





Abstract
rec7 is involved in intra- and intergenic meiotic recombination in all tested regions of the genome of the fission yeast Schizosaccharomyces pombe. Segregational analysis in a rec7 gene disruption mutant revealed frequent occurrence of two-spored asci. Spores giving rise to diploid colonies were shown to derive from skipping of the second meiotic division. Nondisjunction of homologous chromosomes at the first meiotic division was also frequent. The cytological structures and processes, such as formation of linear elements, pairing of homologous chromosomes, and clustering of telomeres and centromeres, are regular in the mutant. Northern blot experiments revealed meiosis-specific expression of rec7. Screening of a meiotic cDNA library also identified transcripts from the opposite strand in the rec7 region. A Rec7-GFP fusion protein was localized in the nucleus of whole cells before karyogamy, during prophase, and after meiosis I. On spreads of prophase nuclei approximately 50 foci of Rec7-GFP were counted. Some of the observed phenotypes of the disruption mutant and the N-terminal sequence homology suggest that Rec7p is a functional homolog of Rec114p of Saccharomyces cerevisiae. The observed phenotypes of the disruption and the appearance of Rec7-GFP in mating haploid cells and after meiosis I are consistent with Rec7p functions before, during, and after meiotic prophase.
MEIOSIS is an essential step in the life cycle of sexually reproducing organisms. It produces haploid gametes from diploid parental cells by two nuclear divisions following a single round of DNA replication. The first (reductional) division differs from the second (equational) division and mitotic divisions in several respects. In most organisms proper completion of the reductional division requires pairing of homologous chromosomes and genetic recombination. Homologous pairing is usually accompanied by the formation of a tripartite, meiosis-specific structure, the synaptonemal complex (SC; Von Wettstein et al. 1984; Giroux 1988). Recombination generates genetic diversity and supplies connections (chiasmata) between the homologous chromosomes, which are indispensable for their correct segregation at the first meiotic division. Failure of these processes leads to the generation of aneuploid meiotic products.
The fission yeast Schizosaccharomyces pombe is a haploid eukaryote in which mating and meiosis are induced upon nitrogen starvation. Cells of opposite mating type conjugate and the zygotes usually undergo meiosis immediately (zygotic meiosis). By transfer of zygotes to growth medium, diploid vegetative strains can be obtained and propagated. By withdrawal of nutrients (in particular, nitrogen) synchronous meiosis is induced in diploid cells that are heterozygous for mating types (azygotic meiosis; Egel 1973; Egel and Egel-Mitani 1974). Meiosis in S. pombe shows some striking features. Unlike most of the eukaryotes, including Saccharomyces cerevisiae, fission yeast forms no SC (Bähler et al. 1993) and shows no crossover interference (Munz 1994). In S. pombe linear elements appear in meiotic prophase. They resemble the axial cores of other eukaryotes and have been proposed to play a role in meiotic chromosome structure and function (Bähler et al. 1993; Kohli 1994; Kohli and Bähler 1994). Investigation of the meiotic recombination-deficient mutant rec8-110 gave the first evidence for the importance of linear elements in chromosome pairing, recombination, and segregation (Molnar et al. 1995). The bouquet structure of chromosomes is usually a transitory prophase stage in other eukaryotes. Fission yeast maintains the bouquet arrangement of chromosomes throughout prophase. In addition, prophase nuclei perform oscillatory movements led by the spindle pole body and the attached telomere cluster (Chikashige et al. 1994). This movement facilitates pairing of the homologous chromosomes and is required for high levels of meiotic recombination (Yamamoto et al. 1999).
Meiotic recombination has been extensively studied both in fission yeast and budding yeast, which are the best-studied model organisms (for recent reviews see Roeder 1997; Fox and Smith 1998; Paques and Haber 1999). Many mutants are available in both species to dissect the meiotic process. Impairment of meiotic recombination leads to effects of different severity in the two yeasts. S. cerevisiae has 16 chromosomes; thus, random chromosome segregation in recombination-deficient mutants yields hardly any viable spores. Meiotic recombination genes in S. cerevisiae have classically been grouped as “early” and “late” genes on the basis of their mutant phenotypes in combination with a spo13 null mutant (Malone 1983; Petes et al. 1991). The spore lethality, conferred by a mutation in an early gene, can be rescued by the elimination of the first division in the spo13 background. In contrast, late mutants cannot be rescued. Early exchange genes are required for the initiation of meiotic recombination (Mao-Draayer et al. 1996). In S. cerevisiae double-strand breaks initiate most, if not all, meiotic recombination events (Sun et al. 1989; Cao et al. 1990). Double-strand breaks have not been detected in any of the early mutants [rad50 and spo11 (Cao et al. 1990); xrs2 (Ivanov et al. 1992); mre11 (Johzuka and Ogawa 1995); mer2 (Rockmill et al. 1995); rec102, rec104, and rec114 (Bullard et al. 1996); mre2 (Nakagawa and Ogawa 1997); and mei4 (Jiao et al. 1999)].
In fission yeast, segregation studies and isolation of recombination-deficient mutants are facilitated by the fact that S. pombe has only three chromosomes (Kohli et al. 1977). Individual chromosomes can be traced through meiosis by the use of appropriate markers and viable spores are obtained, even when chromosomes segregate at random. The latter property was utilized by Ponticelli and Smith (1989) and De Veaux et al. (1992) for the isolation of meiotic recombination-deficient mutants. The rec7-102 mutation severely reduced both intra- and intergenic meiotic recombination in all intervals tested (Ponticelli and Smith 1989; De Veaux and Smith 1994). Lin et al. (1992) isolated the gene and reported meiosis-specific expression for rec7 in the pat1 mutant background. Haploid strains carrying a mutation in the meiosis regulatory gene pat1 initiate haploid meiosis after a temperature shift (Iino and Yamamoto 1985; Nurse 1985).
This study describes the genetic and cytological phenotypes observed in a disruption mutant of rec7. Furthermore, we clarified the gene structure and investigated rec7 expression in wild-type background. Protein localization was examined in whole cells and on nuclear spreads in meiotic time-courses using a Rec7-GFP (green fluorescence protein) construct. Finally, we discuss the results with respect to the possible roles of Rec7p in fission yeast meiosis.
MATERIALS AND METHODS
Strains, media, and standard genetic methods: S. pombe strains used in this study are listed in Table 1. All rec7::ura4+ mutant strains were constructed from SA4 by genetic crosses. Strains carrying the rec7::GFP construct were derived from SA14 by backcrossing with strains carrying rec7::ura4+ and the desired markers.
The standard media and general methods were as described by Gutz et al. (1974). Yeast extract agar (YEA) contained 0.5% Difco (Detroit) yeast extract, 3% glucose, 1.6% agar. Malt extract agar (MEA) contained 3% malt extract, 2% agar. Minimal medium (MMA) consisted of 0.65% Difco nitrogen base without amino acids, 1% glucose, and 1.8% agar. YEA+5 and MEA+5 were supplemented with adenine, uracil, leucine, lysine, and histidine at 100 mg/liter. MMA was supplemented with nutrients according to experimental requirements at 100 mg/liter.
Crosses were carried out on MEA+5 at 25°. YEA+5 was used as a standard growth medium at 30°, unless otherwise indicated. EMM medium (modified Edinburgh minimal) used for complementation analysis of rec7::ura4+ was prepared as described by Schweingruber and Edenharter (1990). For meiotic time-course experiments S. pombe synthetic minimal medium (PM) and PM-N (PM without NH4Cl) were used as described by Beach et al. (1985) and Watanabe et al. (1988), respectively. The classical genetic methods (random spore analysis, dissection of asci, and interrupted mating for construction of diploid strains) were as described by Gutz et al. (1974).
rec7 gene disruption: Two gene disruption mutations were constructed (Figure 1). To create the rec7::ura4+ gene disruption, a 1.6-kb BamHI fragment from pYL45 (Lin et al. 1992) was subcloned into the BamHI site of pUC18 to produce pUC18-rec71.6. In this plasmid a 1.2-kb BglII fragment was replaced by a 1.8-kb BamHI-BglII fragment from pB4-2 (+BglII) containing the ura4+ marker gene. A 2.2-kb BamHI fragment from the resulting gene disruption plasmid pUC18-rec7-2::ura4+ was used to transform strain SA2 to Ura+ by the lithium acetate method (Ito et al. 1983). The second gene disruption, designated rec7-0.2::ura4+ was obtained by replacing a 0.2-kb NsiI-SfuI fragment from pUC18-rec71.6 with a 1.8-kb PstI-ClaI fragment from pB4-1 carrying the ura4+ marker gene. A 3.2-kb BamHI fragment from the resulting gene disruption plasmid pUC18-rec7::ura4+ was used to transform strain SA2 as described above. pB4-2 (+BglII) and pB4-1 are pBluescript vectors (Stratagene, La Jolla, CA) containing the ura4+ gene on a 1.8-kb HindIII fragment (Grimm et al. 1988). Proper integration of the fragments into the genome was confirmed by Southern blot analysis in both disruption constructs.
Segregation analysis: To study chromosome segregation in random spores, rec7 mutant and wild-type strains were crossed as described above. After overnight digestion of the crossing material with snail digestive juice (Société Chimique Pointet-Girard, Villeneuve, France) to inactivate vegetative cells, the surviving spores were plated on YEA+5 medium and incubated at 25° for 7 days. Then colony morphology was recorded, and the size (ploidy) of cells in the colonies was checked microscopically. Approximately 200 colonies per cross were streaked on YEA and YEA+5. YEA was used to reveal the state of chromosome III in the colonies (ade6 markers). YEA has a limiting amount of adenine, and adenine-dependent clones show pigmentation after 4–5 days of growth at 25° as follows: ade6-M210, dark red; ade6-M216, light red; ade6+ or complementing ade6-M210/ade6-M216 colonies are white. After 2 days of growth at 25°, the YEA+5 plates were replicated on appropriately supplemented MMA plates to check for lysine auxotrophy (lys1, marker of chromosome I), and twice on YEA+5 plates. To detect temperature-sensitive clones due to the tps13 mutation (marker on chromosome II), one of the YEA+5 plates was incubated overnight at 37°. The other YEA+5 plate was replicated on MEA+5 together with tester strains to determine the mating type of each clone. The full genotypes of the clones classified as diploid or aneuploid by the microscopic observation of cell size were determined after self-sporulation (h+/h– diploids) or after crossing with haploid wild-type tester strains of opposite mating type (h+/h+ and h–/h– diploids). In this way diploids (aneuploids) showing wild phenotypes (lysine and adenine prototrophy, temperature resistance) were checked for heterozygosity (presence of the recessive mutant alleles lys1–, ade6–, or tps13–).
