





Abstract
Genetic analyses of reproductive barriers represent one of the few methods by which theories of speciation can be tested. However, genetic study is often restricted to model organisms that have short generation times and are easily propagated in the laboratory. Replicate hybrid zones with a diversity of recombinant genotypes of varying age offer increased resolution for genetic mapping experiments and expand the pool of organisms amenable to genetic study. Using 88 markers distributed across 17 chromosomes, we analyze the introgression of chromosomal segments of Helianthus petiolaris into H. annuus in three natural hybrid zones. Introgression was significantly reduced relative to neutral expectations for 26 chromosomal segments, suggesting that each segment contains one or more factors that contribute to isolation. Pollen sterility is significantly associated with 16 of these 26 segments, providing a straightforward explanation of why this subset of blocks is disadvantageous in hybrids. In addition, comparison of rates of introgression across colinear vs. rearranged chromosomes indicates that close to 50% of the barrier to introgression is due to chromosomal rearrangements. These results demonstrate the utility of hybrid zones for identifying factors contributing to isolation and verify the prediction of increased resolution relative to controlled crosses.
STUDENTS of speciation have long been interested in the architecture of barriers to interspecific gene flow. Genetic architecture provides a means for testing theories of speciation (reviewed in Coyne and Orr 1998), offers the opportunity to reconstruct the sequence of genetic changes that accompany or facilitate speciation (Bradshaw et al. 1995, 1998), and provides a predictive framework for interpreting patterns of introgression across hybrid zones (Barton and Hewitt 1985). Additionally, it is hoped that different modes and tempos of speciation will be characterized by different genetic architectures, perhaps providing insights into the frequencies of different speciational processes in nature and the speed with which they occur (e.g., Ungerer et al. 1998).
Hybrid zones provide a unique opportunity for investigating genetic architecture (Barton and Hewitt 1985, Harrison 1990). Unlike controlled hybridization experiments that necessarily are limited to one or a few generations of recombination, hybrid zones contain a wide variety of genotypes that result from hundreds or even thousands of generations of recombination. Thus, it becomes theoretically possible to distinguish between the effects of very closely linked genes even in areas of low recombination. In addition, because the fitness of hybrid genotypes is tested under natural conditions, all components of the barrier to interspecific gene flow or introgression are represented. Finally, hybrid zones may provide an opportunity for genetic study of long-lived or difficult to propagate organisms.
Several different approaches can be used to describe genetic architecture in natural hybrid zones. One of these is based on cline theory and uses estimates of the width of the region of reduced viability, dispersal rate, patterns of linkage disequilibria, and strength of selection against hybrids to determine gene number (Barton and Hewitt 1981, 1985; Szymura and Barton 1991). Although this approach may provide better estimates of gene numbers and overall selection than can typically be obtained from laboratory experiments, it necessarily represents a coarse tool for the study of genetic architecture. It is ill suited, for example, for identifying individual loci that contribute to reduced hybrid fitness, the effects of each locus, and their interactions and chromosomal locations.
A second approach that has been suggested by several authors relies on differential patterns of introgression across hybrid zones (Hunt and Selander 1973; Dowling et al. 1989; Harrison 1990). Introgression of loci (and linked markers) contributing to isolation is expected to be retarded, whereas neutral or positively selected chromosomal blocks (and linked markers) should introgress at higher frequencies (Barton and Hewitt 1985). If the markers have been genetically mapped, the observed patterns of introgression should also make it possible to locate chromosomal blocks contributing to isolation. This approach should allow even small fitness effects to be detected, because block frequencies in natural hybrid zones will be based on the cumulative effects of many generations of selection.
Finally, with the availability of mapped molecular markers, it should also be possible to map specific traits that contribute to isolation in natural hybrid zones such as reduced hybrid fertility or habitat preference (Harrison 1990). In contrast to the previous approach, which relies on introgressed marker frequencies to infer the location of loci under selection, this method searches for correlations between the mapped markers and the trait of interest. This approach can therefore be viewed as a kind of marker-based quantitative trait locus (QTL) analysis, but in natural hybrid zones rather than experimental populations.
The purpose of this article is to study the genetic architecture of the barrier to introgression in three natural hybrid zones between two wild sunflower species, Helianthus annuus and H. petiolaris. The effects of introgressed blocks on hybrid fitness will be inferred from their frequencies. Moreover, by analyzing three independent hybrid zones between the same two species, we hope to more reliably discriminate between patterns of genomic introgression resulting from drift and those due to deterministic forces (i.e., selection). In addition to analyses of the frequency of introgressed blocks, we also have searched for correlations between introgressed blocks and hybrid pollen sterility, perhaps the most important isolating mechanism between these species.
Because the reproductive barrier between H. annuus and H. petiolaris comprises both chromosomal and genic factors (Rieseberg et al. 1995b), genomic patterns of introgression can also be used to address a long-standing controversy concerning the role of chromosomal rearrangements in reproductive isolation and speciation. Some authors have advocated a primary role for chromosomal rearrangments in speciation (e.g., White 1978; King 1993), whereas others have noted that chromosomal differences often have little effect on meiosis or fertility, suggestive of a minimal role in reproductive isolation (John 1981; Sites and Moritz 1987; Coyne et al. 1993). Analyses of patterns of introgression in experimental hybrids between H. annuus and H. petiolaris are consistent with the view that chromosomal rearrangements are a significant impediment to gene flow between these two species (Rieseberg et al. 1995b, 1996a). However, this result has not been replicated in natural hybrid zones. Here we compare rates of introgression across linkage groups that carry rearrangements with those that are completely colinear. Reduced introgression in rearranged linkage groups would imply an important role for chromosomal rearrangements as reproductive barriers, at least in this species pair.
Study species: Helianthus annuus and H. petiolaris are self-incompatible diploid (n = 17) annuals with similar geographic distributions, but H. annuus prefers heavier clay-based soils, whereas H. petiolaris is restricted to drier, sandy soils (Heiser 1947). When these habitats are adjacent, mosaic hybrid zones form, with hybridization frequencies ranging from 4 to 15% (Rieseberg et al. 1998). Although pollen viability in the F1 hybrids averages 5.6% (Ungerer et al. 1998), some backcross and F2 progeny are invariably produced, and hybrid zones include a wide diversity of genotypes, ranging from nearly sterile early generation hybrids to fertile plants resembling one of the parental species (Rieseberg et al. 1998).
