





Abstract
The cellular response to DNA damage that has been most extensively studied is the SOS response of Escherichia coli. Analyses of the SOS response have led to new insights into the transcriptional and posttranslational regulation of processes that increase cell survival after DNA damage as well as insights into DNA-damage-induced mutagenesis, i.e., SOS mutagenesis. SOS mutagenesis requires the recA and umuDC gene products and has as its mechanistic basis the alteration of DNA polymerase III such that it becomes capable of replicating DNA containing miscoding and noncoding lesions. Ongoing investigations of the mechanisms underlying SOS mutagenesis, as well as recent observations suggesting that the umuDC operon may have a role in the regulation of the E. coli cell cycle after DNA damage has occurred, are discussed.
THE environment contains a multitude of deoxyribonucleic acid (DNA)-damaging agents ranging from ultraviolet light (U V) to fungal metabolites like Aflatoxin B1. Furthermore, DNA-damaging agents, such as reactive oxygen species, can be produced by the cell itself as metabolic by-products and intermediates. Together, these agents pose a constant threat to an organism's genome. As a result, organisms have evolved many vitally important mechanisms, such as nucleotide excision repair, to deal with DNA damage in an error-free manner. When DNA damage cannot be repaired in an error-free manner, a mutagenic event may occur. Escherichia coli, among other bacteria, has evolved a coordinated response to challenges to the integrity of its genome. This inducible system, termed the SOS response, includes functions, known collectively as the SOS mutagenesis system, that are required for such mutagenic events to occur (reviewed comprehensively in Friedberg et al. 1995 and also in Walker 1995; Woodgate and Levine 1996). Studies of E. coli as a model system have offered detailed insights into the molecular basis of UV and chemical mutagenesis, just as studies of the bacteriophage T4 model system, in which Jan Drake's laboratory played a leading role (e.g., Drake 1991), have offered insights into numerous aspects of spontaneous and induced mutagenesis. Here we will attempt to provide a concise, updated discussion of the SOS response and the functions of the umuD+C+ gene products in SOS mutagenesis. We will also discuss new discoveries concerning an unexpected role of the umuD+C + operon in the regulation of the E. coli cell cycle after DNA damage and their implications for the evolution of the operon.
The SOS response: The SOS response, a regulon of over 20 unlinked genes, many of which are involved in DNA-damage tolerance and repair, e.g., recA, lexA, umuDC, polB, recN, sulA, uvrA, uvrB, and uvrD, is induced after the cell encounters DNA-damaging agents. One component of this response is an error-prone damage tolerance mechanism called SOS mutagenesis. This process requires the umuD+C+ and recA+ genes and endows the cell with an increased capacity to recover from DNA damage by allowing it to process lesions that cannot be repaired in an error-free manner, e.g., an abasic site opposite a break or a gap in the complementary DNA strand (Friedberg et al. 1995). To avoid a lethal interruption of DNA replication resulting from the inability of the DNA polymerase to replicate through unrepaired lesions in the template DNA, the SOS mutagenesis system allows replication to continue through these lesions (Figure 1). In exchange for increased survival, the cell pays a cost of an elevated mutation rate resulting from this translesion synthesis.
In E. coli, the recA+ (Miura and Tomizawa 1968) and lexA+ (Defais et al. 1971) gene products are required for the regulation and induction of the SOS response (Figure 2). LexA was shown to be a repressor protein that binds to a site (the “SOS box”) in the promoters of the SOS response genes and interferes with the binding of RNA polymerase (Brent and Ptashne 1981; Little et al. 1981). The recA+ gene product plays an important role in homologous recombination and in varied DNA repair pathways, including the repair of daughter-strand gaps and double-strand breaks, as well as SOS mutagenesis. RecA is capable of forming a helical, multimeric nucleoprotein filament around single-stranded DNA (ssDNA) that is central to its activities in the cell (Friedberg et al. 1995). Following DNA damage, RecA is activated when it forms a nucleoprotein filament by binding to ssDNA generated by the cell's partially successful attempts to replicate damaged DNA (Craig and Roberts 1981; Sassanfar and Roberts 1990). The activated form of RecA, referred to as RecA*, functions as a coprotease that mediates LexA cleavage
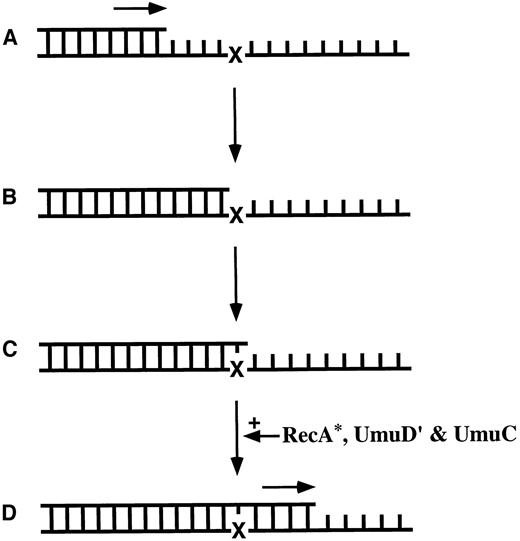
The model of SOS mutagenesis via translesion synthesis. (A) The DNA polymerase is replicating a DNA template normally (active replication is indicated by: →). (B) The polymerase encounters a damaged nucleotide (X). (C) The polymerase incorporates a nucleotide opposite the lesion but cannot replicate through the lesion. (D) RecA*, UmuD′, and UmuC are required for the polymerase to replicate through the lesion (translesion synthesis). If the nucleotide incorporated opposite the lesion was incorrect (C), translesion synthesis will fix the mutation in the organism's genome (D).
