgene (at −185 kb) and the euchromatic/heterochromatic boundary. The distribution likely conforms to a normal population within the euchromatin (P = 0.149; H0 states that the breakpoints were drawn from a normally distributed population). However, no breaks were recovered that gave rise to yellow+ rosy− chromosomes with only a portion of the centromere (Figure 6a). Although the centromere may be progressively deleted from one direction (from the right in Figure 1b), it may not be removed from the other direction (from the left in Figure 1b; χ2 = 6.03, d.f. = 1, P = 0.014; H0 states that yellow+ rosy− derivatives that are the product of breaks to the right of the centromere should be as frequent as yellow− rosy+ SA Ders that are products of breaks in the euchromatin). This observation indicates that although a neocentromere may be activated in euchromatin that juxtaposes the centromere, a neocentromere may not be activated in heterochromatin that juxtaposes the centromere (to the right in Figure 1b). Thus, the centromere may be asymmetric in its activity or ability to spread (see discussion).
The breakpoints leading to yellow− rosy+ derivatives of Dpγ238 showed a nonnormal distribution (Figure 6a). The breakpoints associated with the generation of normally transmitted derivatives (30 of 49, Figure 6a, eucentric) were clustered within the centric (but noncentromeric) heterochromatin. This distribution may represent a bias in repair and recovery or may represent a true frequency of breaks during irradiation (Ashburner 1989; Warters and Lyons 1992; Oleinick et al. 1994; Laurenti et al. 1995; Ahmad and Golic 1998). The majority of breakpoints lie distal to the 1.688/1.672 breakpoint (Figures 1a and 6a) and occur within the 1.688 g/cc 359-bp repeat from the middle of the X heterochromatin (Karpen and Spradling 1990). This suggests that 1.688 g/cc satellite DNA is more permissive to break generation or subsequent chromosome recovery than is the simple repeat 1.672 g/cc (AATATn), which lies proximal to the 1.688/1.672 boundary (χ2 = 2.72, d.f. = 1, P = 0.097; H0 states that 1.688 g/cc and 1.672 g/cc sequences should have an equal density of breakpoints). Since chromosomes with breakpoints on either side of this landmark are transmitted with identical fidelity, we can conclude that the nonrandom recovery of derivatives with breakpoints proximal to or distal to the 1.688/1.672 boundary is not due to differences in chromosome transmission.
Chromosomes with breaks within the structural centromere also showed a breakpoint distribution that deviated from normal (Figure 6a). There were gaps in the breakpoint distribution within what has been defined as the minimal sequence required for full centromere function [+380 to +800 kb of Dp8-23, −30/+0 to +420
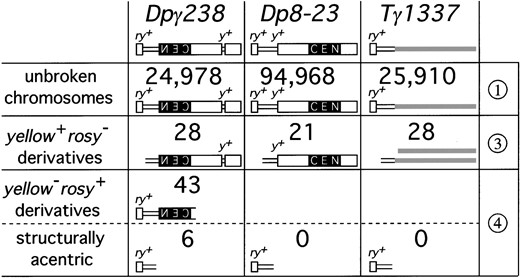
Numbers and classes of chromosome derivatives generated in this study. The figure shows the number of chromosomes screened after irradiation, and the number of chromosomes in each phenotypic class. Numbers to the right refer to classes introduced in Figures 3 and 4. Structurally acentric derivatives lack all centric heterochromatin and represent neocentromere activation events. These represent a subset of derivatives of Dpγ238 and all of the class 4 derivatives of Dp8-23 and Tγ1337.