Comparative genetic linkage mapping indicates that the two species are divergent chromosomally, differing by a minimum of seven interchromosomal translocations and three inversions (Rieseberg et al. 1995c). The three inversions are nested within translocated chromosome blocks. In all, 10 chromosomes differ in structure between the two species, whereas the remaining 7 chromosomes appear to be collinear. Multivalent configurations predicted by the translocations include two trivalents, two quadrivalents, and one hexavalent. Likewise, bridge and fragment formation is predicted in hybrid meiosis as the result of the inversion polymorphisms.
Meiotic observations of interspecific hybrids between H. annuus and H. petiolaris (Heiser 1947; Whelan 1979; Ferriera 1980; Chandler et al. 1986) are largely consistent with predicted configurations based on the comparative mapping studies. All of the studies report multivalent configurations in meiosis, including trivalents, quadrivalents, and hexavalents. In addition, the latter three studies report the presence of one or more chromosome bridges and acentric fragments, a finding consistent with the presence of paracentric inversions.
MATERIALS AND METHODS
Plants: Plants from three mosaic hybrid zones between H. annuus and H. petiolaris were analyzed. All three hybrid zones occur in Keith County (Co.), Nebraska (Figure 1) and were the subject of a previous allozyme investigation of mating patterns (Rieseberg et al. 1998). Rieseberg et al. (1998) also provide detailed locality information. Isozyme and morphological data indicate that the three hybrid zones are very narrow, typically <50 m in width. Genotypes are distributed according to habitat within each hybrid zone, with H. petiolaris-like individuals found in higher, drier microhabitats, and H. annuus-like individuals in lower, wetter microhabitats. All three zones occur on sites created by human disturbance and probably originated when the county was homesteaded in the early 1900s.
For each hybrid zone, we collected achenes from 10 maternal plants that resembled H. annuus morphologically and thus probably represented later generation backcrosses toward H. annuus. This sampling strategy facilitates comparisons with data from experimental introgression lines that involve back-crosses in the same direction (Rieseberg et al. 1995b, 1996a).
Maternal plants were selected at 1- to 2-m intervals along a transect from the center of the hybrid zone, where early generation hybrid plants predominate, to the edge of the hybrid zone, where only parental-like individuals were found. Four to five achenes from each maternal plant were propagated in Indiana University greenhouses. Leaf tissue was collected from a total of 139 juvenile individuals and used for DNA isolations.
To estimate allele frequencies in the parental species, molecular data were also gathered from three “pure” populations of each species (Figure 1). For H. annuus, 15 individuals were sampled from a population in Keith Co., NE (annuus-1; N = 15; locality data in Rieseberg et al. 1998), ∼3 km from the three hybrid zones. Given the possibility that this population might have been exposed to interspecific gene flow with H. petiolaris, the remaining samples (10 from each population) came from more distant, allopatric locations: annuus-2 (4.8 km east of Weed, Hwy. 97, Siskiyou Co., CA; Rieseberg 102) and annuus-3 (40 km east of Fort Stockton, Hwy. 18, Pecos Co., TX; Rieseberg 1095). A similar strategy was taken for H. petiolaris. Fourteen individuals were sampled from a population in Arthur Co., NE (petiolaris-1; locality data in Rieseberg et al. 1998), ∼30 km from the three hybrid zones. The remaining samples (10 from each population) came from more distant locations: petiolaris-2 (1.7 km south of Jct. Hwy. 264 and Hwy. 6, Navaho Co., AZ; Rieseberg 1106) and petiolaris-3 (0.4 km north of Canadian River, I-44, Cleveland Co., OK; Rieseberg 1224).
Laboratory methods: DNAs were isolated and purified as described by Rieseberg et al. (1995b). Briefly, fresh leaf tissue was ground in a CTAB extraction buffer (Whitkus et al. 1992), filtered through a layer of miracloth (Calbiochem, San Diego), and extracted following the method of Doyle and Doyle (1987). Pelleted DNAs were dissolved in TE, further purified using the ELU-QUICK DNA purification kit (Schleicher and Schuell, Keene, NH), and then quantified on a fluorometer.
Eighty-eight randomly amplified polymorphic DNA (RAPD) markers specific to H. petiolaris were chosen from the genetic linkage maps of H. petiolaris and H. anomalus (Rieseberg et al. 1995c; Ungerer et al. 1998); H. anomalus is a stabilized hybrid derivative of H. annuus and H. petiolaris (Rieseberg et al. 1995c). The markers represent all 17 sunflower linkage groups and cover 57.8% of the sunflower genome as mapped by our group (Rieseberg et al. 1995c; Ungerer et al. 1998). Previous mapping studies indicate that these markers are heritable and reproducible (Rieseberg et al. 1995c; Ungerer et al. 1998). To verify that the bands scored in individuals from the hybrid zone were indeed homologous to those previously mapped, homology relationships for each RAPD marker were tested following the methodology of Rieseberg (1996). Putatively homologous RAPD fragments from the three hybrid zones, two adjacent parental populations, and one of the original mapping populations (one individual/band per population) were gel isolated and individually restricted with HaeIII and HinFI. Identical restriction profiles were taken as evidence of fragment homology. Observations of linkage disequilibrium between linked markers in the three hybrid zones provide further confirmation of marker homologies.
RAPD amplifications followed the general procedure of Williams et al. (1990). The amplifications were carried out in a total volume of 25 μl starting with 1 μl (10 ng) of purified template DNA, 10 pmol of primer, and a final concentration of 2 mm MgCl2, 30 mm Tricine, 50 mm KCl, 5% acetamide, 100 μm each dNTP, and 1 unit of Taq DNA polymerase. The reactions were overlaid with mineral oil and placed in an MJ Research (Watertown, MA) Thermal Cycler programmed for an initial denaturization step of 1 min at 94°, 45 cycles of 1 min at 94°, 1 min at 36° (52° for RAPD primers UBC801-900), and 2 min at 72°, and a final extension at 72° for 7 min. Amplification products were separated by electrophoresis in 1.5% TBE agarose gels and detected by staining with ethidium bromide.
Map distances and marker orders: In previous studies of hybrids between H. annuus and H. petiolaris, map distances were based on a map of H. annuus (Rieseberg et al. 1995b, 1996a,b). However, several linkage groups had very small map distances (i.e., low recombination rates) in H. annuus, creating the illusion of tight linkage between markers that were more widely spaced in linkage maps of H. anomalus and H. petiolaris. For this reason, the map distances employed for graphical purposes in the present study (Figures 4 and 5) represent averages over all three maps. However, the order of markers is that of H. annuus because most of the plants from the hybrid zones belong to the same fertility group as H. annuus (Rieseberg et al. 1998) and thus seem likely to share its chromosome structure.