by stimulating the latent ability of the LexA repressor to proteolytically autodigest (Little 1984; Little 1993). The resulting decrease in the cellular pool of LexA results in the induction of the SOS regulon. Additional regulation of SOS induction may be provided by the competition between LexA cleavage and homologous recombination, which are both mediated by the RecA nucleoprotein filament. Structural studies revealed that LexA interacts with a region of the RecA nucleoprotein filament that may also be the binding site for a double-stranded DNA (dsDNA) molecule (Story et al. 1992; Yu and Egelman 1993). This hypothesis is supported by recent biochemical experiments indicating that the LexA cleavage and DNA strand exchange functions of the RecA nucleoprotein filament are competitive reactions (Harmon et al. 1996; Rehrauer et al. 1996).
The present model for the regulation of the SOS response can be summarized as follows (Figure 2): When ssDNA (the cell's internal signal that it has suffered DNA damage) is present, RecA is converted to RecA*. This pool of RecA* stimulates the cleavage of LexA, resulting in the induction of the SOS response and therefore in the increased synthesis of RecA, which remains in the activated RecA* form as long as the inducing signal persists. Although less is known about what happens when the cell recovers from the DNA damage,
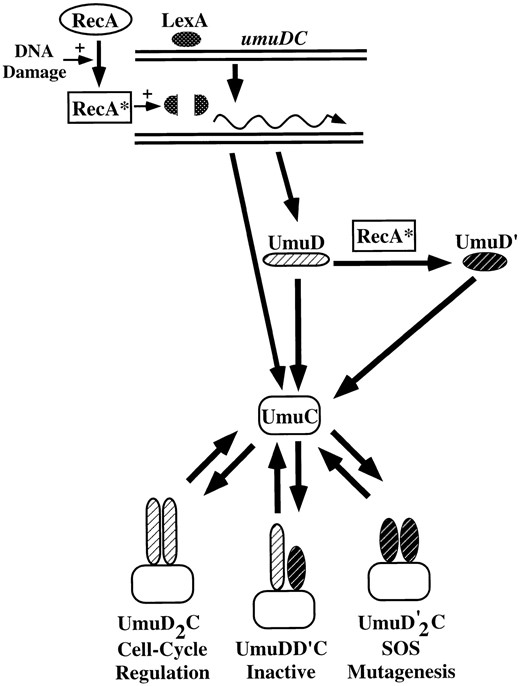
Regulation of the umuDC operon by RecA and LexA. DNA damage generates a signal that converts RecA to RecA*. RecA* mediates the cleavage of the LexA repressor that results in the induction of the umuDC operon as well as the rest of the SOS response genes. RecA* can also mediate the processing of UmuD to the shortened UmuD′ molecule. UmuD and UmuD′ can interact with UmuC in a variety of combinations. The Umu(D)2C complex seems to be involved in regulating the E. coli cell cycle after DNA damage. The Umu(D′)2C complex is active in SOS mutagenesis (translesion synthesis). The third complex, UmuDD′C, does not appear to have an activity, but it may play a role in shutting off SOS mutagenesis by sequestering UmuD′.
the amount of inducing signal is presumed to decrease, causing RecA* to revert to RecA. This results in the reaccumulation of intact LexA, which represses the entire SOS regulon, thereby shutting down the response. Further modulation of the response is provided by the differential affinity of LexA for the promoters of SOS response genes, which allows some genes to be fully induced at a lower level of DNA damage than others (Friedberg et al. 1995).