Many asci with two spores were formed in crosses homozygous for rec7::ura4+. Thus the spores of such dyads were subjected to genetic analysis. After separation of the two spores by micromanipulation, their germination and division (or failure of germination or division) was recorded, in particular for those spores that did not form a colony. The clones deriving from the colony-forming spores were analyzed as described above for colonies deriving from random spores.
Isolation of cDNA clones, RNA extraction, and Northern blot hybridization: To prepare a meiosis-specific cDNA library from S. pombe, we employed two kinds of diploid cells, CD16-1 and CD16-5 (Kitamura and Shimoda 1991). CD16-1 with mating type h+/h– initiates meiosis after nitrogen starvation, whereas the homozygous diploid CD16-5 with mating type h–/h– cannot proceed to meiosis. We collected CD16-1 cells at 1-hr intervals (1–6 hr) after meiosis induction and then combined them for mRNA preparation (see below). From this RNA we prepared a cDNA library in a pAP3neo vector designed to be converted to single-stranded form after transfection with f1 helper phage (Kobori et al. 1998). We also prepared mRNA from CD16-5 cells that were collected at 2-hr intervals (0–8 hr) after nitrogen starvation, and the RNA samples were combined. This mRNA was labeled with biotin using the photobiotin system (Vector Laboratories, Burlingame, CA). It was then mixed in excess with the single-stranded cDNA library from CD16-1 cells in hybridization buffer. In this way a subtracted meiotic cDNA library was obtained as described previously (Kobori et al. 1998). The cDNA library was screened by colony hybridization using 32P-labeled double-stranded DNA as probe (Sambrook et al. 1989). The probe was a 1.2-kb BglII-BglII fragment from the vector pUC18-rec71.6 (described above). Five cDNA clones (two for rec7 and three deriving from the opposite strand, tos) were isolated and analyzed (see Figure 5 and results). Introns were confirmed by sequencing the rec7+ cDNA clones (data not shown).
For RNA isolation CD16-1 and CD16-5 cells were cultivated at 28° in PM medium to logarithmic phase (OD600 = 0.6). Then the cultures were shifted into PM-N medium and incubated under the same conditions. Cells were collected at 2-hr intervals (0–12 hr), mixed with 10% SDS, phenol/chloroform, and RNA extraction buffer, and disrupted with glass beads (Ø = 0.5 mm). The samples were centrifuged and the supernatant was treated with phenol/chloroform and then with chloroform before precipitation with ethanol. The precipitate was dissolved in H2O and again precipitated in the presence of 2 m LiCl. Ten micrograms total RNA was loaded from each time point on an agarose gel. Northern blot analysis with 32P-labeled probes was performed as described by Sambrook et al. (1989).
Complementation of rec7::ura4+: rec7+ and tos cDNA clones were amplified by PCR to introduce SalI and BamHI restriction sites at the 5′ and 3′ ends of the cDNAs, respectively. In addition, for rec7 the 5′ primer contained the six nucleotides ATGAAC coding for the first two amino acids missing in the rec7#4 clone. The resulting fragments were cloned into the SalI-BamHI site of pREP41, a vector designed for moderate expression (Basi et al. 1993). To avoid complementation defects due to a PCR mutation, Pwo polymerase with proofreading activity (Roche Diagnostics) was used and three clones for each pREP41-cDNA construct were assayed for complementation. SA5 and SA6 strains were transformed to Leu+ with the different pREP41-cDNA plasmids by the lithium acetate method (Ito et al. 1983). Cells grew on EMM + adenine medium for 5 days at 30°. Then individual colonies were streaked on the same medium and incubated for an additional 2 days at 30°. A portion of the cells was used for RNA extraction, and the remaining cells were crossed on EMM + adenine and incubated at 30° for 1 day. The five crosses were performed with SA5 and SA6 strains carrying either the empty pREP41 vector, pREP41-rec7, pREP41-tos1, pREP41-tos2, or pREP41-tos3. Northern blot experiments verified the RNA expression in the cells before crossing (data not shown). Mitotic cells carrying the pREP41-rec7 plasmid showed no morphological aberrancies and mated regularly, indicating that moderate expression of rec7 has no deleterious effect on mitotic cells (data not shown). Crossing materials were treated with snail digestive juice and spores were plated on YEA+5. Spore viability was determined after 4 days of growth at 30°. The rec+ and untransformed rec7::ura4+ control strains were crossed on EMM + adenine + leucine plates and treated as described for the transformed strains.
Construction of the rec7::GFP fusion: The sequence coding for GFP-S65T (Heim et al. 1995) was a 0.7-kb NotI fragment cloned in pBluescript (CLONTECH, Palo Alto, CA). To facilitate cloning, a NotI site was generated prior to the stop codon of rec7. First, a 79-bp 5′ and a 306-bp 3′ flanking sequence was PCR-amplified using pUC18-rec71.6 as template. Then a 385-bp StyI-SauI fragment from pUC18-rec71.6 was replaced by the 79-bp StyI-NotI and the 306-bp NotI-SauI fragments from the above amplification. The resulting plasmid was named pUC18-rec71.6+NotI. The pIRT2-rec72.8 plasmid contained a 2.8-kb BamHI restriction fragment with rec7 from pYL2 (Lin et al. 1992) subcloned in pIRT2 (Hindley et al. 1987). A 670-bp SfuI-SauI fragment of this plasmid was replaced by the same fragment from pUC18-rec71.6+NotI to introduce the NotI site. Finally, the 0.7-kb GFP fragment was cloned in the NotI site resulting in a plasmid with a C-terminal rec7::GFP fusion (pIRT2-rec72.8-GFP). Sequencing of this plasmid confirmed the proper integration of GFP. The rec7 protein sequence was changed only minimally in the construct: between the codons for lysine and methionine, which are the last two amino acids before the stop codon, two glycine and one arginine codons were introduced by generation of the NotI site. For genomic integration, first a 2.8-kb SalI-SacI fragment from pIRT2-rec72.8-GFP was subcloned into pBluescript. Then the same 2.8-kb SalI-SacI fragment from the resulting plasmid pBS-rec72.8-GFP and the plasmid pIRT2 (containing the LEU2 gene of S. cerevisiae) were used to cotransform h– leu1-32 rec7-0.2::ura4+ ura4-D18 cells (Figure 1). Leu+ transformants were selected on MMA + ura plates and replica-plated on MMA + ura + 5-fluoroorotic acid (5-FOA) to select for genomic integration (uracil auxotrophic colonies) as described by Grimm et al. (1988). Proper integration was confirmed by PCR and Southern blots (data not shown). All further rec7::GFP strains were obtained by genetic crosses.
Cytological procedures: For meiotic time-course experiments established procedures were applied (Bähler et al. 1993). Diploids SA12, SA17, and SA11 (control) were induced to undergo meiosis. Its progression was monitored by 4′,6-diamidino-2-phenylindole (DAPI) staining for prophase I (horsetail nuclei) and meiosis I (more than one nucleus). The preparation of nuclear spreads and silver staining for examination of linear element formation were described by Bähler et al. (1993). Indirect immunofluorescence experiments on nuclear spreads used the diploid strain SA17 and were carried out as described by Parisi et al. (1999), with exception of the antibodies. Rec7::GFP was detected with rabbit anti-GFP polyclonal antibody (CLONTECH) in 1:50 dilution and goat anti-rabbit IgG FITC conjugate as a secondary antibody (Sigma, St. Louis) in 1:80 dilution. Fluorescence in situ hybridization experiments were performed according to Scherthan et al. (1994).
To observe the localization of Rec7-GFP in zygotic meiosis in living cells, meiosis was induced by transferring haploid, homothallic SA16 cells onto MEA sporulation medium. Plates were incubated overnight at 26°. The next day samples were scratched off the plate at different times and cells were stained with Hoechst 33342 (1 μg/ml, in water, for 10 min) and then resuspended in EMM-N medium and mounted on a coverslip. Zygotes in different stages of the meiosis were identified by the Hoechst staining. Images were taken with a computer-controlled CCD microscope system. A Peltier-cooled CCD camera (Photometrics Ltd, Tucson, AZ) is attached to an Olympus inverted microscope IMT-2. The microscope is controlled by a Silicon Graphics Personal Iris 4D35/TG. For further details of the microscope system see Chikashige et al. (1994) and Hiraoka et al. (1991).
RESULTS
Reduced spore viability and unusual ascus morphology in a rec7 gene disruption: Mutants deficient in meiotic recombination show low spore viability, indicating the essential role of recombination for proper chromosome segregation during the first meiotic division and consequently for the generation of viable offspring. The rec7-102 mutation is probably a point mutation and severely reduces intra- and intergenic recombination throughout the genome (De Veaux and Smith 1994). It showed a spore viability of 22–30% (Ponticelli and Smith 1989). To investigate the function of rec7 in meiotic chromosome segregation, a disruption was constructed by replacement of a large part of the open reading frame (ORF) by the ura4+ marker gene (Figure 1). We have examined spore viability and ascus morphology and performed a detailed segregation analysis using rec7::ura4+ disruption strains.