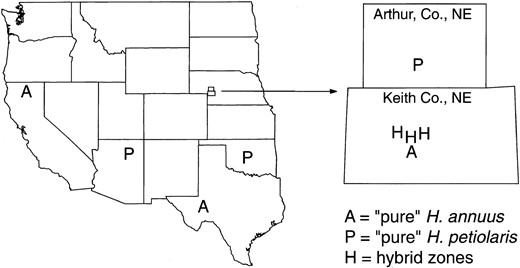
—Collection localities of H. annuus, H. petiolaris, and hybrid zones.
Expected frequencies of introgressed markers: To determine whether individual markers or chromosomal fragments are under significant positive or negative selection, we must estimate expected frequencies of marker introgression under neutral conditions. Because of our imprecise knowledge of the genealogical history of each individual, we cannot use pedigree information to calculate expected marker proportions in natural hybrid zones. However, in previous studies of an experimental introgression line between H. annuus and H. petiolaris (58 individuals tested), it was shown that for linkage groups that were colinear between the two species, proportions of introgressed markers per individual did not differ significantly from neutral expectations (Rieseberg et al. 1995b). If we assume this is the case for natural hybrid zones, we can use marker introgression frequencies from collinear linkages to provide an estimate of expected rates of introgression under neutral conditions. Combining this with the known parental allele frequencies for each locus (from the adjacent parental populations), we can generate a measure of what overall introgression would be for each individual under neutral conditions. This measure is a maximum likelihood estimate of the proportion of alleles from colinear linkages in an individual that are derived from H. petiolaris, based on that individual’s allelic makeup and the known frequencies of the alleles at each locus in the parental reference populations. Because ecological selection is likely in the wild, but not under greenhouse conditions, the frequency of H. petiolaris-derived alleles is likely to be lower in wild hybrid zones than in the experimental hybrids. This approach may lead to an underestimate of the number of underrepresented (negatively selected) loci and an overestimate of overrepresented (positively selected) loci. Thus, estimates provided here of the number and magnitude of chromosomal segments contributing to isolation are conservative.
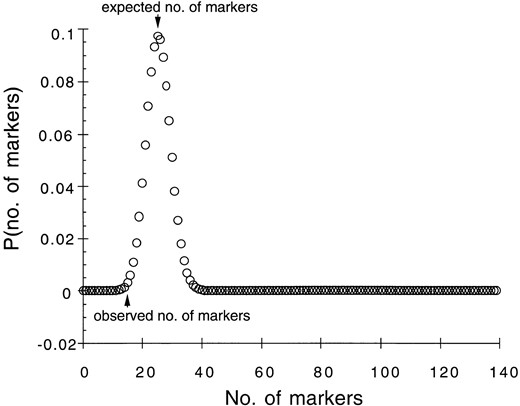
—Probability distribution for number of introgressed markers at locus D4-1.6 (linkage B).
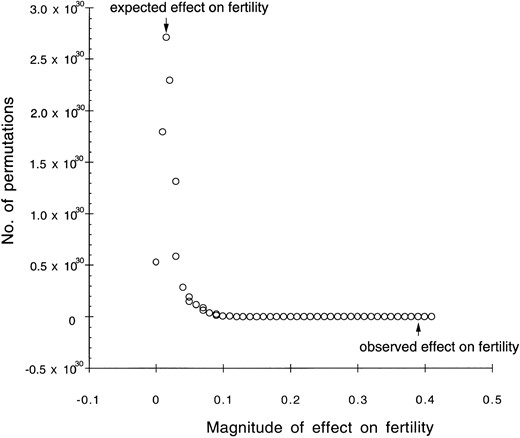
—Probability distribution for number of permutations against magnitude of marker effect on fertility for locus D4-1.6 (linkage B).
Linkage among markers on the same linkage group should lead to correlated states and deviations, and we corrected for these associations so as to estimate the actual effects of each locus. We were unable to estimate the exact effects physical linkage should have on associations because we do not know the number of generations of recombination that created the genotypes analyzed. Thus, we have simply used overall associations among markers on the same linkage group to account for physical linkage, even though this must necessarily include the effects of epistatic selection within linkage groups. For this conservative estimate of linkage effects, we calculated ρ, the Pearson correlation coefficient for discrete variables (Bishop et al. 1975), between all pairwise combinations of linked loci (JMP 3.1.5, SAS Institute Inc.). This provides a statistical measure of the extent to which any two loci covary in this population. The deviation of introgressed marker count from expectation at a locus could then be scaled by the magnitude of the effect of neighboring loci (e.g., a ρ value of 0.5 would change a deviation of +6 to +3) and the likelihood ratio test was recalculated. Individual loci that retained significant deviations after all pairwise effects of linked loci were taken into account were considered to represent a single factor. In addition, other loci that remained significant in the presence of these previously inferred loci, but which had significant (reciprocal) effects on each other were considered a single factor.
Fertility analysis: Pollen viabilities in parental individuals ranged from 0.92 to 1.00, and repeated analyses of pollen from selected plants revealed substantial variance around estimates for individual plants (SE = 2.3/100 grains counted). As a result, hybrids with pollen viabilities between 0.9 and 1.0 were treated as fully fertile. For consistency, the remaining hybrid viability scores were pooled into categories of equal size (0.8-0.9, 0.7-0.8, etc.).
We could not perform a standard marker-based QTL analysis because our sample populations resulted from an unknown number of overlapping hybrid generations. However, we could test marker-trait associations for significance using a permutation test (Good 1994). Given the distribution of pollen viabilities among individuals and introgressed marker frequencies, we generated a probability distribution of mean pollen viabilities for each marker, assuming random associations among markers and pollen viability scores (see Figure 3 for an example of an observed probability curve). The relatively small number of possible fertility scores enabled us to compute all the possible permutations of fertility scores against markers for the introgression frequencies of interest. We then compared the observed mean pollen viability score for each locus to the expected mean using a likelihood ratio test, with two log-likelihood units with Bonferroni correction as the significance cutoff (Sokal and Rohlf 1995). To account for the effects of physical linkage on fertility associations, we again used estimates of the correlation coefficient, ρ, between all pairwise combinations of linked loci, to scale the observed fertility reduction at a locus by the effects of neighboring loci. Because low fertility appears to be associated with early generation hybrids, we were concerned that the significant effects of some loci might be spurious due to associations with other unlinked loci (i.e., genome-wide linkage disequilibria). Thus, we also repeated the above test for pairwise interactions between all unlinked loci in the genome.