SOS mutagenesis: The umuD and umuC genes, which form an operon, are required for the process of SOS mutagenesis (Figure 1; Kato and Shinoura 1977; Steinborn 1978). Disruptions of the umuD+C+ operon eliminate most UV mutability (Bagg et al. 1981; Woodgate 1992) and chemical mutability by a variety of compounds such as 4-nitroquinoline 1-oxide (4-NQO), methyl methanesulfonate (MMS), and neocarcinostatin (Friedberg et al. 1995). This lack of mutability is accompanied by a moderate increase in the cell's sensitivity to killing by DNA-damaging agents such as UV light (Bagg et al. 1981; Woodgate 1992). UmuD and UmuC have molecular weights of approximately 15 kD and 45 kD, respectively (Elledge and Walker 1983; Shinagawa et al. 1983; Kitagawa et al. 1985; Perry et al. 1985).
DNA sequence analysis of the umuD+C + operon revealed that the UmuD protein shares homology with the carboxyl-terminal domain of LexA, which contains the latent autodigestive activity (Perry et al. 1985). This prompted the finding that the coprotease activity of RecA*is also able to mediate the autodigestion of UmuD to UmuD′, removing the first 24 amino acids of the protein (Burckhardt et al. 1988; Shinagawa et al. 1988) and making it active in SOS mutagenesis (Figure 2; Nohmi et al. 1988). Recent evidence supports the hypothesis that UmuD and LexA interact with the RecA nucleoprotein filament in a similar, yet not entirely identical, manner and are then stimulated to autodigest (Konola et al. 1997; Nastri et al. 1997). It is possible that the differences in the interactions of UmuD and LexA with the RecA nucleoprotein filament are responsible, in part, for the fact that the rate of RecA*-mediated cleavage of UmuD is slower than that of LexA (Burckhardt et al. 1988; Woodgate and Ennis 1991). In vivo, this delayed cleavage is manifested as a delay of maximal SOS mutagenic activity by approximately 30 min after UV irradiation (Defais et al. 1976). Recently, evidence of further post-translational regulation of SOS mutagenesis has been reported. Lon, an ATP-dependent protease, has been shown to play a role in the degradation of UmuC and UmuD, and the ClpXP protease is involved in the degradation of UmuD′ when it is in a heterodimer with UmuD (Frank et al. 1996).
Because a lexA(Def) recA(Def) strain expressing UmuD′ and UmuC is not UV-mutable, RecA must play another, perhaps more direct, role in SOS mutagenesis besides its coprotease activity (Nohmi et al. 1988). This role seems to depend on the ability of RecA to form nucleoprotein filaments. The recA1730 protein is incapable of forming normal nucleoprotein filaments (Dutreix et al. 1992) and is defective in SOS mutagenesis in a lexA (Def) background even though it can produce UmuD′ from UmuD (Dutreix et al. 1989; Bailone et al. 1991). Evidence from umuD′C+overexpression experiments indicates that UmuD′ and UmuC can inhibit Hfr × F− recombination. Devoret and his colleagues (Sommer et al. 1993; Boudsocq et al. 1997) have speculated that this inhibition is the result of the interaction of UmuD′ and UmuC with the growing end of a RecA-nucleoprotein filament. Furthermore, in vitro experiments have shown that crude extracts from cells that overproduce UmuD′ and UmuC inhibit RecA-mediated homologous pairing (Szpilewska et al. 1995). Kowalczykowski and his colleagues (Harmon et al. 1996; Rehrauer et al. 1996) have suggested that this apparent switching of the RecA nucleoprotein filament from recombinational repair to SOS mutagenesis may be due to the aforementioned competition between coprotease substrate, i.e., UmuD, and the secondary dsDNA molecule for the same binding site on the RecA nucleoprotein filament. A slightly different model of RecA's role in SOS mutagenesis involves a targeting mechanism in which the RecA-nucleoprotein filament serves as a lure to target the Umu proteins to damaged ssDNA (Woodgate and Sedgwick 1992; Frank et al. 1993). In support of both models, affinity chromatography has been used to suggest an interaction of UmuD, UmuD′ (Frank et al. 1993), and UmuC (Freitag and McEntee 1989) with RecA. In light of these hypotheses, it is interesting to note that a purified Umu(D′)2C complex was shown to bind to naked ssDNA, as well as to RecA-ssDNA nucleoprotein filaments (Bruck et al. 1996). However, the exact nature of this more direct role of RecA in SOS mutagenesis is still unclear.