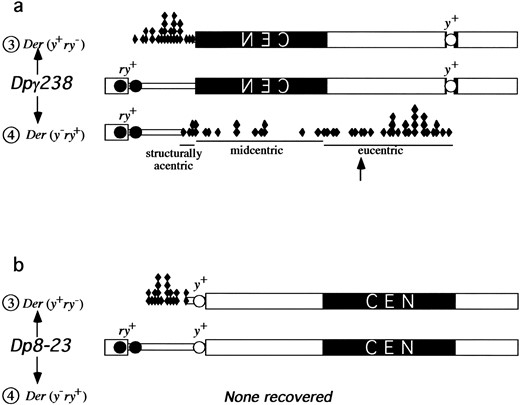
Distribution of breakpoints along Dpγ238 and Dp8-23. (a) The distribution of breaks of Dpγ238. yellow+ rosy− derivatives are shown above the Dpγ238 chromosome, and diamonds represent breakpoints. yellow− rosy+ derivatives are shown below the Dpγ238 chromosome. The 1.688/1.672 boundary (Figure 1a) is marked with an arrow. (b) The distribution of breaks of Dp8-23. yellow+ rosy− derivatives are shown above the Dp8-23 chromosome, and diamonds represent breakpoints. No yellow− rosy+ derivatives were recovered. Symbols are as in Figure 1.
kb of Dpγ238 (Le et al. 1995; Sun et al. 1997)]. These gaps may be due to suppressed breakage within the centromere, or they may be due to preferential recovery of the products of breaks within the centromere. Since two separate studies (here and Murphy and Karpen 1995b) observed decreased frequency of breaks in this region, a nonnormal distribution, and an overlap in the regions where breakpoints were not recovered, it is likely that these regions of the centromere are refractory to being broken or recovered at the end of a chromosome (as in Ahmad and Golic 1999). Our study cannot determine what features of the centromere are incapable of supporting breaks or the recovery of broken chromosomes.
Analysis of the transmission of structurally acentric derivative chromosomes: Six structurally acentric chromosomes were generated from the irradiation of Dpγ238, supporting the findings of a previous study (Murphy and Karpen 1995b). These chromosomes lacked all centric and centromeric heterochromatin associated with centromere activity, yet exhibited single mitosis transmission fidelity of approximately 90% (Williams et al. 1998; Maggert 2000). These structurally acentric fragments contain kinetochore- and centromere-specific proteins (Williams et al. 1998; Lopez et al. 2000; Blower and Karpen 2001), indicating that a neocentromere has formed on normally noncentromeric DNA.
Previous studies demonstrated that the size of a neocentromere-containing structurally acentric derivative (SA Der) correlates with its transmission (Williams et al. 1998). By analyzing the size-stability correlation of SA Ders, we found that it follows the relationship f = a + h ln L, where f is the fidelity of single mitotic transmission, L is the size of the chromosome in kilobases, a is a constant with value −2.965, and h is the regression coefficient with numeric value 0.667. This function and the value of h are derived from transmission data from Dpγ238 derivatives between 220 and 400 kb (Figure 7, range under arrow) and regresses well with the data (coefficient of determination, R2 = 0.919). This coefficient of determination shows that 91.9% of the stability of neocentromere-containing test fragments can be explained as a function of chromosome size alone. Chromosomes that contain centromere-derived DNA follow this relationship as well as chromosomes without centromere-derived DNA, suggesting that the size dependency described by f = −2.965 + 0.667 ln L is irrespective of the DNA constitution. A chromosome increases in stability as it increases in size, but not more so if that added chromatin is derived from a centromere, up to the point of 100% fidelity. Extrapolating this trend, a SA Der of sufficient size (approximately one-half megabase) would be expected to be fully stable through mitosis and should not differ in transmission from an endogenous centromere. This prediction should hold true for derivatives that contain centromeric DNA (as observed in Williams et al. 1998) or that are completely devoid of centromeric DNA. Direct determination of this latter prediction is impossible in this system, as SA Ders of >290 kb include centromeric
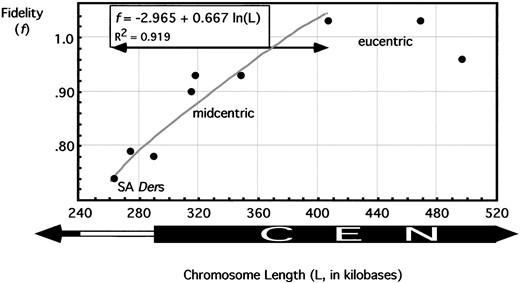
Stabilities of chromosome derivatives through single mitoses. Brooding analyses were done on structurally acentric derivatives, as well as on derivatives with partially deleted centromeres and derivatives with fully intact centromeres. Exponential, logarithmic, and linear regressions were performed to give the best fit curve of data within the range of arrow (gray line, f = −2.965 + 0.667 ln L; R2 = 0.919). x-axis shows chromosome size in kilobases, and y-axis gives probability of a chromosome successfully transmitting through a single germline stem cell division.