RESULTS
Marker frequencies in parental populations: The 88 RAPD markers employed in this experiment occurred at high frequencies in the three presumably pure populations of H. petiolaris (mean frequencies were 0.82, 0.82, and 0.83 in petiolaris-1, petiolaris-2, and petiolaris-3, respectively) and were completely absent in the two allopatric populations of H. annuus (annuus-2 and annuus-3). However, 17 of the 88 markers occurred at low frequencies (mean introgression = 0.017) in the parapatric H. annuus population, suggestive of limited introgression into this population.
Linkage disequilibrium and marker orders along linkage groups: Patterns of linkage disequilibrium along linkage groups were largely consistent with previously determined marker orders (Rieseberg et al. 1995c, Ungerer et al. 1998). That is, marker associations were highest between adjacent markers and declined with map distance as would be predicted. In several instances, however, markers spanning translocation breakpoints showed little disequilibria (a predicted result since these markers are not linked in H. petiolaris). Another exception was observed for linkage group U, in which markers, although clearly linked, exhibited patterns of associations that were not consistent with marker orders. This may be due to an inversion polymorphism within one of the parental species. Alternatively, marker orders for linkage group U may have been incorrect in the original mapping populations for some unknown reason.
Introgression: All but four of the tested individuals from the three hybrid zones had a hybrid ancestry, with the frequency of introgressed markers per individual ranging from 0 to 0.65. Taking into account allele frequencies of the parental populations, this translates into hybrid index scores of 0-0.59, with a mean hybrid index of 0.063. These data are consistent with morphological observations, suggesting that most plants represent advanced generation backcrosses toward H. annuus.
A broad comparison of the frequency of marker introgression in the 7 colinear linkages vs. the 10 rearranged linkages shows that the average frequency of introgressed markers in the rearranged linkages is approximately half that of colinear linkages for all three hybrid zones (hybrid zone 1: 0.102 colinear vs. 0.063 rearranged, t = 3.30, P = 0.0018; hybrid zone 2: 0.178 colinear vs. 0.102 rearranged, t = 5.04, P < 0.0001, hybrid zone 3: 0.116 colinear vs. 0.059 rearranged, t = 3.64, P = 0.0007; all zones: 0.131 colinear vs. 0.074 rearranged, t = 6.89, P < 0.0001). This indicates that chromosomal rearrangements represent a substantial impediment to introgression. Because the three inversions are nested within translocated chromosome blocks, it was difficult to distinguish between the effects of translocations and inversions on introgression. Rates of introgression across inverted chromosomal blocks were lower than rates across chromosomal blocks with translocation breakpoints only, but the difference was not significant. Likewise, no significant correlations were observed between the complexity of predicted multivalent configurations in meiosis (trivalents vs. quadrivalents vs. hexavalents) and rates of introgression.
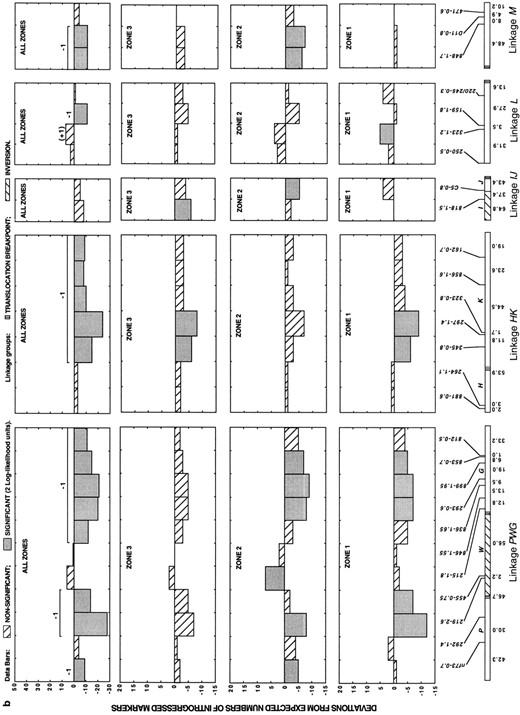
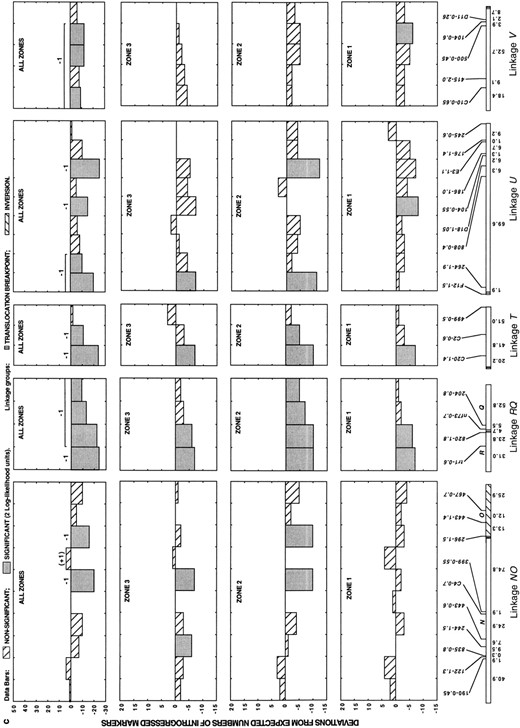
In addition to differences in the degree of introgression between colinear and rearranged linkage groups, our analysis revealed substantial heterogeneity in the introgression rates of markers within each of these genomic regions (Figure 4). Nonetheless, patterns of marker introgression were strikingly consistent across the three hybrid zones (Figure 4). In no instance did the same marker introgress at significantly higher than expected frequencies in one hybrid zone and at significantly lower than expected frequencies in a second hybrid zone. Furthermore, of the 88 markers, 55 (63%) introgressed at frequencies that deviated from expectations in the same direction in all three hybrid zones (the average pairwise correlation between the deviation scores in the three hybrid zones is 0.68; P < 0.0001 for all comparisons). The consistency of introgression across the three zones indicates that much of the genome is under selection and that drift has had a minor effect on the frequencies of most markers.
In all three hybrid zones, almost all overrepresented markers were in the colinear part of the genome, whereas most underrepresented markers mapped to rearranged linkages (Table 1; Figure 4). Pooled data for the three hybrid zones (Figure 4) revealed that 42 markers introgressed at significantly lower than expected frequencies (11 from colinear and 31 from rearranged linkages). A total of 8 markers from the pooled data set introgressed at significantly higher than expected frequencies and map to colinear linkages.
Because markers that deviate significantly from neutrality are sometimes on the same linkage group, the effects of linkage must be considered before chromosomal segments contributing to isolation can be counted. Thus, we calculated a maximum likelihood estimate of the pairwise associations (ρ) between all markers on each linkage group as described in materials and methods. Marker deviations that remained significant after the effects of linkage were removed were considered to represent independent chromosomal blocks.