To compare the spore viability of the gene disruption to that of the rec7-102 mutation, random spore analysis was carried out in crosses SA3 × SA4 and GP421 × GP431, respectively (Figure 2). The advantage of random spore analysis over analysis of tetrads (in rec7::ura4+ with varying morphology and numbers of spores) is that a representative sample of spores is selected for study without bias. Crossing of strains SA1 and SA2 served as a rec+ control. For the detailed genotype of these strains see Table 1. The spore viabilities of the gene disruption and of the rec7-102 mutation showed no significant difference (23 vs. 24%). This suggests that the rec7-102 point mutation leads to a null phenotype. Intergenic recombination in rec7::ura4+ was measured in two intervals by random spore analysis and scoring of about 200 viable spores. In the lys4-his4 interval the recombination frequency was 14.9% in wild-type strains (SA18 × SA19 cross, average of four experiments) and <0.5% in rec7 disruption strains (SA22 × SA23 cross, two experiments). For the lys3-ura1 interval 18.5% recombination frequency was obtained by crossing the SA20 and SA21 control strains (average of three experiments) and <0.5% was measured in the disruption mutant (SA24 × SA25, two experiments).
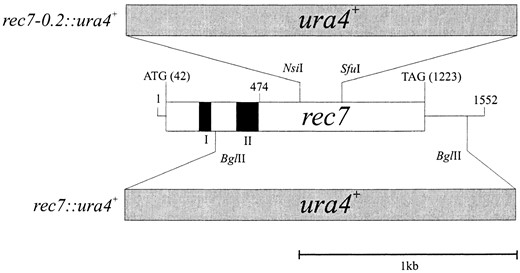
—Diagram of the rec7 gene and two disruptions. The 1.6-kb BamHI fragment shown was sequenced by Lin et al. (1992). The open box shows the ORF of the rec7 gene. Translation start (A corresponds to nucleotide position 42) and stop (G corresponds to nucleotide position 1223) are indicated. Solid boxes represent experimentally confirmed introns. Their positions are: intron I from nucleotide 195 to 251, intron II from 366 to 470. The originally published ORF of rec7 was from 474 to 1223. Nucleotide positions are according to the numbering of the DNA sequence deposited in the GenBank database (accession no. M85297; Lin et al. 1992). In the rec7::ura4+ construct the ura4+ marker gene replaced the BglII-BglII restriction fragment. This disruption mutation was used to analyze genetic and cytological phenotypes. In the rec7-0.2::ura4+ construct the marker gene was inserted between the 5′ NsiI and the 3′ SfuI restriction sites. This disruption mutation was used for the construction of the rec7::GFP strain.
The morphology of rec7::ura4+ asci is frequently aberrant (Figure 3, A and B). They contain spores of variable size, and the arrangement of spores is frequently irregular.
Strains
| Strain | Genotype | Origin |
|---|---|---|
| L972 | h – | Bern collection |
| L975 | h + | Bern collection |
| GP421 | h– ade6-52 pro2-1 ura4-595 rec7-102 | Ponticelli and Smith (1989) |
| GP431 | h+ ade6-M26 arg3-124 ura4-294 rec7-102 | Ponticelli and Smith (1989) |
| SA1 | h– leu1-32 ura4-D18 | Bern collection |
| SA2 | h+ leeu1-32 ura4-D18 | Bern collection |
| SA3 | h– leu1-32 rec7::ura4+ ura4-D18 | This study |
| SA4 | h+ leu1-32 rec7::ura4+ ura4-D18 | This study |
| SA5 | h– ade6-M216 leu1-32 rec7::ura4+ ura4-D18 | This study |
| SA6 | h+ ade6-M210 leu1-32 rec7::ura4+ ura4-D18 | This study |
| SA7 | h– ade6-M210 tps13-24 ura4-D18 | This study |
| SA8 | h+ ade6-M216 lys1-131 ura4-D18 | This study |
| SA9 | h– ade6-M216 lys1-131 rec7::ura4+ ura4-D18 | This study |
| SA10 | h+ ade6-M210 tps13-24 rec7::ura4+ ura4-D18 | This study |
| SA11 | h+/h– ade6-M210/ade6-M216 (diploid) | This study |
| SA12 | h+/h– ade6-M210/ade6-M216 leu1-32/leu1-32 rec7::ura4+/rec7::ura4+ ura4-D18/ura4-D18 (diploid) | This study |
| CD16-1 | h+/h– ade6-M210/ade6-M216 +/cyh1 +/lys5-391 (diploid) | Kitamura and Shimoda (1991) |
| CD16-5 | h–/h– ade6-M210/ade6-M216 +/cyh1 +/lys5-391 (diploid) | Kitamura and Shimoda (1991) |
| SA13 | h– leu1-32 rec7-0.2::ura4+ ura4-D18 | This study |
| SA14 | h – leu1-32 rec7::GFP ura4-D18 | This study |
| SA15 | h+ leu1-32 rec7::GFP ura4-D18 | This study |
| SA16 | h90 leu1-32 rec7::GFP ura4-D18 | This study |
| SA17 | h+/h– ade6-M210/ade6-M216 leu1-32/leu1-32 rec7::GFP/rec7::ura4+ ura4-D18/ura4-D18 (diploid) | This study |
| SA18 | h– lys4-95 ura4-D18 | Bern collection |
| SA19 | h+ his4-239 ura4-D18 | Bern collection |
| SA20 | h+ lys3-37 | Bern collection |
| SA21 | h– ura1-171 | Bern collection |
| SA22 | h– lys4-95 rec7::ura4+ ura4-D18 | This study |
| SA23 | h+ his4-239 rec7::ura4+ ura4-D18 | This study |
| SA24 | h– lys3-37 rec7::ura4+ ura4-D18 | This study |
| SA25 | h+ ura1-171 rec7::ura4+ ura4-D18 | This study |
| Strain | Genotype | Origin |
|---|---|---|
| L972 | h – | Bern collection |
| L975 | h + | Bern collection |
| GP421 | h– ade6-52 pro2-1 ura4-595 rec7-102 | Ponticelli and Smith (1989) |
| GP431 | h+ ade6-M26 arg3-124 ura4-294 rec7-102 | Ponticelli and Smith (1989) |
| SA1 | h– leu1-32 ura4-D18 | Bern collection |
| SA2 | h+ leeu1-32 ura4-D18 | Bern collection |
| SA3 | h– leu1-32 rec7::ura4+ ura4-D18 | This study |
| SA4 | h+ leu1-32 rec7::ura4+ ura4-D18 | This study |
| SA5 | h– ade6-M216 leu1-32 rec7::ura4+ ura4-D18 | This study |
| SA6 | h+ ade6-M210 leu1-32 rec7::ura4+ ura4-D18 | This study |
| SA7 | h– ade6-M210 tps13-24 ura4-D18 | This study |
| SA8 | h+ ade6-M216 lys1-131 ura4-D18 | This study |
| SA9 | h– ade6-M216 lys1-131 rec7::ura4+ ura4-D18 | This study |
| SA10 | h+ ade6-M210 tps13-24 rec7::ura4+ ura4-D18 | This study |
| SA11 | h+/h– ade6-M210/ade6-M216 (diploid) | This study |
| SA12 | h+/h– ade6-M210/ade6-M216 leu1-32/leu1-32 rec7::ura4+/rec7::ura4+ ura4-D18/ura4-D18 (diploid) | This study |
| CD16-1 | h+/h– ade6-M210/ade6-M216 +/cyh1 +/lys5-391 (diploid) | Kitamura and Shimoda (1991) |
| CD16-5 | h–/h– ade6-M210/ade6-M216 +/cyh1 +/lys5-391 (diploid) | Kitamura and Shimoda (1991) |
| SA13 | h– leu1-32 rec7-0.2::ura4+ ura4-D18 | This study |
| SA14 | h – leu1-32 rec7::GFP ura4-D18 | This study |
| SA15 | h+ leu1-32 rec7::GFP ura4-D18 | This study |
| SA16 | h90 leu1-32 rec7::GFP ura4-D18 | This study |
| SA17 | h+/h– ade6-M210/ade6-M216 leu1-32/leu1-32 rec7::GFP/rec7::ura4+ ura4-D18/ura4-D18 (diploid) | This study |
| SA18 | h– lys4-95 ura4-D18 | Bern collection |
| SA19 | h+ his4-239 ura4-D18 | Bern collection |
| SA20 | h+ lys3-37 | Bern collection |
| SA21 | h– ura1-171 | Bern collection |
| SA22 | h– lys4-95 rec7::ura4+ ura4-D18 | This study |
| SA23 | h+ his4-239 rec7::ura4+ ura4-D18 | This study |
| SA24 | h– lys3-37 rec7::ura4+ ura4-D18 | This study |
| SA25 | h+ ura1-171 rec7::ura4+ ura4-D18 | This study |
Strains
| Strain | Genotype | Origin |
|---|---|---|
| L972 | h – | Bern collection |
| L975 | h + | Bern collection |
| GP421 | h– ade6-52 pro2-1 ura4-595 rec7-102 | Ponticelli and Smith (1989) |
| GP431 | h+ ade6-M26 arg3-124 ura4-294 rec7-102 | Ponticelli and Smith (1989) |
| SA1 | h– leu1-32 ura4-D18 | Bern collection |
| SA2 | h+ leeu1-32 ura4-D18 | Bern collection |
| SA3 | h– leu1-32 rec7::ura4+ ura4-D18 | This study |
| SA4 | h+ leu1-32 rec7::ura4+ ura4-D18 | This study |
| SA5 | h– ade6-M216 leu1-32 rec7::ura4+ ura4-D18 | This study |
| SA6 | h+ ade6-M210 leu1-32 rec7::ura4+ ura4-D18 | This study |
| SA7 | h– ade6-M210 tps13-24 ura4-D18 | This study |
| SA8 | h+ ade6-M216 lys1-131 ura4-D18 | This study |
| SA9 | h– ade6-M216 lys1-131 rec7::ura4+ ura4-D18 | This study |