chromatin in addition to any resident neocentromere activity. Hence, we cannot use the activation and liberation of neocentromeres from Dpγ238 to directly determine whether a neocentromere can drive inheritance of a chromosome as well as a bona fide centromere. However, the recruitment of centromere proteins (Williams et al. 1998; Blower and Karpen 2001) argues that neocentromeres do possess normal centromere function and that their instability is due predominantly to defects in antipoleward forces (Murphy and Karpen 1995a), sister chromatid cohesion (Lopez et al. 2000), and other inheritance functions (Murphy 1998; Dobie et al. 2001).
Irradiation of Dp8-23: Dp8-23 was subject to irradiation in parallel to Dpγ238 (Figure 3) to assess the frequency of neocentromere recovery from a test segment separated from an active centromere by 400 kb of heterochromatin. Of the 94,968 chromosomes screened, we recovered 21 yellow+ rosy− derivatives (Figure 5, Dp8-23 column). Two free Dp substrate chromosomes (Dpγ238 and Dp8-23) were differentially mutable, with Dpγ238 producing yellow+ rosy− derivatives at approximately 5-fold higher frequency. The reason for this difference is not known, but may reflect different features of the chromatin structure within euchromatin vs. neocentromere-competent chromatin. To compensate for this difference in γ-induced chromosome breakage frequency, we irradiated and scored 3.7-fold more Dp8-23 chromosomes to recover approximately similar numbers of yellow+ rosy− derivatives. These chromosome derivatives were expected to represent breaks between yellow and the proximal PZ{ry+} transgene, analogous to the yellow+ rosy− derivatives from Dpγ238. The structures of these derivatives were confirmed using pulsed-field gel electrophoresis and Southern analysis (Maggert 2000).
The distribution of breakpoints of the yellow+ rosy− derivatives of Dp8-23 were analyzed in the same fashion as those of Dpγ238 (Figure 6b). The euchromatic breakpoints of Dp8-23 follow a normal distribution that is statistically indistinguishable from the distribution of breakpoints in Dpγ238-derived chromosomes (Mann-Whitney U = 255, P = 0.438; Student's t = 0.179, d.f. = 44, P = 0.859; H0 states that the breakpoints are drawn from populations with identical distributions).
No yellow− rosy+ derivatives of Dp8-23 representing neocentromere activation events were recovered. These events may not have been detected because the yellow− rosy+ derivatives may have segregated to the same cell as yellow+ rosy− derivatives. To abrogate the possibility that neocentromeres were activated on the test fragment, we outcrossed 25,000 yellow+ rosy+ F1 females to yellow− rosy− males and scored for the appearance of new derivatives in F2. Previous work has shown that SA Ders and yellow+ rosy− derivatives segregate at random in females (Karpen et al. 1996). No new yellow− rosy+ or yellow+ rosy− derivatives were detected, indicating that the failure to recover yellow− rosy+ derivatives from the screen of Dp8-23 was not due to failure to detect them in the presence of reciprocal chromosomes. Instead, the failure to recover SA Ders from Dp8-23 is most likely due to their inability to manifest neocentromere activity.
Irradiation of Tγ1337: Since small chromosome size compromises the stability of neocentromere-containing chromosomes (Williams et al. 1998; Maggert 2000) and could have played a role in our failure to recover neocentromeres from Dp8-23, we reasoned that we could alleviate the size restriction of neocentromere liberation by increasing the potential target size of the neocentromere-containing test segment. Since the test segment in Tγ1337 is separated from the endogenous chromosome 2 centromere by ~28 Mb (Adams et al. 2000), the entire chromosome arm was a target for γ-induced breakage (Mason et al. 1997). Since larger neocentromere-containing fragments have greater stability, any inherent neocentromere activity on the test segment of Tγ1337 should give rise to stable test fragments consisting of the test fragment plus additional chromosome 2 material. The proximal limit of potential breaks may be as much as one-half of 2L, the amount of this arm that can be hyperploid without affecting viability (Lindsley et al. 1972).