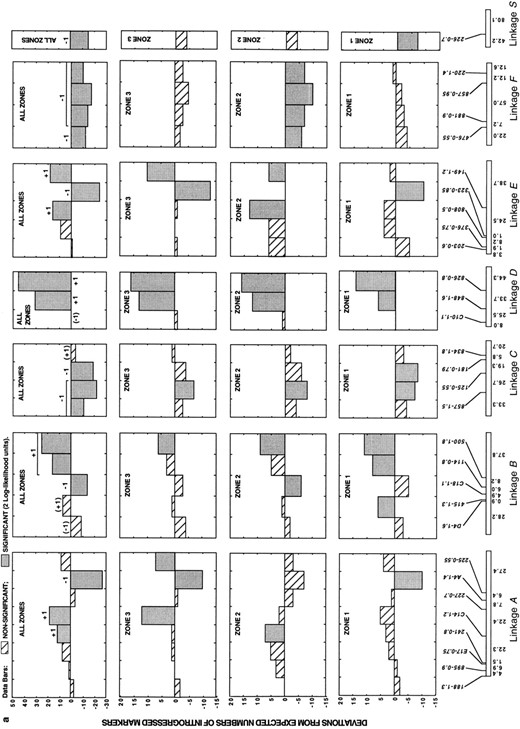
—Direct count deviations from the expected numbers of introgressed markers in three natural hybrid zones between H. annuus and H. petiolaris, as well as from a pooled data set. For the pooled data set, independently selected chromosomal blocks (after consideration of linkage effects) are indicated by +1 or -1, depending on the direction of selection. Parentheses indicate additional segments that appear to be affected by selection after consideration of linkage effects. The sizes of the independently selected chromosomal segments are indicated by a line above the data bars.
Map distances are given below each linkage group and represent averages across three genetic maps for wild sunflowers (Rieseberg et al. 1995c; Ungerer et al. 1998). Markers are given above each linkage group and are shown in the same order as found in H. annuus. Marker nomenclature includes, from bottom to top, the primer designation and the size in base pairs of the segregating fragment scored (primers A1-A20, B1-B20, C1-C20, D1-D20, E1-E20, and F1-F20 are from Operon Technologies; primers 100-900 are from the University of British Columbia Biotechnology Center). (a) Colinear linkage groups. (b and c) Rearranged linkage groups.
—(continued).
—(continued).
For the colinear linkages, the 11 markers that introgressed at significantly lower than expected frequencies represent a minimum of eight negatively selected chromosomal blocks. Presumably, each block contains one or more genes that are disadvantageous in hybrids, but the possibility they contain small or “cryptic” chromosomal rearrangements cannot be ruled out. An additional two blocks are tightly linked to positively selected loci (linkages B and C; Figure 4) and appear to be disadvantageous when the effects of linkage are considered.
For the 10 rearranged linkage groups, 16 negatively selected chromosomal blocks are required to explain patterns of introgression. Of these, 2 occur within inversions (linkage blocks W and O; Figure 4), suggesting that they may result from inversion polymorphisms. Several other negatively selected blocks include markers that are closely adjacent (<25 cM) to translocation breakpoints. However, in only 3 such blocks (on linkages T, RQ, and U; Figure 5) are the markers closest to the translocation breakpoint most strongly negatively selected. Presumably, these 3 blocks are negatively selected due to translocations. The remaining 11 negatively selected blocks within the rearranged linkage groups most likely carry genes that reduce hybrid fitness (although their low frequency in the hybrid zones may result from both genic and chromosomal factors). Summarizing across the entire genome, 21 genic and at least 5 chromosomal factors were detected that isolate these species (Figure 4; Table 1).
Although the genetic basis of isolation was the focus of this study, 11 chromosomal segments appeared to be advantageous in hybrids (Figure 4; Table 1). However, this result should be viewed with caution because our analytical methods may overestimate positive selection.
Analyses of pollen fertility: The most obvious isolating mechanism between H. annuus and H. petiolaris is the reduction in pollen viability observed in early generation hybrids (Heiser 1947). Searches of the pooled data set for correlations between pollen viability and the 88 molecular markers revealed that 60 markers representing 21 independent chromosomal blocks were significantly associated with reduced pollen fertility (Figure 5). Ten of the fertility blocks were found on colinear linkage groups, suggesting that at least part of the reduction observed in hybrid male fertility results from genic factors. The remaining 11 blocks are from rearranged linkages. However, fertility blocks are so large within the rearranged linkage groups that it was not possible to distinguish between the contributions of chromosomal and genic factors for these blocks.
Of the 21 fertility blocks, 14 were associated with one or more significantly underrepresented blocks identified in the introgression analysis, while only 3 were associated with significantly overrepresented blocks. Of these 3, only 1 was associated solely with a positively selected block. Most likely, the discovery of 7 fertility segments not associated with negatively selected chromosomal blocks indicates that our analytical methods are underestimating negative selection in the hybrid zone. Alternatively, these blocks may have small effects on fertility or are linked to loci that are advantageous in hybrids.
In addition, several blocks that appear to be strongly negatively selected in the hybrid zones were not significantly correlated with reduced hybrid fertility. This may be due to the lack of power associated with the low frequency of some of these blocks in the hybrid zones (i.e., they may have already been purged from the population). For example, markers on linkages R (tr1-0.6) and T (C20-1.4 and C2-0.6) are absent in all introgressed plants, making a measure of correlation with pollen viability impossible. Alternatively, some of the negatively selected blocks identified by the introgression analysis may contain genes that affect isolating mechanisms that were not studied here, such as pollen competition (Rieseberg et al. 1995a) and habitat selection (Heiser 1947).
The analysis discussed above takes into account linkage disequilibria among markers on the same linkage group. However, significant linkage disequilibrium was sometimes observed among unlinked loci as well, particularly for those loci most highly correlated with reduced fertility. This might be a result of epistatic interactions among these loci. However, low fertility is also associated with early generation hybrids, possibly resulting in genome-wide disequilibria. If this latter explanation is true, it implies that the significant effects of some blocks might be spurious. Because we could not distinguish between the two possible explanations, we have also analyzed the data using the conservative approach of removing all linkage disequilibrium effects from the analysis in the same manner as for linked loci. After this, 14 of the 21 chromosomal blocks associated with reduced pollen viability remain significant (Figure 5).