The protein-protein interactions in which UmuD, UmuD′, and UmuC proteins participate are varied (Figure 2). Genetic and biochemical evidence exists for the formation of UmuD2 and UmuD′2 homodimers (Bruck et al. 1996; Jonczyk and Nowicka 1996; Woodgate et al. 1989). UmuD-UmuD′ heterodimers form preferentially relative to the two homodimer species and inhibit SOS mutagenesis, presumably by titrating out the active UmuD′ (Battista et al. 1990; Jonczyk and Nowicka 1996). This inhibitory effect on SOS mutagenesis by UmuD was confirmed in an in vitro translesion synthesis reaction (Rajagopalan et al. 1992). An interaction between UmuC and UmuD′has been shown using immunoprecipitation, glycerol gradient analysis, and yeast two-hybrid experiments (Woodgate et al. 1989; Jonczyk and Nowicka 1996). Although not evident in immunoprecipitation or two-hybrid studies, a UmuC-UmuD interaction can be observed in vitro (Woodgate et al. 1989). An association between the UmuD′-UmuD heterodimer and UmuC has been demonstrated by affinity chromatography experiments (Woodgate et al. 1989).
The UmuD′ crystal structure has been solved (Peat et al. 1996a), and the monomer, which is globular with an extended amino terminus, participates in two major modes of UmuD′-UmuD′interaction in the crystal. Peat and his colleagues (1996a) proposed that one of these modes represents the dimer interface (Figure 3A) while the other interface is involved in polymerizing UmuD′2 homodimers into an extended filament (Figure 3B). Further investigation into the possible biological relevance of the UmuD′ filament indicated that UmuD′ multimers could be detected in vitro following glutaraldehyde crosslinking (Peat et al. 1996b). However, recent nuclear magnetic resonance (NMR) studies of UmuD′ indicate that the protein is a dimer, not a filament, in solution even at the high concentrations (~1 mm) required for NMR analysis. Furthermore, the interface of the UmuD′2 homodimer in solution involves extensive
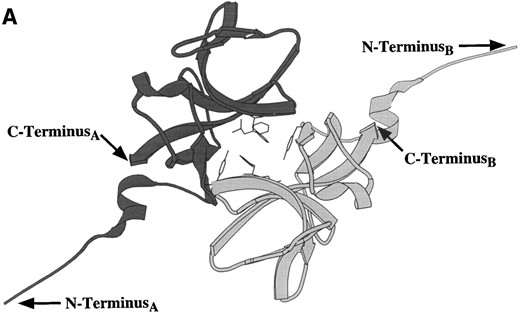
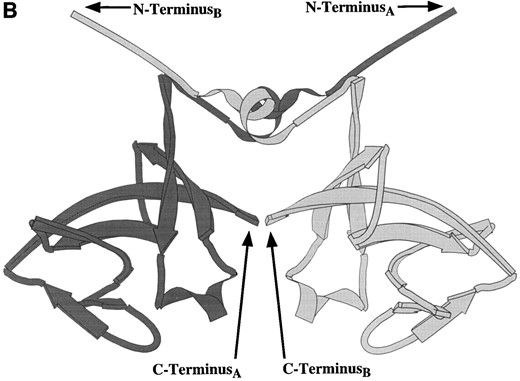
Ribbon diagrams of the two UmuD′2 homodimer interfaces seen in the crystal structure (Peat et al. 1996a). (A) This interface, which involves interactions between aromatic amino acids, was described as the UmuD′2 homodimer interface by Peat and his colleagues (1996a). Note the separation of the carboxyl-termini of the two monomers. (B) The second interface, termed the “filament dimer” interface by Peat and his colleagues (1996a,b), involves aliphatic interactions that are particularly extensive between the two carboxyl-termini. NMR analysis of the UmuD′2 homodimer indicates that the dimer interface in solution involves extensive carboxyl-termini interactions and, therefore, is this aliphatic interface (Ferentz et al. 1997). In addition, the multimeric UmuD′2 filaments seen in the crystal by Peat and his colleagues (1996a,b) do not occur in solution (Ferentz et al. 1997).
interactions between the carboxyl termini of the two monomers (Ferentz et al. 1997). Thus, the interface of UmuD′2 in solution most closely resembles the interface in the crystal structure that was referred to as the “filament dimer” interface by Peat and his colleagues (1996a,b; Figure 3B). Although the entire structure of the UmuD′ monomer is not yet fully solved by NMR, the globular domain of UmuD′ has been confirmed. However, the extended amino terminus seen in the crystal is highly flexible in solution (Ferentz et al. 1997).