| SA10 | h+ ade6-M210 tps13-24 rec7::ura4+ ura4-D18 | This study |
| SA11 | h+/h– ade6-M210/ade6-M216 (diploid) | This study |
| SA12 | h+/h– ade6-M210/ade6-M216 leu1-32/leu1-32 rec7::ura4+/rec7::ura4+ ura4-D18/ura4-D18 (diploid) | This study |
| CD16-1 | h+/h– ade6-M210/ade6-M216 +/cyh1 +/lys5-391 (diploid) | Kitamura and Shimoda (1991) |
| CD16-5 | h–/h– ade6-M210/ade6-M216 +/cyh1 +/lys5-391 (diploid) | Kitamura and Shimoda (1991) |
| SA13 | h– leu1-32 rec7-0.2::ura4+ ura4-D18 | This study |
| SA14 | h – leu1-32 rec7::GFP ura4-D18 | This study |
| SA15 | h+ leu1-32 rec7::GFP ura4-D18 | This study |
| SA16 | h90 leu1-32 rec7::GFP ura4-D18 | This study |
| SA17 | h+/h– ade6-M210/ade6-M216 leu1-32/leu1-32 rec7::GFP/rec7::ura4+ ura4-D18/ura4-D18 (diploid) | This study |
| SA18 | h– lys4-95 ura4-D18 | Bern collection |
| SA19 | h+ his4-239 ura4-D18 | Bern collection |
| SA20 | h+ lys3-37 | Bern collection |
| SA21 | h– ura1-171 | Bern collection |
| SA22 | h– lys4-95 rec7::ura4+ ura4-D18 | This study |
| SA23 | h+ his4-239 rec7::ura4+ ura4-D18 | This study |
| SA24 | h– lys3-37 rec7::ura4+ ura4-D18 | This study |
| SA25 | h+ ura1-171 rec7::ura4+ ura4-D18 | This study |
| Strain | Genotype | Origin |
|---|---|---|
| L972 | h – | Bern collection |
| L975 | h + | Bern collection |
| GP421 | h– ade6-52 pro2-1 ura4-595 rec7-102 | Ponticelli and Smith (1989) |
| GP431 | h+ ade6-M26 arg3-124 ura4-294 rec7-102 | Ponticelli and Smith (1989) |
| SA1 | h– leu1-32 ura4-D18 | Bern collection |
| SA2 | h+ leeu1-32 ura4-D18 | Bern collection |
| SA3 | h– leu1-32 rec7::ura4+ ura4-D18 | This study |
| SA4 | h+ leu1-32 rec7::ura4+ ura4-D18 | This study |
| SA5 | h– ade6-M216 leu1-32 rec7::ura4+ ura4-D18 | This study |
| SA6 | h+ ade6-M210 leu1-32 rec7::ura4+ ura4-D18 | This study |
| SA7 | h– ade6-M210 tps13-24 ura4-D18 | This study |
| SA8 | h+ ade6-M216 lys1-131 ura4-D18 | This study |
| SA9 | h– ade6-M216 lys1-131 rec7::ura4+ ura4-D18 | This study |
| SA10 | h+ ade6-M210 tps13-24 rec7::ura4+ ura4-D18 | This study |
| SA11 | h+/h– ade6-M210/ade6-M216 (diploid) | This study |
| SA12 | h+/h– ade6-M210/ade6-M216 leu1-32/leu1-32 rec7::ura4+/rec7::ura4+ ura4-D18/ura4-D18 (diploid) | This study |
| CD16-1 | h+/h– ade6-M210/ade6-M216 +/cyh1 +/lys5-391 (diploid) | Kitamura and Shimoda (1991) |
| CD16-5 | h–/h– ade6-M210/ade6-M216 +/cyh1 +/lys5-391 (diploid) | Kitamura and Shimoda (1991) |
| SA13 | h– leu1-32 rec7-0.2::ura4+ ura4-D18 | This study |
| SA14 | h – leu1-32 rec7::GFP ura4-D18 | This study |
| SA15 | h+ leu1-32 rec7::GFP ura4-D18 | This study |
| SA16 | h90 leu1-32 rec7::GFP ura4-D18 | This study |
| SA17 | h+/h– ade6-M210/ade6-M216 leu1-32/leu1-32 rec7::GFP/rec7::ura4+ ura4-D18/ura4-D18 (diploid) | This study |
| SA18 | h– lys4-95 ura4-D18 | Bern collection |
| SA19 | h+ his4-239 ura4-D18 | Bern collection |
| SA20 | h+ lys3-37 | Bern collection |
| SA21 | h– ura1-171 | Bern collection |
| SA22 | h– lys4-95 rec7::ura4+ ura4-D18 | This study |
| SA23 | h+ his4-239 rec7::ura4+ ura4-D18 | This study |
| SA24 | h– lys3-37 rec7::ura4+ ura4-D18 | This study |
| SA25 | h+ ura1-171 rec7::ura4+ ura4-D18 | This study |
DAPI staining of asci shows rather unequal distribution of nuclear material between spores. Generally, the larger the spores are, the smaller is their number in the asci. The bigger spores contain more DAPI-stainable material than do the smaller ones. Figure 3C shows a quantification of rec7::ura4+ asci according to their spore number. In this experiment zygotic asci produced in SA5 × SA6 (rec7::ura4+ strains) and standard h+ × h– crosses were classified by their spore number. A similar phenotype was observed in azygotic sporulation of diploid rec7 disruption strains (data not shown). Asci with two large spores were abundant in rec7::ura4+ crosses. Their morphology suggested that they might carry diploid spores.
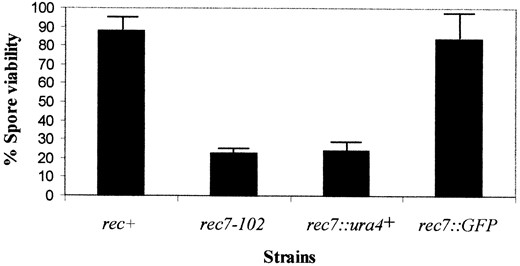
—Spore viabilities measured in rec+, rec7-102, rec7::ura4+, and rec7::GFP crosses. All the data presented are the average of five independent random spore analysis experiments. The error bars indicate the standard deviation.
rec7 disruption strains frequently form spores giving rise to colonies with diploid cells homozygous for centromere markers: The low spore viability and irregular ascus formation in the mutant imply a high level of chromosomal missegregation during the meiotic divisions. To gain more information, the segregation analysis described by Molnar et al. (1995) was applied to rec7::ura4+ in a modified form. In fission yeast most aneuploid spores are not able to form colonies with maintenance of the initial aneuploid state. When colonies are formed, the resulting cells tend to have euploid genomes derived from the aneuploid state by chromosome loss. Only disomics for chromosome III proliferate for many generations. They form small colonies with frequent segregation of haploid cells (Niwa and Yanagida 1985; Molnar et al. 1995). Therefore, direct detection of aneuploid progeny is based on marking chromosome III of the strains to be crossed with two different ade6 alleles. ade6-M210 confers dark red colony color and ade6-M216 light red colony color. Colonies containing haploid cells with two different ade6 alleles arise from aneuploid spores. Heteroallelic diploids are prototrophic due to interallelic complementation and form white colonies. Centromere markers allow the distinction of first and second division segregation patterns in diploid spores. The markers lys1, tps13, and ade6 map 4, 2, and 13 cM from their respective centromeres on the chromosomes I, II, and III (Kohli et al. 1977). Reduction of recombination in crosses homozygous for rec7 mutations (Ponticelli and Smith 1989; results above) is likely to tighten the centromere linkage of these markers.
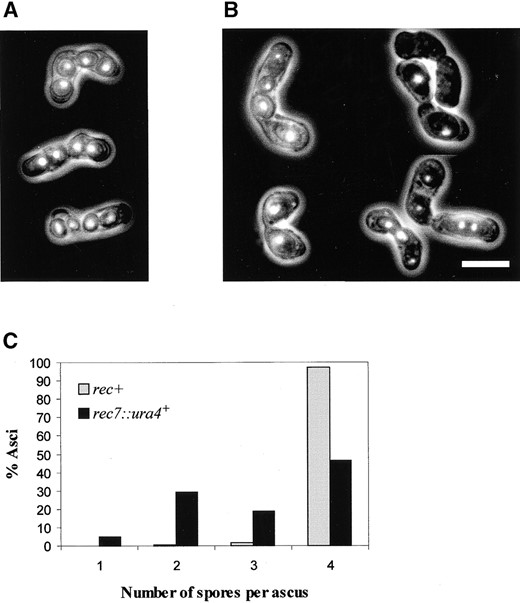
—Irregular ascus formation in rec7 disruption mutants. Morphology of zygotic asci of (A) rec+ and (B) rec7::ura4+ strains. The DNA content of the spores was visualized by DAPI staining. Photographs were taken with double illumination (UV and visible light). In contrast to the uniform spore size and DNA staining in the control, the rec7::ura4+ asci vary in spore number and spore size, and their spores have different DNA amounts. (C) Spore number in rec+ and rec7::ura4+ zygotic asci. Strains were crossed by standard genetic methods and spore numbers were determined by phase contrast microscopy in ∼200 mature asci. The averages of three experiments are given for the control and mutant crosses.
Chromosome segregation was analyzed in random spores by crossing of SA7 with SA8 (rec+) and SA9 with SA10 (rec7::ura4+). The spores were plated on full medium. The morphologies of the resulting colonies (size and shape) were recorded as well as the size of the cells in the colonies: small cells are diagnostic for haploidy, large cells for diploidy. Then the full genotype of the cells of each colony was determined as described in materials and methods. Table 2 shows the classification of colonies according to genotype (centromere marker patterns), cell size, and deduced ploidy of the spores that gave rise to the colonies.