Frequency of over- and underrepresented markers in the colinear and rearranged parts of the genome
| Colinear (30 markers) | Rearranged (58) | Total (88) | ||||
|---|---|---|---|---|---|---|
| Markers | Blocks | Markers | Blocks | Markers | Blocks | |
| Zone 1 | ||||||
| > expected | 5 | 3 | 1 | 1 | 6 | 4 |
| < expected | 5 | 5 | 12 | 8 | 17 | 13 |
| Zone 2 | ||||||
| > expected | 6 | 6 | 1 | 1 | 7 | 7 |
| < expected | 6 | 4 | 18 | 12 | 24 | 16 |
| Zone 3 | ||||||
| > expected | 6 | 6 | 0 | 0 | 6 | 6 |
| < expected | 3 | 3 | 9 | 8 | 12 | 11 |
| Pooled | ||||||
| > expected | 8 (+ 2)a | 7 (+ 2) | 0 (+ 2) | 0 (+ 2) | 8 (+ 4) | 7 (+ 4) |
| < expected | 11 (+ 2) | 8 (+ 2) | 31 | 16 | 42 (+ 2) | 24 (+ 2) |
| Colinear (30 markers) | Rearranged (58) | Total (88) | ||||
|---|---|---|---|---|---|---|
| Markers | Blocks | Markers | Blocks | Markers | Blocks | |
| Zone 1 | ||||||
| > expected | 5 | 3 | 1 | 1 | 6 | 4 |
| < expected | 5 | 5 | 12 | 8 | 17 | 13 |
| Zone 2 | ||||||
| > expected | 6 | 6 | 1 | 1 | 7 | 7 |
| < expected | 6 | 4 | 18 | 12 | 24 | 16 |
| Zone 3 | ||||||
| > expected | 6 | 6 | 0 | 0 | 6 | 6 |
| < expected | 3 | 3 | 9 | 8 | 12 | 11 |
| Pooled | ||||||
| > expected | 8 (+ 2)a | 7 (+ 2) | 0 (+ 2) | 0 (+ 2) | 8 (+ 4) | 7 (+ 4) |
| < expected | 11 (+ 2) | 8 (+ 2) | 31 | 16 | 42 (+ 2) | 24 (+ 2) |
Numbers in parentheses represent additional markers or blocks that became significant after linkage was taken into consideration.
Frequency of over- and underrepresented markers in the colinear and rearranged parts of the genome
| Colinear (30 markers) | Rearranged (58) | Total (88) | ||||
|---|---|---|---|---|---|---|
| Markers | Blocks | Markers | Blocks | Markers | Blocks | |
| Zone 1 | ||||||
| > expected | 5 | 3 | 1 | 1 | 6 | 4 |
| < expected | 5 | 5 | 12 | 8 | 17 | 13 |
| Zone 2 | ||||||
| > expected | 6 | 6 | 1 | 1 | 7 | 7 |
| < expected | 6 | 4 | 18 | 12 | 24 | 16 |
| Zone 3 | ||||||
| > expected | 6 | 6 | 0 | 0 | 6 | 6 |
| < expected | 3 | 3 | 9 | 8 | 12 | 11 |
| Pooled | ||||||
| > expected | 8 (+ 2)a | 7 (+ 2) | 0 (+ 2) | 0 (+ 2) | 8 (+ 4) | 7 (+ 4) |
| < expected | 11 (+ 2) | 8 (+ 2) | 31 | 16 | 42 (+ 2) | 24 (+ 2) |
| Colinear (30 markers) | Rearranged (58) | Total (88) | ||||
|---|---|---|---|---|---|---|
| Markers | Blocks | Markers | Blocks | Markers | Blocks | |
| Zone 1 | ||||||
| > expected | 5 | 3 | 1 | 1 | 6 | 4 |
| < expected | 5 | 5 | 12 | 8 | 17 | 13 |
| Zone 2 | ||||||
| > expected | 6 | 6 | 1 | 1 | 7 | 7 |
| < expected | 6 | 4 | 18 | 12 | 24 | 16 |
| Zone 3 | ||||||
| > expected | 6 | 6 | 0 | 0 | 6 | 6 |
| < expected | 3 | 3 | 9 | 8 | 12 | 11 |
| Pooled | ||||||
| > expected | 8 (+ 2)a | 7 (+ 2) | 0 (+ 2) | 0 (+ 2) | 8 (+ 4) | 7 (+ 4) |
| < expected | 11 (+ 2) | 8 (+ 2) | 31 | 16 | 42 (+ 2) | 24 (+ 2) |
Numbers in parentheses represent additional markers or blocks that became significant after linkage was taken into consideration.
DISCUSSION
Hybrid zones are often viewed as natural experiments that serve as windows on evolutionary processes such as speciation (Barton and Hewitt 1985; Harrison 1990, Arnold 1997). In particular, the highly recombinant genotypes found in hybrid zones offer a unique opportunity to dissect the architecture of barriers between species. However, only recently have genetic mapping tools become available that allow the genotypic diversity in hybrid zones to be fully exploited. In fact, as far as we know, the present study represents the first detailed application of these tools to the investigation of genetic architecture in natural hybrid zones.
Our results verify the predicted utility of hybrid zones for analyzing genetic architecture. First, patterns of introgression across the three hybrid zones were largely concordant (Figure 4), indicating not only that most chromosomal blocks are under similar selective regimes in all three hybrid zones, but that this approach is robust and repeatable. Second, we were able to identify 26 chromosomal segments that appear to be negatively selected in hybrids (Figure 4). This is the largest number of segments that have been shown to affect hybrid fitness in a plant species pair, confirming the predicted resolving power of this approach. Third, by searching for correlations between reduced pollen viability and introgressed markers, we were able to demonstrate that pollen sterility is significantly associated with 16 of these 26 segments (Figure 5). This result not only provides a straightforward explanation for why this subset of blocks is disadvantageous in hybrids, but also demonstrates the feasibility of using hybrid zones for marker-assisted QTL analyses. Finally, comparison of rates of introgression across colinear and rearranged chromosomes provides a simple but direct method for estimating the relative contributions of chromosomal rearrangements and genic factors to genetic isolation.
Although our data demonstrate the potential utility of hybrid zones for studies of genetic architecture, several drawbacks to this method are identified as well. From a practical standpoint, it can be difficult to find enough taxon-specific markers to conduct such a study, particularly for closely related taxa. Experimental crosses require markers that differentiate only the individuals being crossed, not most or all individuals from each taxon, so the marker requirements are much less formidable. Although markers need not be diagnostic for hybrid zone studies (Rieseberg et al. 1998), accurate estimates of linkage disequilibrium between markers and traits of interest do require substantial interspecific differentiation for the markers in question.