In an independent approach, the structure of the UmuD2 and UmuD′2 homodimers has been probed using a series of engineered functional derivatives of the UmuD or UmuD′ protein that contain a single cysteine residue at defined positions in the amino acid sequence. By using oxidizing agents to stimulate disulfide-bond formation and a cysteine-specific homobifunctional crosslinking reagent, inferences have been made about the structures of the two homodimers. Although the presence of the amino-terminal 24 amino acid residues on UmuD compared with UmuD′ is an obvious indicator that the structures of the two dimers are different, crosslinking data indicate that there are additional differences in the tertiary structures of the domains common to both proteins (Lee et al. 1994; Guzzo et al. 1996; A. Guzzo and G. C. Walker, unpublished results). Further biochemical and structural experimentation will be required to elucidate the differences between the UmuD′2 and UmuD2 homodimers. These differences are likely to be extremely important considering recent observations (discussed below) concerning the differing roles of UmuD and UmuD′ in cellular physiology.
The nature of the UmuC protein has remained much more elusive than that of its partners UmuD and UmuD′. The initial purification of an active protein was challenging and required denaturation followed by a gradual refolding in the presence of equimolar amounts of ribosomal S9 protein (Woodgate et al. 1989). This purified UmuC was shown to have a ssDNA binding activity in vitro (Petit et al. 1994), which was also seen with apurified, soluble Umu(D′)2C complex (Bruck et al. 1996). The ssDNA binding activity of the Umu(D′)2C complex was shown to be cooperative with a Hill coefficient, n = 3, and it was estimated that one Umu(D′)2C complex bound per 20 nucleotides of ssDNA. However, activity of this complex in translesion synthesis has not been reported. The biological consequences of the ssDNA binding activity are not yet clear, but the ability of UmuC and the Umu(D′)2C complex to bind ssDNA could be involved in interactions with the DNA polymerase at the replication fork or in the direction of the Umu proteins to damaged DNA.
In vivo, there is a genetic requirement for the groEL+ and groES+ genes for SOS mutagenesis, which appears to result from a reduction of the cellular UmuC levels in a groE mutant strain (Donnelly and Walker 1989). SOS mutagenesis in a groE background can be restored by overexpression of UmuD′ and UmuC, which increases the stability of UmuC in those mutant strains (Donnelly and Walker 1992). This requirement for the GroEL and GroES chaperonins was confirmed by the in vitro restoration of activity of inactive UmuC after successive treatments with the Hsp70 (DnaK, DnaJ, and GrpE) and Hsp60 (GroEL and GroES) chaperone complexes (Petit et al. 1994).
Molecular mechanism of SOS mutagenesis: A general model for SOS mutagenesis proposed by Bridges and Woodgate (1984; Figure 1) divides the process into two distinct steps: misincorporation of a nucleotide opposite the lesion, followed by continued replication past the lesion. Bridges and Woodgate (1984) used in vivo evidence to argue that while the misincorporation step is independent of UmuD′, UmuC, and RecA, continued replication, which fixes the mutation induced in the misincorporation step, requires those three proteins (Bridges and Woodgate 1984, 1985; Bridges 1988; Bates and Bridges 1991). This model is supported by experiments using N-2-acetylaminofluorene (AAF)-adducted plasmid DNA that suggest that SOS mutagenesis results from an increase in the frequency of replication through the lesion rather than an increase in the misincorporation rate of the polymerase (Koffel-Schwartz et al. 1996), and that the umuD+C+ gene products are required for replication of AAF-adducted DNA only when the 3′ terminal nucleotide of the nascent strand is directly opposite the adduct on the template strand (Napolitano et al. 1997). Studies of premutagenic DNA lesions that are processed by the SOS mutagenesis system have indicated the following: (1) that lesions such as abasic sites, UV-induced thymine-thymine cyclobutane dimers, and pyrimidine-pyrimidone (6-4) photoproducts are virtually complete blocks to DNA replication; (2) that replication through these lesions and the mutation frequencies at these sites are greatly enhanced in SOS-induced cells; and (3) that the translesion synthesis that occurs in SOS-induced cells processes some premutageneic lesions differently from the basal level that is present in uninduced cells, providing further evidence that a modified DNA polymerase is functioning in SOS-induced cells (Banerjee et al. 1988; Banerjee et al. 1990; Lawrence et al. 1990; LeClerc et al. 1991).
The specific molecular details of umuD+C+-mediated translesion synthesis, the underlying cause of SOS mutagenesis, are still not understood. A number of models have been proposed for the molecular mechanism of this process, including a suppression of a DNA polymerase's proofreading ability, an increase in its processivity, or a relaxation of its requirement for a proper Watson-Crick DNA structure for continued replication (Friedberg et al. 1995). The identity of the DNA polymerase (DNA Pol) that participates in translesion synthesis has been examined, and it has been shown that two of E. coli's three DNA polymerases, DNA Pol I (Bates et al. 1989) and DNA Pol II (Iwasaki et al. 1990), are not required for SOS mutagenesis. DNA Pol III, the major replicative polymerase in E. coli, has been implicated in SOS mutagenesis (Hagensee et al. 1987). Both in vivo and in vitro, translesion synthesis requires UmuC and UmuD′ or homologous mutagenesis proteins as well as RecA, even when a DNA Pol III lacking ε, the 3′→5′ exonuclease proofreading subunit, is used (Slater and Maurer 1991; Rajagopalan et al. 1992). Therefore, it appears that the suppression of the proofreading function of DNA Pol III is not the mechanism by which translesion synthesis occurs.