The screen for recovery of neocentromere-containing test fragments from Tγ1337 or of reciprocal break events is detailed in Figure 4. In this screen, the rosy+ test segment and the yellow+ gene were on chromosome 2 homologues and so the phenotype of flies bearing neocentromere-containing fragments would be yellow+ rosy+, while complementary chromosomes would be found in yellow− rosy− organisms. We screened 25,910 chromosomes and recovered 28 terminal deficiencies of Tγ1337 that were rosy−, indicating loss of all or a portion of the test segment (Figure 5, Tγ1337 column). Of these, 3 were confirmed to be terminal deficiencies with breakpoints within the test segment. No rosy+ neocentromere-containing fragments were recovered. The absence of neocentromere-containing test fragments from Tγ1337 was even more significant considering that the target size was larger than in Dpγ238 or Dp8-23 and that liberated fragments would have a higher stability due to their larger size. The failure to recover any neocentromeric fragments from Tγ1337 also eliminates the possibility that the test segment of both Dp8-23 and Dpγ238 contain a latent centromere that is repressed by the centric heterochromatin in Dp8-23.
DISCUSSION
Neocentromere activation requires centromere juxtaposition: Here we describe irradiation-mutagenesis experiments designed to identify the mechanism of neocentromere formation in D. melanogaster. Prior to this study, two models existed to explain the generation of neocentromeres in Drosophila and Homo sapiens—derepression of latent centromere-competent euchromatic sequences vs. centromere spreading (Choo 1997a, 1998). We distinguished between these models through a genetic assay for neocentromere activation and recovery. Three substrate chromosomes were irradiated, and an identical 290-kb test segment was liberated and genetically assayed for neocentromere activity. The three test segments were identical in molecular structure and differed only in their chromosomal context. In Dpγ238, the test segment was juxtaposed to an active centromere; in Dp8-23, the test segment was juxtaposed to centric, but centromerically inert DNA; in Tγ1337, the test segment was juxtaposed to euchromatin. Neocentromeres were activated only from Dpγ238, where the test fragment was derived from a position abutting the centromere. In contrast, no neocentromere-containing fragments were recovered from Dp8-23 or Tγ1337, despite the ability to generate and recover reciprocal, centromere-containing derivatives. The absence of neocentromeres from Dp8-23 and Tγ1337 is different from the Dpγ238 data (V 2 = 4.084, d.f. = 1, P = 0.0435; H0 states that Dpγ238 and Dp8-23 will liberate neocentromere-containing fragments at the same rate), suggesting that fragments derived from these different chromatin contexts do not have the neocentromere-forming properties of Dpγ238-derived fragments, despite their structural identity.
After irradiation and liberation, fragments that are capable of manifesting neocentromere activity do so only if they were juxtaposed to an active centromere prior to liberation. This result demonstrates that the chromatin context of a fragment affects its centromere activity before or after liberation and that neocentromere activity is not an innate characteristic of a DNA sequence. Additionally, if noncentromeric heterochromatin is repressive for neocentromere activity, then Tγ1337 represents a situation where repressive heterochromatin is separated from the test segment by 20 Mb. Any breaks within that 20 Mb would separate an inherent neocentromere in the test segment from the heterochromatin and allow activation. The absence of neocentromere activation after irradiation of Tγ1337 rules out models in which neocentromere formation occurs due to derepression of latent centromeres in the test fragment. We conclude that some aspect of chromatin structure or DNA modification (Lyko et al. 2000) was conferred by the centromere to the juxtaposed euchromatic region in Dpγ238 and that acquisition of centromere-specific properties was sufficient to impart stable neocentromere activity onto the test fragment. We further propose that neocentromere activation involves spreading of centromere identity factors in cis, in a manner similar to that observed for dosage compensation complexes in Drosophila (Kelley et al. 1999) or centromere proteins in Schizosaccharomyces pombe (Partridge et al. 2000).