The control cross yielded uniformly medium-sized, regularly shaped colonies with small cells. Thus, the great majority of the spores were haploid. The only missegregations detected were three disomics for chromosome III (1.4%). In contrast, the size and shape of
Random spore analysis of chromosome segregation in rec7::ura4+ and rec+ crosses
| Segregation of markersa | No. of colonies | |||||
|---|---|---|---|---|---|---|
| lys1 | tps13 | ade6 | Cell size | Deduced ploidy of spore | rec7 | rec+ |
| + or – | + or – | M210 or M216 | Haploid | Haploid | 87 | 213 |
| + or – | + or – | M210/M216 | Haploid | Disomic chromosome IIIb | 7 | 3 |
| + + or –– | ++ or –– | M210/M210 or M216/M216 | Diploid | Diploid, homozygous for centromere markers | 84 | 0 |
| + / – | +/– | M210/M216 | Diploid | Diploid, heterozygous for centromere markers | 1 | 0 |
| + + or –– | ++ or –– | M210/M210 and M216/M216 | Diploid | Aneuploid (2n + 1 or 2) chromosome IIIb | 24 | 0 |
| + / – | ++or –– | M210/M210 or M216/M216 | Diploid | Aneuploid (2n + 1 or 2) chromosome Ib | 6 | 0 |
| + + or – – | +/– | M210/M210 or M216/M216 | Diploid | Aneuploid (2n + 1 or 2) chromosome IIb | 5 | 0 |
| + / – | ++or –– | M210/M216 | Diploid | Aneuploid (2n + 1 or 2) chromosome I and IIIb | 1 | 0 |
| + / – | +/– | M210/M210 or M216/M216 | Diploid | Aneuploid (2n + 1 or 2) chromosome I and IIb | 1 | 0 |
| Total: 216 | 216 | |||||
| Segregation of markersa | No. of colonies | |||||
|---|---|---|---|---|---|---|
| lys1 | tps13 | ade6 | Cell size | Deduced ploidy of spore | rec7 | rec+ |
| + or – | + or – | M210 or M216 | Haploid | Haploid | 87 | 213 |
| + or – | + or – | M210/M216 | Haploid | Disomic chromosome IIIb | 7 | 3 |
| + + or –– | ++ or –– | M210/M210 or M216/M216 | Diploid | Diploid, homozygous for centromere markers | 84 | 0 |
| + / – | +/– | M210/M216 | Diploid | Diploid, heterozygous for centromere markers | 1 | 0 |
| + + or –– | ++ or –– | M210/M210 and M216/M216 | Diploid | Aneuploid (2n + 1 or 2) chromosome IIIb | 24 | 0 |
| + / – | ++or –– | M210/M210 or M216/M216 | Diploid | Aneuploid (2n + 1 or 2) chromosome Ib | 6 | 0 |
| + + or – – | +/– | M210/M210 or M216/M216 | Diploid | Aneuploid (2n + 1 or 2) chromosome IIb | 5 | 0 |
| + / – | ++or –– | M210/M216 | Diploid | Aneuploid (2n + 1 or 2) chromosome I and IIIb | 1 | 0 |
| + / – | +/– | M210/M210 or M216/M216 | Diploid | Aneuploid (2n + 1 or 2) chromosome I and IIb | 1 | 0 |
| Total: 216 | 216 | |||||
Location of markers: lys1, tps13, and ade6 are centromere-linked markers on chromosome I, II, and III, respectively. The genotypes of the cells in the colonies resulting from the spores are shown.
The colonies in these classes were mostly small and of irregular shape. Colonies of the other classes were mostly large and of regular shape.
Random spore analysis of chromosome segregation in rec7::ura4+ and rec+ crosses
| Segregation of markersa | No. of colonies | |||||
|---|---|---|---|---|---|---|
| lys1 | tps13 | ade6 | Cell size | Deduced ploidy of spore | rec7 | rec+ |
| + or – | + or – | M210 or M216 | Haploid | Haploid | 87 | 213 |
| + or – | + or – | M210/M216 | Haploid | Disomic chromosome IIIb | 7 | 3 |
| + + or –– | ++ or –– | M210/M210 or M216/M216 | Diploid | Diploid, homozygous for centromere markers | 84 | 0 |
| + / – | +/– | M210/M216 | Diploid | Diploid, heterozygous for centromere markers | 1 | 0 |
| + + or –– | ++ or –– | M210/M210 and M216/M216 | Diploid | Aneuploid (2n + 1 or 2) chromosome IIIb | 24 | 0 |
| + / – | ++or –– | M210/M210 or M216/M216 | Diploid | Aneuploid (2n + 1 or 2) chromosome Ib | 6 | 0 |
| + + or – – | +/– | M210/M210 or M216/M216 | Diploid | Aneuploid (2n + 1 or 2) chromosome IIb | 5 | 0 |
| + / – | ++or –– | M210/M216 | Diploid | Aneuploid (2n + 1 or 2) chromosome I and IIIb | 1 | 0 |
| + / – | +/– | M210/M210 or M216/M216 | Diploid | Aneuploid (2n + 1 or 2) chromosome I and IIb | 1 | 0 |
| Total: 216 | 216 | |||||
| Segregation of markersa | No. of colonies | |||||
|---|---|---|---|---|---|---|
| lys1 | tps13 | ade6 | Cell size | Deduced ploidy of spore | rec7 | rec+ |
| + or – | + or – | M210 or M216 | Haploid | Haploid | 87 | 213 |
| + or – | + or – | M210/M216 | Haploid | Disomic chromosome IIIb | 7 | 3 |
| + + or –– | ++ or –– | M210/M210 or M216/M216 | Diploid | Diploid, homozygous for centromere markers | 84 | 0 |
| + / – | +/– | M210/M216 | Diploid | Diploid, heterozygous for centromere markers | 1 | 0 |
| + + or –– | ++ or –– | M210/M210 and M216/M216 | Diploid | Aneuploid (2n + 1 or 2) chromosome IIIb | 24 | 0 |
| + / – | ++or –– | M210/M210 or M216/M216 | Diploid | Aneuploid (2n + 1 or 2) chromosome Ib | 6 | 0 |
| + + or – – | +/– | M210/M210 or M216/M216 | Diploid | Aneuploid (2n + 1 or 2) chromosome IIb | 5 | 0 |
| + / – | ++or –– | M210/M216 | Diploid | Aneuploid (2n + 1 or 2) chromosome I and IIIb | 1 | 0 |
| + / – | +/– | M210/M210 or M216/M216 | Diploid | Aneuploid (2n + 1 or 2) chromosome I and IIb | 1 | 0 |
| Total: 216 | 216 | |||||
Location of markers: lys1, tps13, and ade6 are centromere-linked markers on chromosome I, II, and III, respectively. The genotypes of the cells in the colonies resulting from the spores are shown.
The colonies in these classes were mostly small and of irregular shape. Colonies of the other classes were mostly large and of regular shape.
colonies derived from spores of the mutant cross were variable. As judged from microscopic and genetic analysis, most of the large colonies contained large diploid cells. Almost always the medium-sized colonies contained small haploid cells. Small colonies were shown to derive mostly from aneuploid spores. Haploid spores formed a minority (40%) among the meiotic products of rec7::ura4+. Most striking were the diploid colonies (large cells) homozygous for all three centromere markers (39% of the colonies). It was concluded that the original spores giving rise to this class were diploid. The second remarkable observation was the frequent occurrence of colonies with large cells (diploid) and segregation of colony colors, and/or heterozygosity for chromosome I, and/or heterozygosity for chromosome II markers (17%). These colonies were often small. It was concluded that they originated from 2n + 1 or 2n + 2 aneuploid spores. It could not be decided whether the original spores carried one or two extra chromosomes over the diploid chromosome set. For further interpretation see discussion. The frequency of missegregation in rec7::ura4+ is underestimated in this study, because the chromosomal configurations that allow survival, and thus experimental examination, represent only a fraction of the missegregation products.
Skipping of meiosis II and nondisjunction I in rec7::ura4: Some of the types of chromosomal missegregation can be examined directly by dyad or tetrad dissection. The different segregation types are illustrated in Figure 4. Our genetic assay for chromosome III allows the detection of nondisjunction at meiosis I (Figure 4B) and also of precocious separation of sister chromatids (Figure 4C). Nondisjunction at meiosis II (Figure 4D) and chromosome loss (failure of attachment to spindle) cannot be diagnosed with our assay. The frequent occurrence of two-spored asci (Figure 3) and the high frequency of diploid and hyperdiploid spores (Table 2) suggested that for rec7::ura4+, dyad rather than tetrad dissection would be the more effective method for diagnosing segregation errors. Under the assumption that dyads result from omission of meiosis II, the middle row of Figure 4 shows the relevant configurations.
For this study, 85 dyads were dissected from the cross SA9 × SA10 (Table 3). The spores were monitored for germination and the growing colonies were analyzed for random spores as described in materials and methods and above. From 42 dyads two colonies were obtained. In 39 dyads out of these 42, both spore colonies revealed homozygosity for all centromere markers. Obviously in these asci meiosis was interrupted after the first division (Figure 4A, middle row). From a single ascus one diploid colony showed homozygosity, the other diploid heterozygosity for all centromere markers. This may be the result of a rare triploid or tetraploid zygote undergoing meiosis interruption followed by chromosome loss during colony formation. Two asci yielded a haploid and a diploid colony, the latter homozygous for centromere markers. In these asci chromosome loss and interruption of the meiotic process might have occurred.
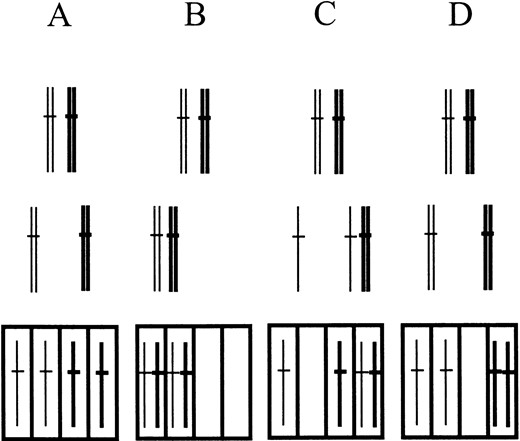
—Types of chromosome segregation. The diagrams refer to any of the three S. pombe chromosomes, but in the genetic assay applied, only chromosome III is considered. Thick lines relate to dark red colony color conferred by the ade6-M210 allele. Thin lines represent chromosomes III with the ade6-M216 allele, yielding light red colony color. Top, pairing of homologous chromosomes in meiotic prophase. Middle, two nuclei after meiosis I. Bottom, four nuclei after meiosis II. (A) Regular meiotic divisions. (B) Nondisjunction I. (C) Precocious separation of sister chromatids. (D) Nondisjunction II.