A second problem concerns estimates of expected rates of introgression under neutral conditions in hybrid zones. In experimental crosses, Mendelian rules of inheritance can be used to calculate expectations, but this is not possible in natural hybrid zones. This is a serious problem, because overestimates of rates of neutral introgression will lead to overestimates of the number of negatively selected chromosomal segments. Biases in the opposite direction will occur if rates of neutral introgression are underestimated. However, it would seem that the latter bias is preferable to the former because we prefer a conservative estimate of gene numbers. The analyses employed in this article use data from experimental hybridization studies to estimate expected rates of neutral introgression (see materials and methods). Most likely, these analyses underestimate rates of neutral introgression and, as a result, probably underestimate the number and magnitude of negatively selected chromosomal segments in these hybrid zones.
A third problem concerns the detection of QTL that contribute to isolation in hybrid zones. Because these QTL are likely to be negatively selected, chromosomal segments that carry them will occur at low frequencies in hybrid zones, reducing the power of QTL detection. This problem was fairly severe in this study. In fact, several H. petiolaris chromosomal segments were completely absent in all individuals sampled, and several additional blocks occurred in such low frequencies that detection of significant effects on pollen viability was not possible.
A related problem is that the few negatively selected chromosomal blocks that remain in the hybrid zone will be found predominantly in early generation hybrids. Thus, the advantage of highly recombinant genotypes will be largely lost for QTL analyses of reproductive barriers such as fertility. This problem is evident in the present study, in which the effects of linkage were much greater in the QTL analysis of pollen viability than in the analysis of block frequencies (Figures 4 and 5).
Nonetheless, even with these problems, it is clear that hybrid zones provide unique opportunities for genetic analyses of wild plant and animal species. Although we employed markers that had been previously mapped in experimental populations, linkage between markers could also be inferred from analyses of pairwise associations. Thus, marker-assisted quantitative genetic studies should be feasible in natural hybrid zones even in the absence of preexisting linkage maps.
Comparisons with experimental introgression lines: Overall, patterns of introgression in natural hybrid zones are similar to those previously reported for three experimental introgression lines (Rieseberg et al. 1995b, 1996a,b), but several differences were observed as well. The biggest difference was expected: genotypes in the natural hybrid zone were more highly recombinant than in the experimental hybrids, particularly when rearranged linkage groups are compared. Greater recombination enabled detection of negatively selected blocks that were missed in the experimental hybrids (e.g., linkage B, C18-1.1), as well as the subdivision of large negatively selected blocks into smaller ones in the rearranged linkage groups. Also, it appears that enhanced recombination led to somewhat higher rates of introgression across the rearranged linkages generally, although overall patterns were essentially identical.
The other important difference is that several markers that were significantly overrepresented in the experimental hybrids are underrepresented in the natural hybrid zones: linkage C (marker 181-0.79), linkage F (markers 476-0.55 and 220-1.4), and linkage S (226-0.7). A possible explanation for this discrepancy is that these markers are linked to ecological differences between the parental species that would not be selected against in the greenhouse, but do represent a significant disadvantage in the wild. For all other blocks, the experimental and hybrid populations largely agree.
Patterns of introgression in both studies suggest a complex basis for reproductive isolation, involving both individual genes and chromosomal rearrangements. The prior study identified 14 negatively selected chromosomal segments within the colinear linkage (Rieseberg et al. 1996a), compared to 10 in the present study. This difference is primarily due to reduced genomic coverage in the present study (58 vs. 80% of the genome). For colinear regions assayed in both studies, 7 negatively selected blocks were detected in the experimental hybrids vs. 10 in the present study (once again emphasizing the increased power obtainable with increased recombination).
Inferences about numbers of negatively selected blocks differ more dramatically for the rearranged linkage groups. In the previous study, little recombination or introgression was observed in the rearranged linkages, and individual blocks under negative selection could not be identified. By contrast, the highly recombinant genotypes from the three hybrid zones allowed us to resolve fairly small chromosomal blocks, to identify the subset of these (16 total) that were negatively selected, and to differentiate between chromosomal and genic effects. Nonetheless, even in the natural hybrid zones there was little recombination across three large linkage blocks (G, K, and V). Perhaps there is strong epistatic selection to retain these blocks. Alternatively, recombination may be disrupted in these genomic regions in interspecific hybrids. If the latter hypothesis is correct, studies of more ancient hybrid zones may enable these blocks to be dissected.
Inferences about the nature of species and species barriers: Patterns of introgression observed in these wild sunflower hybrid zones allow us to make several inferences or generalizations about the nature of species and species barriers. Although many of these can be derived from the hybrid zone literature generally (Harrison 1990), they may be most clearly demonstrated by the current data set.
First, our data illustrate the distinction between genetic and reproductive isolation. H. annuus and H. petiolaris are not fully reproductively isolated (i.e., hybrids are common in nature), yet introgression is limited outside the immediate area of contact. In fact, all markers in this study exhibited a substantial decrease in frequency just 10-20 m from the center of the hybrid zone. Thus, genetic isolation may be a more reliable criterion for species status than reproductive isolation.
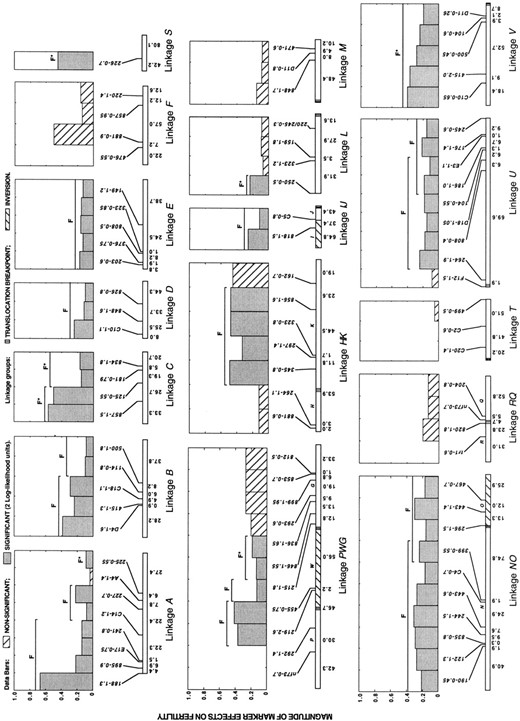
—Magnitude of individual marker effects on mean fertility reduction for pooled data from three natural hybrid zones between H. annuus and H. petiolaris. Independent chromosomal blocks (after consideration of linkage effects) that significantly affect pollen viability are indicated by the letter F. The sizes of these segments are indicated by a line above the data bars. Asterisks indicate blocks that no longer have a significant effect on fertility after genome-wide linkage disequilibrium is taken into account. Map distances and marker nomenclature are as in Figure 4.