Overexpression of the umuD+C+ operon from a multicopy plasmid in lexA(Def) cells, which constitutively express the SOS response genes, causes a cold-sensitive growth phenotype at 30°. This umuD+C +-mediated cold sensitivity for growth is accompanied by a rapid, reversible, UmuDC-dependent inhibition of DNA synthesis at the restrictive temperature (Marsh and Walker 1985) that is dependent on the cell being in lag phase (T. Opperman, S. Murli and G. C. Walker, unpublished results). In addition, overexpression of either ε (Jonczyk et al. 1988) or β, the dimeric processivity element that encircles the template DNA and tethers the core of DNA Pol III to the DNA (Tadmor et al. 1992), partially suppresses SOS mutagenesis, perhaps by titrating out the Umu proteins or by competing with the Umu proteins for a binding site on the DNA Pol III holoenzyme. These data suggest an interaction between the Umu proteins and the E. coli DNA replication machinery.
Echols and his colleagues (Rajagopalan et al. 1992) were able to reconstitute translesion synthesis in vitro using purified UmuC, UmuD′, RecA, DNA Pol III, single-stranded DNA binding protein (SSB), and a linear ssDNA template with a synthetic abasic lesion engineered at a defined location. In these studies, ~5% of the primers were extended through the abasic site. In the absence of RecA, UmuC, or UmuD′, the background level of translesion synthesis was ~0.5%. These values are similar to those reported in studies of translesion synthesis in vivo with either an abasic site-containing M13mp7-based vector (Lawrence et al. 1990) or an AAF-adducted plasmid (Koffel-Schwartz et al. 1996), which indicated a 0.1–0.7% or a 0.42% translesion synthesis frequency in the absence of SOS induction and a 5–7% or a 12.4% frequency after the SOS response is induced, respectively. An alternative in vitro system for dissecting translesion synthesis using crude bacterial cell extracts of varying genotypes has shown a requirement of recA+ and umuC+ for UV mutagenesis (Cohen-Fix and Livneh 1992, 1994). These in vitro systems will be useful in dissecting the complex set of molecular interactions between the Umu proteins, RecA, DNA Pol III, and the damaged DNA template that are required for translesion synthesis. Understanding the mechanisms by which a DNA polymerase can replicate through regions of damaged DNA will undoubtedly teach us much about the manner by which DNA Pol III monitors itself and the template DNA strand during normal DNA replication.
Homologs of umuD+C+: There are numerous prokaryotic homologs of the umuD+C+ operon and multiple eukaryotic umuC+ homologs as well (Tables 1 and 2). A set of highly homologous operons located on bacterial chromosomes or on naturally occurring plasmids has been cloned and sequenced, and one study has indicated that homologous operons may be present in many other bacterial species as well (Sedgwick et al. 1991). The best studied of the prokaryotic homologs is the mucA+B+ operon that was discovered on the plasmid pKM101, a derivative of the naturally occurring R46 plasmid that is used to enhance the susceptibility of Salmonella
E. coli UmuD homologs
| UmuD homolog | Similaritya(%) | P (N)b |
|---|---|---|
| S.t. UmuD | 72.7 | 2.2 × 10−59 |
| R391 RumA | 54.7 | 6.8 × 10−40 |
| TP110 ImpA | 44.6 | 3.2 × 10−33 |
| S.t. 60-MDa SamA | 44.6 | 2.2 × 10−30 |
| pKM101 MucA | 39.6 | 5.9 × 10−29 |
| Bacteriophage P1 HumDc | 34.6 | 1.0 × 10−15 |
| P.s. pPSR1 Ru1A | 30.9 | 6.0 × 10−11 |
| UmuD homolog | Similaritya(%) | P (N)b |
|---|---|---|
| S.t. UmuD | 72.7 | 2.2 × 10−59 |
| R391 RumA | 54.7 | 6.8 × 10−40 |
| TP110 ImpA | 44.6 | 3.2 × 10−33 |
| S.t. 60-MDa SamA | 44.6 | 2.2 × 10−30 |
| pKM101 MucA | 39.6 | 5.9 × 10−29 |
| Bacteriophage P1 HumDc | 34.6 | 1.0 × 10−15 |
| P.s. pPSR1 Ru1A | 30.9 | 6.0 × 10−11 |
Accession numbers for the nucleotide or amino acid sequences: E. coli UmuD, M13387; S.t. UmuD, M57431, M35010; 60-MDa plasmid SamA, D90202; R391 RumA, U13633; TP110 ImpA, X53528; pKM101 MucA, D90147; P1 HumD, M95666 (nucleotides 1661–2050); P.s. pPSR1 Ru1A, U43696. Abbreviations for organism names: E. coli, Escherichia coli; S.t, Salmonella typhimurium; P.s, Pseudomonas syringae.