In 23 dyads only one spore was able to form a colony. Nine of these spores were classified as aneuploids for chromosome III. The first three of them yielded three different diploid segregants upon restreaking the original spore colony: ade6-M210/ade6-M216 (white), ade6-M210/ade6-M210 (dark red), ade6-M216/ade6-M216 (light red). The siblings of these colony-forming spores were not able to divide (microscopic examination), most probably due to the lack of chromosome III. These dyads give direct evidence for nondisjunction I, combined with omission of meiosis II (Figure 4B, middle row). The colonies from six other spores were homogeneously white upon restreaking, but turned out to be heterozygous for ade6 after backcrossing (dark red and light red segregants). The sibling spores of three of them did not divide at all, whereas those of the three others divided at least once but were unable to form a colony. These six cases may derive from nondisjunction I or precocious sister chromatid separation (Figure 4C, middle row). The missegregation is again combined with omission of a meiotic division and subsequent loss of chromosomes during colony development. Four dyad spore colonies with heterozygosity for chromosome II (one self-sporulating tps13/+ clone) or chromosome I (three lys1/+ clones revealed by backcrossing) were also observed. The colonies from the remaining 10 dyad spores were either diploid (homozygous for all centromere
Classification of rec7::ura4+ dyads
| Deduced genotypes of colony-forming spores | No. of dyads |
|---|---|
| Dyads with two colony-forming spores | 42 |
| 2n/2n, both homozygous for centromere markers | 39 |
| 2n/2n, one homo-, other heterozygous for centromere markers | 1 |
| n/2n, diploid homozygous for centromere markers | 2 |
| Dyads with one colony-forming spore | 23 |
| 2n + 2 chromosome III, spore colonies heterogeneousa | 3 |
| 2n + 2or2n + 1 chromosome III, spore colonies homogeneous | 6 |
| 2n + 2or2n+ 1 chromosome II, spore colony homogeneous | 1 |
| 2n + 2or2n+ 1 chromosome I, spore colonies homogeneous | 3 |
| 2n, homozygous for centromere markers | 9 |
| n or aneuploid, spore colony haploid | 1 |
| Dyads with no colony-forming spore | 20 |
| Total | 85 |
| Deduced genotypes of colony-forming spores | No. of dyads |
|---|---|
| Dyads with two colony-forming spores | 42 |
| 2n/2n, both homozygous for centromere markers | 39 |
| 2n/2n, one homo-, other heterozygous for centromere markers | 1 |
| n/2n, diploid homozygous for centromere markers | 2 |
| Dyads with one colony-forming spore | 23 |
| 2n + 2 chromosome III, spore colonies heterogeneousa | 3 |
| 2n + 2or2n + 1 chromosome III, spore colonies homogeneous | 6 |
| 2n + 2or2n+ 1 chromosome II, spore colony homogeneous | 1 |
| 2n + 2or2n+ 1 chromosome I, spore colonies homogeneous | 3 |
| 2n, homozygous for centromere markers | 9 |
| n or aneuploid, spore colony haploid | 1 |
| Dyads with no colony-forming spore | 20 |
| Total | 85 |
These colonies were found to consist of cells with three different genotypes at the ade6 locus: ade6-M210/ade6-M216, ade6-M210/ade6-M210, ade6-M216/ade6-M216.
Classification of rec7::ura4+ dyads
| Deduced genotypes of colony-forming spores | No. of dyads |
|---|---|
| Dyads with two colony-forming spores | 42 |
| 2n/2n, both homozygous for centromere markers | 39 |
| 2n/2n, one homo-, other heterozygous for centromere markers | 1 |
| n/2n, diploid homozygous for centromere markers | 2 |
| Dyads with one colony-forming spore | 23 |
| 2n + 2 chromosome III, spore colonies heterogeneousa | 3 |
| 2n + 2or2n + 1 chromosome III, spore colonies homogeneous | 6 |
| 2n + 2or2n+ 1 chromosome II, spore colony homogeneous | 1 |
| 2n + 2or2n+ 1 chromosome I, spore colonies homogeneous | 3 |
| 2n, homozygous for centromere markers | 9 |
| n or aneuploid, spore colony haploid | 1 |
| Dyads with no colony-forming spore | 20 |
| Total | 85 |
| Deduced genotypes of colony-forming spores | No. of dyads |
|---|---|
| Dyads with two colony-forming spores | 42 |
| 2n/2n, both homozygous for centromere markers | 39 |
| 2n/2n, one homo-, other heterozygous for centromere markers | 1 |
| n/2n, diploid homozygous for centromere markers | 2 |
| Dyads with one colony-forming spore | 23 |
| 2n + 2 chromosome III, spore colonies heterogeneousa | 3 |
| 2n + 2or2n + 1 chromosome III, spore colonies homogeneous | 6 |
| 2n + 2or2n+ 1 chromosome II, spore colony homogeneous | 1 |
| 2n + 2or2n+ 1 chromosome I, spore colonies homogeneous | 3 |
| 2n, homozygous for centromere markers | 9 |
| n or aneuploid, spore colony haploid | 1 |
| Dyads with no colony-forming spore | 20 |
| Total | 85 |
These colonies were found to consist of cells with three different genotypes at the ade6 locus: ade6-M210/ade6-M216, ade6-M210/ade6-M210, ade6-M216/ade6-M216.
markers) or haploid. They are not informative with respect to the nature of the missegregation events. For a full interpetation of the different classes of spore colonies see discussion.
Structure and expression of the rec7 gene: A renewed analysis of the rec7 sequence revealed two putative introns and led to a 5′ extension of the ORF (Y. Lin and G. R. Smith, personal communication; Figure 1). To clarify the gene structure, a meiotic cDNA library [primed with oligo(dT)] was screened by colony hybridization for rec7+ clones. With a 1.2-kb BglII fragment of pUC18-rec71.6, 11 hybridization-positive colonies were identified by screening 2 × 105 colonies. Five independent cDNA clones were recovered (Figure 5) and their sequences were determined. All of them carried runs of A deriving from poly(A)-RNA at one end. The rec7#2 and rec7#4 cDNA clones correspond to the ORF of the rec7+ gene. Analysis of these cDNA clones confirmed the two introns in the 5′ region. These clones carried exon I but did not fully extend to the putative initiation codon. The smaller cDNA clones tos1, tos2, and tos3 were interpreted to derive from transcripts of the complementary strand of the rec7+ gene (tos: transcript from opposite strand). They do not contain open reading frames longer than 110 amino acids.
Lin et al. (1992) reported an unusually long (5 kb) transcript for rec7 in pat1 mutant-induced meiosis. To examine the gene expression in wild-type meiosis, the diploid strain CD16-1 h+/h– was induced to undergo synchronous meiosis. As a control the nonsporulating diploid CD16-5 h–/h– was used. Total RNA was extracted at regular intervals and the expression of rec7 was analyzed by Northern hybridization with double-stranded probes derived from different locations in the rec7 region (Figure 5). At 4–12 hr into meiosis four transcripts were detected in the sporulating strain (CD16-1). In addition to the major signal at 8.2 kb, three minor signals appeared at 12, 2.7, and 1.6 kb with probe 4 and probe rec7#2. A similar expression pattern was observed with rec7-specific double-stranded DNA probe when mRNA was blotted from the time-course of strain SA11 (data not shown). To determine the origin of the long transcript, the Northern blot was hybridized with probes located 5′ of the ORF. Probes 1, 2, and 3 did not show the pattern of signals obtained with probes 4 and rec7#2 (Figure 5). This indicates that the 12- and the 8.2-kb RNAs do not derive from the upstream region of rec7, but from the rec7 gene itself and possibly its downstream region. The four transcripts shown in Figure 5 (probes 4 and rec7#2) were not found in Northern blots of RNA from the control strain CD16-5 h–/h– (data not shown).
Complementation of rec7::ura4+ by rec7+ but not by tos cDNAs: To test whether the rec7 ORF and/or the tos trancripts are responsible for rec7 function, complementation of the rec7 disruption by defined cDNA clones was attempted. Obviously, the deletion of the 1.2-kb BglII-BglII restriction fragment in rec7::ura4+ (Figure 1) should abolish or severely shorten the transcript with the rec7 ORF and also the tos1 and tos2 RNAs from the opposite strand (Figure 5). Expression of the tos3 RNA may still occur. Full-length rec7 cDNA and the tos cDNAs were cloned into the pREP41 vector (Basi et al. 1993). The orientation of the inserted DNA assured expression of the presumed rec7 mRNA and the presumed tos RNAs indicated in Figure 5. The SA5 and SA6 rec7 disruption strains were transformed with the pREP41 vector and the different pREP41-cDNA constructs. Transformants of different mating type were crossed and the resulting spores were checked for viability. The disruption control (SA5 × SA6) and the pREP41-transformed disruption strains showed 24 and 25% spore viability, respectively (averages of three experiments). This indicates that the vector alone had no effect. Transformation of pREP41-rec7 fully restored spore viability (91%, average of three experiments). The rec+ control cross (h+ × h–) showed on average 97% spore viability in two experiments. In contrast, transformation with the pREP41-tos constructs did not result in significant increases of spore viability. Viabilities of 30% for tos1, 28% for tos2, and 26% for tos3 were observed (averages of three experiments). This suggests that the studied meiotic phenotype (spore viability) is due to lack of Rec7 protein encoded by the rec7 mRNA. The tos RNAs are not sufficient for wild-type spore viability. But, their requirement for meiosis was not excluded by the experiments.
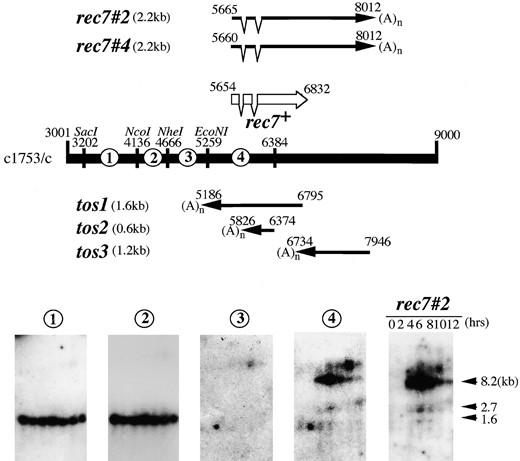
—Transcription at the rec7 gene region. The bar represents the sequence of cosmid c1753 (GenBank accession no. AL 035210) between nucleotides 3001 and 9000. The arrows show the localization of the five sequenced clones isolated from a meiotic cDNA library. The clones rec7#2 and rec7#4 missed 11 and 6 nucleotides of the ORF starting with ATG at nucleotide 5654 (42 in Figure 1). DNA fragments denoted by 1, 2, 3, 4, and the rec7#2 cDNA clone were used to probe the Northern blot shown below. RNA extracted from h+/h– diploid cells undergoing meiosis (time points 0–12 hr) was blotted and four RNA species of 1.6, 2.7, 8.2, and 12 kb in size were revealed with the probes 4 and the rec7#2. RNA isolated from a G1 arrested h–/h– diploid did not reveal these four RNAs, but showed the RNA species identified with probes 1 and 2 (data not shown).