It is possible to determine whether a neocentromere was active prior to irradiation or became activated shortly after liberation. Of 28 yellow+ rosy− derivatives of Dpγ238, 6 had breakpoints that overlapped with the range of breakpoints of the yellow− rosy+ structurally acentric derivatives (SA Ders). These 6, then, were complementary to the 6 SA Ders and represented breaks in the same region of DNA and recovery of the structurally centromeric half of the Dpγ238 chromosome. The frequencies of recovery of either side of the chromosome were statistically indistinguishable and, although we cannot state that the neocentromere activity existed on the test fragment prior to liberation, we can conclude that the manifestation of neocentromere activity was rapid and efficient following liberation. It is possible that neocentromere activity existed before the chromosome was broken and separation merely allowed the neocentromere to behave independently from the endogenous Dpγ238 centromere.
Franz Schrader commented on others' (Upcott 1937; Darlington 1939) work, “ … chromosomes are occasionally broken in such a way that the fracture takes its path through the kinetochore. Kinetochore fragments thus produced evidently may be functional at times for the resulting chromosome fragments continue to participate regularly in the mitotic process” (Schrader 1939). The activity on these fragments was manifest immediately, similar to our data for the breakage products derived from Dpγ238. These observations are consistent with an iterated model for centromere structure (Zinkowski et al. 1991; M. D. Blower, B. A. Sullivan and G. H. Karpen, unpublished results) where the centromere may be defined by repeated centromere subunits. These subunits may cooperate when close together (Page and Shaffer 1998), may compete if separated in cis (Sullivan and Willard 1998; Agudo et al. 2000), or may be independently active when present on separated fragments. Thus, it is likely that centromere activity existed on the test segment of Dpγ238 prior to irradiation and liberation, in response to the inversion that moved the centromere next to the euchromatin. The different frequencies of neocentromere recovery from Dpγ238 and Dp8-23 show that centromere competence of the test segment is not inherent to the DNA sequence of the test segment itself. Thus, some feature of Dpγ238, and not of the resultant test fragment, is important in eliciting neocentromere activation.
Epigenetic identity of the centromere: The neocentromeres are stable through mitotic and meiotic cell divisions. Factors responsible for conferring centromere identity and activity to neocentromeric DNA could be capable of continuous association with the DNA, or alternatively the DNA could actively recruit these factors each cell division. Our data support the former model, since the latter model predicts that a centromeric sequence would be sufficient for centromere activity, which our results from the irradiations of Dp8-23 and Tγ1337 clearly exclude. The sequence independence of neocentromere formation and propagation in Dpγ238 strongly suggests that centromere identity is determined and propagated by an epigenetic mechanism that may rely on localized DNA or protein modification or the recruitment of centromere-specific factors.
Recently, a constitutive centromere-specific protein has been identified in Drosophila (Henikoff et al. 2000). This molecule, CID, is a histone H3 variant, similar to the human centromere-specific protein CENP-A (Palmer et al. 1991; Sullivan et al. 1994). The means of centromere-specific recruitment of CENP-A proteins is not well understood, but is not likely to be due to sequence-specific binding (Shelby et al. 1997; Barry et al. 1999, 2000) or temporal differences between the replication of centromeric chromatin and bulk chromatin (Csink and Henikoff 1998; Shelby et al. 2000; Sullivan and Karpen 2001), and is instead likely due to recruitment by previously associated CENP-A (Shelby et al. 1997; Maggert 2000; Maggert and Karpen 2000). The localization of CENP-A at the human centromere is not sufficient to induce centromere activity, as ectopic CENP-A incorporation along the entire chromosome is not sufficient to induce holocentric behavior (Shelby et al. 1997; K. F. Sullivan, personal communication). The presence of CID at the neocentromere of the SA Ders generated in this study (Blower and Karpen 2001) shows that neocentromeres can acquire a kinetochore-specific chromatin factor. Furthermore, outer kinetochore proteins such as L(1)ZW10 and DYNEIN are also present at these neocentromeres (Williams et al. 1998; Starr et al. 1998; Lopez et al. 2000; Blower and Karpen 2001). Thus, the regulation of Drosophila neocentromere identity is likely to be equivalent to that of a normal Drosophila centromere, as observed in humans (Saffery et al. 2000).