Percent similarity to E. coli UmuD was determined from amino acid sequences aligned with the Lasergene software package (DNASTAR, Inc., Madison, WI) using the CLUSTAL algorithm with the PAM250 residue weight table.
P(N) values are a measure of the probability that the similarity between the two sequences occurs by chance. The smaller the P(N) value, the less likely the similarity has occurred by chance. For the comparison of amino acid sequences, P(N) values <10−6 are considered statistically significant (Karlin and Altschul 1993). These P(N) values were calculated by the BLASTP v.1.4.9MP database search program using the E. coli UmuD amino acid sequence as the query.
The bacteriophage P1 humD gene was identified in a search of DNA sequence databases for LexA binding sites, and it encodes a putative, precleaved UmuD′-like protein that lacks a cognate UmuC-like partner (Lewis et al. 1994).
E. coli UmuD homologs
| UmuD homolog | Similaritya(%) | P (N)b |
|---|---|---|
| S.t. UmuD | 72.7 | 2.2 × 10−59 |
| R391 RumA | 54.7 | 6.8 × 10−40 |
| TP110 ImpA | 44.6 | 3.2 × 10−33 |
| S.t. 60-MDa SamA | 44.6 | 2.2 × 10−30 |
| pKM101 MucA | 39.6 | 5.9 × 10−29 |
| Bacteriophage P1 HumDc | 34.6 | 1.0 × 10−15 |
| P.s. pPSR1 Ru1A | 30.9 | 6.0 × 10−11 |
| UmuD homolog | Similaritya(%) | P (N)b |
|---|---|---|
| S.t. UmuD | 72.7 | 2.2 × 10−59 |
| R391 RumA | 54.7 | 6.8 × 10−40 |
| TP110 ImpA | 44.6 | 3.2 × 10−33 |
| S.t. 60-MDa SamA | 44.6 | 2.2 × 10−30 |
| pKM101 MucA | 39.6 | 5.9 × 10−29 |
| Bacteriophage P1 HumDc | 34.6 | 1.0 × 10−15 |
| P.s. pPSR1 Ru1A | 30.9 | 6.0 × 10−11 |
Accession numbers for the nucleotide or amino acid sequences: E. coli UmuD, M13387; S.t. UmuD, M57431, M35010; 60-MDa plasmid SamA, D90202; R391 RumA, U13633; TP110 ImpA, X53528; pKM101 MucA, D90147; P1 HumD, M95666 (nucleotides 1661–2050); P.s. pPSR1 Ru1A, U43696. Abbreviations for organism names: E. coli, Escherichia coli; S.t, Salmonella typhimurium; P.s, Pseudomonas syringae.
Percent similarity to E. coli UmuD was determined from amino acid sequences aligned with the Lasergene software package (DNASTAR, Inc., Madison, WI) using the CLUSTAL algorithm with the PAM250 residue weight table.
P(N) values are a measure of the probability that the similarity between the two sequences occurs by chance. The smaller the P(N) value, the less likely the similarity has occurred by chance. For the comparison of amino acid sequences, P(N) values <10−6 are considered statistically significant (Karlin and Altschul 1993). These P(N) values were calculated by the BLASTP v.1.4.9MP database search program using the E. coli UmuD amino acid sequence as the query.
The bacteriophage P1 humD gene was identified in a search of DNA sequence databases for LexA binding sites, and it encodes a putative, precleaved UmuD′-like protein that lacks a cognate UmuC-like partner (Lewis et al. 1994).
typhimurium cells to mutagenesis in the Ames Test (McCann et al. 1975). S. typhimurium itself contains two operons homologous to umuD+C+, the chromosomal umuD+C+ST and samA+B+, which are borne on a 60-megadalton (MDa) cryptic plasmid. In spite of the presence of two umu-like operons, both of which can complement E. coli umuDC mutations (although samA+B+ must be on a high-copy-number plasmid), S. typhimurium is poorly mutable by UV, and it appears that the chromosomal umuD+C+ST is responsible for all of the mutagenesis seen (Koch et al. 1992; Nohmi et al. 1992).