Localization of Rec7p in whole cells and on nuclear spreads: To localize the protein in the cell, we tagged rec7 with the green fluorescence protein (GFP-S65T; Heim et al. 1995) at the C terminus and integrated the construct into the genome. Crosses homozygous for rec7::GFP had wild-type levels of spore viability, indicating that Rec7p is fully active in the construct (crosses of SA14 and SA15, Figure 2).
To observe the localization of Rec7-GFP in zygotic meiosis, strain SA16 (h90 haploid) was transferred to sporulation medium. Samples of living cells taken at different time points were first observed after staining with Hoechst dye for definition of the state of mating or meiosis. Then, images of GFP fluorescence were taken in 10 optical sections covering the whole nucleus and, after deconvolution, were projected into two dimensions. After mating of cells and before karyogamy Rec7-GFP foci were already visible (Figure 6a). Single foci were visible in the two unfused nuclei of the zygote. The number of foci increased up to three per haploid nucleus during karyogamy (Figure 6b) and then further in the horsetail nuclei (Figure 6c). At the end of the horsetail movement the number of foci decreased (Figure 6d), but small numbers of foci were clearly present after the first meiotic division (Figure 6, e and f).
Another approach to study the expression and localization of Rec7p is indirect immunofluorescence on nuclear spreads of azygotic meiosis. Time-course experiments were performed with the h+/h– diploid strain SA17 bearing the rec7::GFP construct (Figure 7). Fluorescence microscopy revealed that Rec7p was localized to distinct foci within the nucleus (Figure 7A). No staining above background was observed when nuclear spreads of the untagged SA11 strain were probed with anti-GFP antibody in control time-course experiments (data not shown). The nuclei contained on average ∼50 foci at 6, 8, and 10 hr after meiosis induction (Figure 7B). At 12 hr the average number of foci decreased (27 foci per nucleus), suggesting degradation of the protein later in meiosis. Later time points were difficult to examine due to poor spreading of the sporulating cells. Figure 7C correlates the presence of Rec7p foci with the cytological landmarks of meiosis. Foci first appeared at 6 hr when the number of horsetail nuclei began to increase. The horsetail shape is a consequence of the intense nuclear movement during meiotic prophase (Chikashige et al. 1994). Therefore, these nuclei are good indicators of meiotic prophase. The abundance of nuclei with foci correlated well with the number of horsetail nuclei during the time-course.
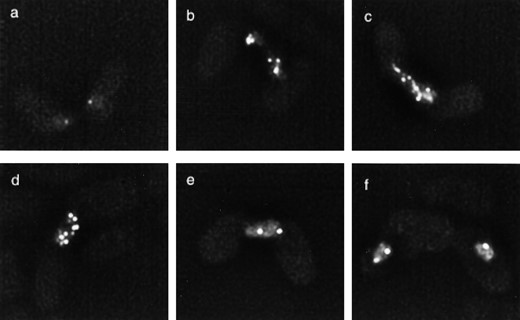
—Localization of Rec7-GFP in zygotic meiosis. Living homothallic cells of strain SA16 undergoing mating and meiosis were observed at different stages by fluorescence microscopy. (a) Early karyogamy. (b) Karyogamy. (c) Typical horsetail nucleus. (d) Nucleus at end of horsetail movements. (e) Nucleus at meiosis I. (f) Nucleus at meiosis II. For further explanation see the text.
DISCUSSION
S. pombe is a highly recombination-proficient eukaryote whose meiosis shows some features not seen in many eukaryotes (see Introduction). Recombination-deficient mutants have been isolated (Ponticelli and Smith 1989; De Veaux et al. 1992). Analysis of their phenotypes and characterization of the corresponding genes and their products are expected to contribute to a better understanding of meiosis. Here we report the construction and characterization of the rec7::ura4+ gene disruption, the examination of the rec7 gene structure and expression, the analysis of the chromosome segregation defect, and tagging of the Rec7 protein and its localization in the cells.
The rec7-102 point mutation showed low spore viability and severely reduced meiotic recombination (Ponticelli and Smith 1989; De Veaux and Smith 1994). De Veaux and Smith (1994) reported that some rec– mutants reduced meiotic recombination to a greater extent in certain genomic regions than in others and called the phenomenon region specificity. The rec7-102 mutation showed no region specificity. The disruption studied here showed similar phenotypes: low spore viability and strongly reduced intergenic recombination in the intervals lys4-his4 on chromosome II and lys3-ura1 on chromosome I.
rec7 gene structure and expression: By isolation and sequencing of rec7 cDNA clones, the existence of two introns at the 5′ end of the gene was confirmed (Figure 1). The two cDNA clones missed a few nucleotides and did not extend to the predicted translation initiation codon (Figure 5). But expression of a cDNA clone completed with the missing nucleotides restored wild-type spore viability to the rec7::ura4+ disruption strain. This suggests that the nucleotide position 42 is the true translation initiation point (Figure 1). Sequence comparison with the corrected ORF showed a similarity of the predicted Rec7 protein to the Rec114 protein of S. cerevisiae (Fox and Smith 1998).
In addition, three cDNA clones derived from RNA transcribed from the opposite strand were sequenced (Figure 5). No ORF longer than 110 amino acids was found in the cDNA clones tos1, tos2, and tos3. None of the pREP41-tos constructs resulted in rescue of spore viability when transformed into the rec7::ura4+ disruption strain, while transformation with the completed rec7 cDNA resulted in full complementation. This shows that Rec7 protein is required for wild-type meiosis. The individual tos RNAs are not sufficient for rescue of the spore viability defect. Nevertheless, an in vivo function for the tos RNAs cannot be excluded.
Multiple transcripts were observed in a diploid wild-type strain undergoing azygotic meiosis (Figure 5). The longest transcripts, which are likely to extend downstream of rec7, may be related to the unusually long transcript reported by Lin et al. (1992) in pat1-induced haploid meiosis. The S. pombe genome sequencing project reported no ORF downstream of rec7 (http://www.sanger.ac.uk/Projects/S_pombe/). The long transcripts may arise from incomplete transcription termination. Multiple transcripts and/or unusually long mRNAs have been detected for several early recombination genes in S. cerevisiae, including REC114 (Pittman et al. 1993), MEI4 (Menees et al. 1992), MEK1 (Rockmill and Roeder 1991), and MER1 (Engebrecht et al. 1991). The biological significance of this phenomenon is not clear.
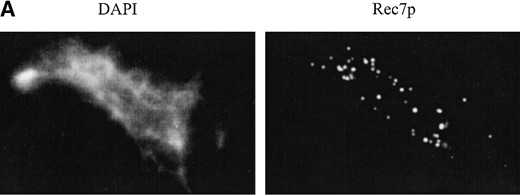
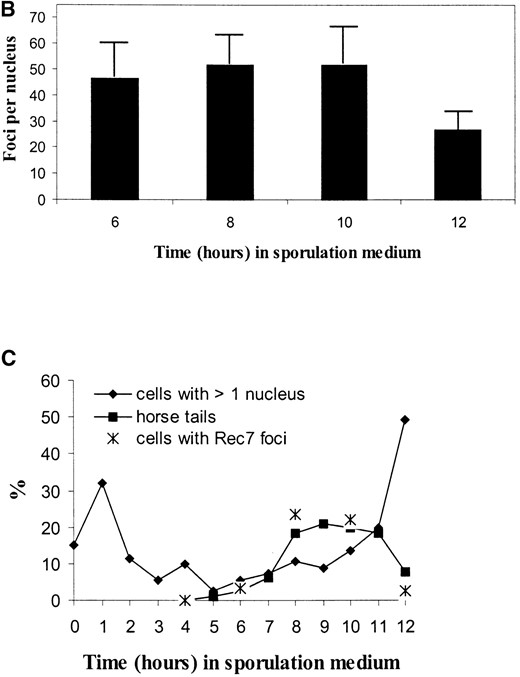
—Immunolocalization of Rec7p-staining foci in a meiotic time-course experiment. Nuclear spreads were prepared from rec7::GFP diploid cells (SA17) 0, 2, 4, 6, 8, 10, and 12 hr after transfer to meiotic medium. Spread nuclei were indirectly stained for Rec7p using polyclonal anti-GFP antibody. Timing of cytological events during meiosis is compared to the appearance of Rec7p foci. (A) Example of a spread nucleus 8 hr after meiotic induction. (B) Quantitation of Rec7p-staining foci during meiosis. Twenty well-spread nuclei were examined at each time point. The error bars indicate standard deviations. (C) Timing of cytological events during meiosis. Horsetail nuclei indicate meiotic prophase. Cells with more than one nucleus have completed the first meiotic division. The increase of cells with two nuclei at 1 hr after induction of meiosis is due to the final mitotic division, before the cells enter meiosis from the G1 phase.
Cytological analysis of rec7::ura4: Meiotic time-course experiments (azygotic meiosis) with wild-type and rec8 mutant strains of S. pombe for the study of nuclear structures were carried out before (Bähler et al. 1993; Scherthan et al. 1994; Molnar et al. 1995). The same experiments with spreading of meiotic nuclei followed by silver staining and electron microscopy (visualization of linear elements) or by fluorescence in situ hybridization (evaluation of sister chromatid cohesion, of chromosome pairing, and of the bouquet structure by checking for clustering of telomeres and centromeres) were carried out with a rec7::ura4+ disruption strain (SA12) and compared to a wild-type control (SA11). This series of experiments was carried out twice. No differences between rec7::ura4+ and wild type were found with respect to all the mentioned features (data not shown). Linear element formation was also checked in zygotic meiosis (mating of heterothallic strains), and again no changes were found. In addition, the timing of meiotic events, as monitored by DAPI staining of cells at different time points (appearance of horsetail nuclei, first meiotic division) was identical for rec7::ura4+ and wild type (data not shown).