The spreading of Drosophila centromere activity: CID-directed CID recruitment may also serve as a mechanism for spreading of centromere activity. Perhaps the establishment of the neocentromere is a by-product of the maintenance mechanism of centromere identity factors. Recruitment of CID and other factors to newly synthesized DNA may be inexact, resulting in deposition of centromere identity-determining factors into adjacent regions (Figure 8). Thus, the single activity of CID-dependent CID recruitment could account for both the epigenetic inheritance and processive spreading of the centromere (Maggert 2000). In a normal situation, this could result in “filling” of gaps that may arise in normal centromeres, protecting their coherence though multiple cell divisions.
The question then becomes one of why spreading does not occur more often. Centromeres are not seen to migrate or grow during the life of an organism (White 1954). Whatever keeps a centromere from spreading in a normal chromosome has been defeated in Dpγ238, while it remains intact in Dp8-23 and Tγ1337. Discrete elements, either genetic or epigenetic, may flank the centromere and limit spreading. Alternatively, it may be heterochromatin and repeat-sequence DNA itself that prevents a centromere from spreading. In Dpγ238, one side of that boundary is removed through chromosome inversion, juxtaposing the centromere to euchromatin. This condition is different from the usual placement of centromeres within large blocks of heterochromatin and may be unique within the collection of chromosome aberrations available in Drosophila (Lindsley and Zimm 1992). Although 31 yellow− rosy+ derivatives of Dpγ238 were recovered that broke in the proximal heterochromatin of the right arm, no cognate neocentromeric yellow+ rosy− derivatives were recovered. Hence, centromere activity can spread into euchromatin, but is apparently incapable of spreading into or through heterochromatin in both Dp8-23 and Dpγ238.
Although heterochromatic mass contributes to centromere kinetic strength (Novitski 1951, 1955; Wines and Henikoff 1992), it was also shown that this is not a function of the centromere itself (Lindsley and Novitski 1957). We propose that heterochromatin is not
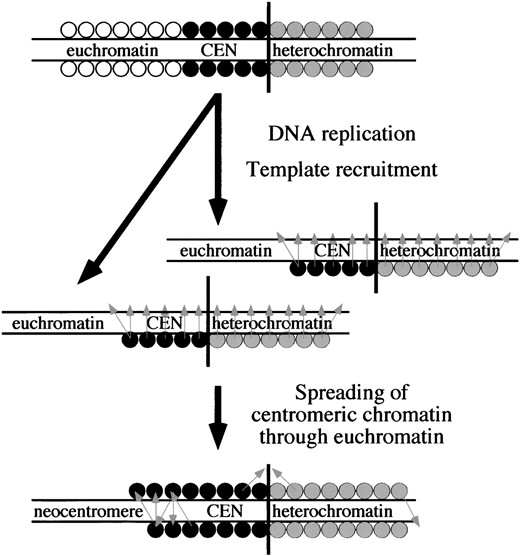
Model for epigenetic maintenance and spreading. The top chromosome shows different regions of DNA packaged as euchromatin (open circles), centromere (solid circles), and heterochromatin (shaded circles). Vertical black line indicates the heterochromatin/centromeric chromatin boundary. After replication (twin arrow), some factors remain associated with the DNA. Templating occurs (light gray arrows), as these factors recruit similar factors to the nascent DNA. This results in properly packaged DNA on both daughter chromosomes. Errors in recruitment can result in factors being recruited slightly out of register, which could cause an expansion of either the centromere or the heterochromatin. Boundaries between centromeres and heterochromatin could be maintained by an equilibrium between these competing forms of chromatin spreading, whereas euchromatin is not able to block centromere spreading and can acquire neocentromere function.
causative in centromere identity, but rather serves to stabilize centromere placement and keeps centromeres from moving or spreading along a chromosome. Only subsequent maturation of the kinetochore, microtubule binding, and/or chromatid cohesion would affect the measured centromere strength (Vig 1982).