Second, our data demonstrate the semipermeable nature of hybrid zones. Although much of the H. annuus genome is protected by selection from interspecific gene flow, several genomic regions do appear to accept foreign alleles or chromosomal segments (Figure 4). These observations are consistent with theory, which suggests that reduced hybrid fitness serves as a barrier to negatively selected loci and linked alleles, but not necessarily to neutral or advantageous alleles (Barton and Hewitt 1985). Thus, as argued by Harrison (1990), genetic isolation should be viewed as a property of individual genes or chromosomal segments, not as a characteristic of entire genomes.
Third, our ability to assess the relative contributions of genic and chromosomal factors to the genetic barrier between these species contributes to the ongoing debate regarding the role of chromosomal rearrangements in genetic isolation and speciation. Because the majority of plant and animal species that have been analyzed karyotypically appear to differ in terms of chromosome structure (White 1978; Jackson 1985), some authors have argued that chromosomal rearrangements play a primary role in reproductive isolation and speciation (Stebbins 1950; White 1978; King 1993). However, there are increasing numbers of reports in which rearrangements have been shown to have little or no effect on meiotic pairing or fertility (Shaw 1981; Sites and Moritz 1987; Coyne et al. 1993), leading Coyne et al. (1993) to conclude that “it can no longer be assumed without proof that fixed or polymorphic chromosomal rearrangements are underdominant in nature” (p. 495). Unfortunately, it is difficult to prove that chromosomal rearrangements themselves are responsible for meiotic abnormalities or reduced fertility because it is difficult to rule out genic effects (although see Darlington 1932; Tadmor et al. 1987; Quillet et al. 1995). Even when the rearrangements are shown to contribute to reduced fertility, their overall contribution to genetic isolation in nature has rarely been assessed (although see Shaw et al. 1986). Map-based studies of introgression in natural hybrid zones such as this one provide a means by which the contributions of chromosomal rearrangements and genic incompatibilities to isolation can be teased apart. Here we demonstrate that approximately half the barrier to introgression between H. annuus and H. petiolaris appears to result from inversions and translocations. This corroborates earlier studies of artificial crosses that indicate an important role for chromosomal rearrangements as barriers between these two species (Rieseberg et al. 1995b).
Fourth, the identification of 26 negatively selected chromosomal blocks (Figure 4; Table 1) is consistent with other studies that report a complex genetic basis for traits that contribute to reproductive isolation (reviewed in Coyne and Orr 1998). Because the 26 blocks identified undoubtedly contribute to several different components of isolation, this number is best compared to indirect estimates of the numbers of factors reducing hybrid fitness in hybrid zones of grasshopper (50-500, Barton and Hewitt 1981) and toads (26-88; Szymura and Barton 1991). Our estimates of factor numbers from these three sunflower hybrid zones probably should not be viewed as substantially different from that of either animal hybrid zone, considering that (1) less than 60% of the sunflower genome was sampled, (2) some blocks showed little recombination, (3) the fitness effects of blocks from only one species (H. petiolaris) were tested, and (4) our analytical methods are likely to have understimated the number of negatively selected blocks.
Our analyses of pollen fertility allow more direct comparisons with the large body of experimental genetic studies reviewed by Coyne and Orr (1998). Specifically, the identification of 21 chromosomal segments that contribute to reduced pollen viability is consistent with other studies that have identified a large number of factors that affect male sterility (e.g., True et al. 1996, Wu et al. 1996) and accords well with theoretical expectations that the genetics of hybrid sterility will grow quickly in complexity as species diverge (i.e., the snowball effect; Orr 1995). Linkage disequilibria among fertility loci from different linkage groups (not shown) also suggest the existence of complex higher order interactions among chromosomal segments that contribute to reduced male fertility as has been reported in Drosophila (Palopoli and Wu 1994; Davis and Wu 1996), but a full exploration of these interactions is beyond the scope of the present study.
Polymorphism levels for isolating factors: The congruent patterns of introgression in the three sunflower hybrid zones might be viewed as evidence for low levels of intraspecific polymorphism for factors that contribute to isolation. However, the fact that all three hybrid zones, although apparently independent, occur in the same large county in western Nebraska, reduces the strength of this result. Any conclusions about levels of polymorphism for isolating factors must await comparisons with hybrid zones from more widely separated parts of the geographic range of these species.
Advantageous chromosomal blocks: Our tentative identification of 11 chromosomal blocks that appear to have positive fitness consequences in natural hybrid zones might be viewed as additional support for the view that certain interspecific gene combinations are actually advantageous in hybrids (Parsons et al. 1993, Rieseberg et al. 1996b; Burke et al. 1998). However, we are reluctant to make this assertion from the present data set, because it seems probable that our data analysis methods overestimate the number and magnitude of positively selected blocks. Nonetheless, deviations from neutral expectations for several of these blocks are extreme, and it is possible that these H. petiolaris blocks may be positively selected in an H. annuus genetic background. Moreover, evidence for positive selection was also detected in a study of experimental hybrids between these species (Rieseberg et al. 1996b). Thus, the presence of a small number of H. petiolaris blocks that are advantageous in hybrid or backcross plants might be expected.
Conclusions: In this article, we have used genetic mapping tools to explore the utility of hybrid zones as venues for studying the nature of genetic differences between species. We have verified several of the predicted advantages of hybrid zones for genetic study, such as the ability to (1) identify chromosomal segments carrying QTL in the absence of artificial crosses, (2) distinguish among closely linked loci, and (3) dissect the relative contributions of genic and chromosomal factors to isolation. We also discovered some potential shortcomings of this approach such as reduced power for detecting highly disadvantageous QTL in hybrid zones due to their low frequency and the difficulty of estimating expected introgression patterns under neutral conditions. Nonetheless, we were able to demonstrate a complex basis for reproductive isolation between these species, identify 21 blocks associated with reduced male fertility, and show that much of the barrier to introgression between these two species is due to translocations and inversions. We predict that hybrid zones will prove to be a valuable tool for studying the quantitative genetics of wild species, particularly those species that are long lived or difficult to propagate in the lab or greenhouse.
Acknowledgement
We thank Alex Buerkle, Shanna Carney, Rhonda Rieseberg, Mike Wade, and Diana Wolf for their critical reading of the manuscript, Stuart Baird for helpful comments on the statistical methods, and Jeff Regan for technical assistance. This research was supported by National Science Foundation grant BSR-9419206.
Footnotes
Communicating editor: J. A. Birchler
LITERATURE CITED

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}