Cervantes et al. (2000) showed that double-strand break formation is strongly reduced in a rec7 mutant. This is also the case in mutants of REC114, the homologous early recombination gene of S. cerevisiae (Pittman et al. 1993; Bullard et al. 1996). S. cerevisiae rec114 mutants and other early recombination mutants undergo meiosis I earlier than wild type. It is likely that this phenotype is related to a function of the early recombination genes in the coordination of recombination and meiosis I (Jiao et al. 1999). Shortening of meiotic prophase was not observed in rec7::ura4+. Mutants of the cohesin gene rec8 are defective in sister chromatid cohesion and chromosome pairing and in the formation of linear elements (Parisi et al. 1999). None of these phenotypes or other irregularity was found in rec7:: ura4+, indicating that the region-specific recombination genes rec8, rec10, and rec11 (De Veaux and Smith 1994) have functions that differ from the function of rec7.
Intranuclear localization of the Rec7 protein: The study of living cells carrying the rec7-GFP fusion and undergoing conjugation and zygotic meiosis resulted in an unexpected observation. The Rec7 protein was found to be present in the nuclei before karyogamy (Figure 6). At karyogamy up to three foci were detected in each haploid nucleus. This number corresponds intriguingly to the chromosome number in haploid nuclei. Multiple signals were found during prophase I (horsetail nuclei, Figure 6c). Before meiosis I occurred, the number of foci was reduced again. Some Rec7-GFP signals persisted after meiosis I (Figure 6f).
These observations suggest that Rec7 has an early function before meiosis and recombination. But this role is not essential, since rec7 mutants do mate and undergo zygotic meiosis. The early appearance of the protein could not be detected on nuclear spreads in meiotic time-course experiments of azygotic meiosis (Figure 7, see below). Differences may exist between zygotic and azygotic meiosis. Yamamoto et al. (1999) measured intergenic recombination frequencies in zygotic and azygotic meiosis of wild-type strains and found an approximately twofold higher frequency in zygotic meiosis. The obvious differences between the two modes of meiosis concern conjugation and karyogamy, which are absent in azygotic meiosis. Surprising also is the persistence of Rec7 protein up to meioisis II. This again indicates a function of the protein in processes after completion of recombination, possibly in chromosome segregation. Further analysis of the distribution and state of the Rec7 protein in the different stages of meiosis is necessary. More information on the functions of Rec7p may also be revealed by identification of nuclear structures that it associates with.
Spreading of nuclei leads to separation of Rec7 foci over an extended area (Figure 7). With the help of an anti-GFP antibody ∼50 foci were counted per nucleus. Observation of living cells (Figure 6) does not allow resolution of foci to the same extent. The frequency of spread nuclei showing foci correlated well with the percentage of horsetail nuclei at the different stages of the time-course (Figure 7C). Intriguingly, the number of foci corresponds well with the average number of 45 crossovers occurring in S. pombe meiosis (Munz 1994). Thus, the Rec7 protein is likely to have functions in protein complexes assembling at sites of recombination initiation or at intermediates of recombination.
The role of Rec7p in chromosome segregation and meiotic divisions: We analyzed meiotic chromosome segregation in the rec7 disruption mutant. Different defects in meiosis are expected to lead to different missegregation types. Absence of crossovers is expected to lead to random segregation of homologous chromosomes, resulting in the nondisjunction I missegregation type for some of the chromosomes (Figure 4B). Failure of sister chromatid cohesion leads to precocious separation of sister chromatids (PSSC; Figure 4C). These missegregation types can be identified in our segregation system, but nondisjunction at meiosis II (e.g., two identical copies of chromosome III, Figure 4D) and chromosome loss are not detectable by this approach.
Random spore analysis revealed the frequent occurrence of two irregular meiotic products in rec7::ura4+ that were not detected in the control (Table 2). A large fraction of the spores (39%) grew into large diploid colonies with first division segregation for all the centromere markers. We interpret these as direct diploid products of meiosis. A total of 17% of the colonies were mostly small due to slow cell division or frequent cell death, but carried diploid cells in their mature stage. The centromere markers showed mixed patterns: first and second division segregations for different chromosomes were observed in the same spore. We classify these spores as aneuploids and propose that they arose from the combination of either nondisjunction I (2n + 2) or PSSC (2n + 1) with the skipping of one of the meiotic divisions. These results, taken together with the frequent occurrence of two- and three-spored asci in the mutant (Figure 3B), suggest that in rec7 at least two defects occur frequently, one of which is failure of the second meiotic division.
Dyad analysis confirmed the frequent occurrence of normal meiosis I combined with skipping of meiosis II. In total, 46% of the dyads were of this type, containing two normally growing diploid colonies homozygous for centromere markers (Table 3). About half of the dyads with only one colony-forming spore were diagnostic of missegregation. The three genetically heterogeneous spore clones are best explained by nondisjunction I of chromosome III and failure of meiosis II. During growth of the resulting 2n + 2 spores the supernumerary chromosomes were then lost. The spontaneous loss of chromosomes from aneuploid genotypes during mitotic growth is well documented in S. pombe (Kohli et al. 1977). Ten other spore colonies were genetically homogeneous, but heterozygous for centromere markers of individual chromosomes. It is likely that these clones also derive from a missegregation event, either nondisjunction I (2n + 2 spore) or PSSC (2n + 1 spore), in combination with failure of meiosis II. Unlike for rec8-110 (Molnar et al. 1995), PSSC was not detected by fluorescence in situ hybridization in rec7::ura4+ (see above), suggesting that nondisjunction I is the more likely explanation for the missegregation events in question. An alternative origin of diploid spores with mixed centromere marker pattern (homozygosity for some, heterozygosity for other chromosomes) could be mixed segregation of chromosomes in a single meiotic division. Hugerat and Simchen (1993) reported mixed division patterns in the single division occurring in spo13 meiosis of S. cerevisiae. If mixed segregation occurs in rec7::ura4+, it should be detectable in full dyads. Although dyads with two viable spores were frequent (49%), mixed segregation patterns were not detected. Therefore, we favor nondisjunction I as the main type of missegregation in rec7::ura4+.
With lower frequency than for chromosome III, nondisjunction I was also detected for chromosomes I and II (Table 3). Disomics for chromosome III were demonstrated to be stable enough so that disomic colonies can be obtained (Niwa and Yanagida 1985). This is not the case for aneuploidy of chromosomes I or II. But our results indicate that some spores hyperdiploid for chromosome I or II are able to divide and to lose chromosomes until euploidy is reached. By microscopic observation, it was also found that many spores presumed to be aneuploid did not germinate or died after a few divisions. This is in accordance with a study on polyploid S. pombe strains: tri- and tetraploid strains are mitotically unstable and segregate diploid and haploid progeny (Molnar and Sipiczki 1993).
While the occurrence of nondisjunction I was expected for a strongly recombination-deficient mutant, it was surprising to find frequent omission of meiosis II (30% of asci, Figure 3C). This was not the case in rec8 mutants, which produce only a low number of diploid spores (Parisi et al. 1999; Molnar et al. 1995). In contrast to rec8, rec7 mutants show a strong reduction of recombination throughout the genome and absence of double-strand breaks (Cervantes et al. 2000). Failure of initiation of recombination by double-strand break formation, or a later event in such an abnormal meiosis, may trigger a block for meiosis II. The Rec7 protein may be involved in this regulation of meiosis II. Other early recombination mutants of fission yeast also show high frequencies of dyads (e.g., rec15, our unpublished results).
Also surprising is the high frequency of dyads with two viable spores showing first division segregation for all three chromosomes (46%, Table 3). On the basis of random segregation only 12.5% were expected. This indicates that an alternative mechanism for meiosis I chromosome segregation not requiring crossover formation may exist in fission yeast. Mechanisms for achiasmate segregation of chromosomes have been described in other organisms (Hawley and Theurkauf 1993; Loidl et al. 1994). Achiasmate segregation and omission of meiosis II improve spore viability in the absence of crossovers.
The possible roles of Rec7p in meiosis: There are no known sequence motifs in Rec7p that would shed light on its biochemical activities. The homology of Rec7p to Rec114p of S. cerevisiae suggests a conserved role for these proteins in the initiation of meiotic recombination. This is confirmed by the absence of double-strand break formation in a rec7 mutant (Cervantes et al. 2000). Rec7p is localized to ∼50 foci in the prophase nuclei (Figure 7). This number roughly corresponds to the number of crossovers per S. pombe meiosis (45; Munz 1994). If Rec7p localizes to the initiation sites and is involved in the generation of both conversion and crossover recombination products, a two- to threefold higher number of Rec7p foci per nucleus would be expected. This discrepancy may be due to technical problems, or Rec7p may have an additional function at crossover sites, after double-strand break formation. The discovery that Rec7p is already present in haploid nuclei before karyogamy (Figure 6) may indicate an additional, albeit nonessential, role before meiosis. A late role after prophase I and up to meiosis II is indicated by the prolonged presence of Rec7p (Figure 6). This coincides with the possible role of Rec7p in the regulation of meiosis II discussed above. The presence throughout meiosis is in accordance with a possible involvement of Rec7p in the regulation of several meiotic processes, including meiosis II.
Footnotes
Communicating editor: G. R. Smith
Acknowledgement
We thank Jürg Bähler for the GFP-S65T-containing plasmid. This work was supported by grants from the Swiss National Science Foundation, the Human Frontier Science Program and the Japan Science and Technology Corporation.
LITERATURE CITED
Author notes
These authors contributed equally to this study.
Present address: Novartis Crop Protection AG, Biochemistry, CH-4002 Basel, Switzerland.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}