Heterochromatin possesses the ability to act as a boundary to DNA replication (Leach et al. 2000) and may act similarly to block centromere movement or spreading. Ironically, the activity of heterochromatin that regulates positioning of the centromere on the chromosome may be its own ability to be epigenetically maintained and spread (Spofford 1976; Wakimoto 1998; Wallrath 1998; Farkas et al. 2000; Talbert and Henikoff 2000; Figure 8). Although spreading of heterochromatin and gene inactivation need not be processive (Talbert and Henikoff 2000), it is clear that genes inappropriately linked to heterochromatin are more likely to be inactivated than are unlinked genes (Spofford 1976; Talbert and Henikoff 2000; Tolchkov et al. 2000). If both centromeric chromatin and heterochromatin are maintained efficiently and independently, unable to spread into each other's territory, the boundaries of a centromere would be expected to remain stable over long periods of time (Maggert 2000). Although in general euchromatin poses no such barrier, some sequences of euchromatic origin do show epigene regulation [e.g., Polycomb response elements (Cavalli and Paro 1998)]. It is possible that these sequences will also pose a barrier to neocentromere spreading. Whole-chromosome kinetochores (holocentric chromosomes) may represent a case where the block to spreading has been defeated throughout the genome, perhaps in response to loss of substantial blocks of heterochromatin.
Forty different neocentromeres have been catalogued in human clinical cases. Their etiology is unknown, but many models have been proposed (Choo 1997a, 1998; Karpen and Allshire 1997; Murphy and Karpen 1998; Wiens and Sorger 1998; Willard 1998; Abad et al. 2000; Koch 2000; Maggert and Karpen 2000). The structures of the neocentromeric “marker” chromosomes appear to rule out a simple in cis-spreading model (Choo 1997a; Depinet et al. 1997; Warburton et al. 2000). However, genomic sensing effects in trans have been demonstrated between homologous loci (Donaldson and Karpen 1997; Colot et al. 1996; Dernburg et al. 1996; Dorer and Henikoff 1997; Henikoff 1997), and spreading of proteinaceous or ribonucleoprotein complexes in cis (Partridge et al. 2000) and in trans (Kelley et al. 1999) have been directly observed. We propose that centromere identity may be able to spread in trans as well as in cis and that human neocentromeres may have arisen through the accidental association of normally noncentromeric DNA with an endogenous centromere during centromere templating. Infrequently, neocentromere activation may also arise spontaneously through the inappropriate recruitment of centromere identity determinants at ectopic sites along the chromosome (Maggert and Karpen 2000). The demonstration that some human neocentromeres are correlated with chromosome aberrations (Warburton et al. 2000) may provide a similarity with the Drosophila minichromosome system and an experimental system in which to investigate these models.
Regardless of the mechanism of neocentromere formation in humans, once Drosophila and human neocentromeres are formed, their centromere identity is propagated epigenetically through cell division. Epigenetic regulation may not only occur at this neocentromere of Drosophila, but is likely be a common feature of bona fide regional centromeres of Drosophila and of other organisms (Karpen and Allshire 1997). The role of epigenetic regulation in other aspects of chromosome biology and gene regulation are well established (Henikoff 1997; Cavalli and Paro 1998; Wakimoto 1998; Ahmad and Golic 1999). We are now challenged to identify other aspects of chromosome biology that are regulated in a sequence-independent fashion and to determine the biochemical mechanisms responsible for epigenetic propagation of basic chromosomal functions.
Acknowlegement
We thank Pam Geyer, Jim Mason, Mike McKeown, Dan Lindsley, Barbara Wakimoto, John Newport, Lawrence Goldstein, Terence Murphy, and Kathryn Donaldson for very helpful and fruitful discussions, and Christopher M. Yan for technical assistance. Structural analysis of Tγ1337 was initiated by Kathryn Donaldson and structural analysis of Dpγ238 was initiated by Huong Le. We also thank Dan Lindsley, Barbara Wakimoto, Suzanne Eckert, Gary Huckell, Jennifer Armstrong, Mike Blower, Kim Finley, Kumar Hari, Ophelia Papoulas, Beth Sullivan, and Robin Truelove for critical review of this manuscript. This work was done in partial fulfillment of a doctorate of philosophy in biology at the University of California San Diego. Keith Maggert was a Lucille P. Markey Charitable Trust Fellow and a Chapman Charitable Trust Fellow. This work was funded by grants from the American Cancer Society (DB1200) and the National Institutes of Health (R01-GM54549).
Footnotes
Communicating editor: R. S. Hawley
LITERATURE CITED

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}