Interestingly, E. coli has another chromosomally encoded umuC+homolog, dinB+, which was initially identified in a screen for loci whose expression is stimulated by DNA damage (Kenyon and Walker 1980). When this gene was later independently identified, sequenced, and described as dinP+, the homology to umuC+ was revealed (Ohmori et al. 1995). DinB is involved in mutagenesis because a mutant strain is defective in the untargeted UV mutagenesis of bacteriophage λ (Brotcorne-Lannoye and Maenhaut-Michel 1986), and overexpression of the protein, in the absence of DNA-damaging agents, enhances mutagenesis of an F′lac plasmid (Kim et al. 1997). In contrast to umuC+, dinB+does not have an adjacent umuD-like partner. Numerous DNA sequences encoding proteins with homology to UmuC and DinB have been found in all three kingdoms of life (Table 2): eukarya [Saccharomyces cerevisiae (Larimer et al. 1989) and Caenorhabditis elegans (Kulaeva et al. 1996)], archaea [Sulfolobus solfataricus (Kulaeva et al. 1996)], and bacteria [e.g., Bacillus subtilis (Kulaeva et al. 1996), S. typhimurium (Nohmi et al. 1991), and Mycobacterium tuberculosis (Kulaeva et al. 1996)]. Although the functions of some of these homologous proteins have yet to be determined, the family of UmuC-like proteins appears to be an ancient one. The reason for the apparent lack of widespread UmuD homologs is not clear, but if DinB does not require a cognate partner to function, the relationship between the functions of UmuC and DinB may shed additional light on the role(s) of UmuD and UmuD′ in the cell.
The mechanism of damage-induced mutagenesis has also been studied in eukaryotic systems, with the best-characterized model system being the yeast S. cerevisiae. The nonessential S. cerevisiae gene REV1 is required for UV mutagenesis (Lemontt 1971) and has a 152-aminoacid-residue internal section that is 25% identical (42% similar) to UmuC (Larimer et al. 1989). Two other genes, REV3 and REV7, are also required for damage-induced mutagenesis in S. cerevisiae. In vitro experiments have shown that Rev3p and Rev7p interact to form a DNA polymerase, termed DNA Pol ζ, that can replicate through UV-damaged DNA with limited efficiency (Nelson et al. 1996b). Nelson and his colleagues (1996a,b) suggested that other proteins such as Rev1p, which have been shown genetically to be required for UV mutagenesis, are likely required for the proper functioning of DNA Pol ζ in vivo. In an intriguing set of experiments in another eukaryotic model system, it was found that the replication arrest of UV-damaged M13 ssDNA molecules in Xenopus oocytes could be relieved by the injection of messenger RNA molecules encoding either UmuD′ and UmuC or MucA′ and MucB (Oda et al. 1996). The exact mechanism by which the bacterial proteins were able to relieve the replication arrest is unclear. Oda and her colleagues (1996) have speculated that the bacterial proteins might mimic factors that are expressed in progesterone-matured oocytes, which can replicate damaged ssDNA, by interacting directly with the Xenopus replication machinery or with an unknown factor(s) that inhibits the replication of damaged ssDNA in oocytes. There is also evidence for translesion synthesis of damaged DNA templates in mammalian cells. Studies of UV mutagenesis in Chinese hamster ovary (Spivak and Hanawalt 1992), HeLa (Thomas and Kunkel 1993; Thomas et al. 1993), and xeroderma pigmentosum variant (Wang et al. 1993) cell lines indicate that the resultant mutagenic events are caused by error-prone replication through UV-induced lesions in DNA. It appears that S. cerevisiae, and perhaps
E. coli UmuC and DinB homologs
| E. coli UmuC | E. coli DinB | |||
|---|---|---|---|---|
| Homolog | Similaritya (%) | P (N)b | Similaritya (%) | P (N)b |
| E. coli UmuC | — | — | 17.4 | 3.0 × 10−11 |
| E. coli DinBc | 17.4 | 3.0 × 10−11 | — | — |
| S.t. UmuC | 83.6 | 1.1 × 10−257 | 16.5 | 9.4 × 10−9 |
| S.t. 60-MDa SamB | 60.7 | 2.3 × 10−182 | 15.7 | 7.4 × 10−6 |
| R391 RumB | 57.3 | 3.7 × 10−174 | 17.7 | 6.5 × 10−8 |
| TP110 ImpB | 55.5 | 3.4 × 10−167 | 15.4 | 9.1 × 10−8 |
| pKM101 MucB | 53.1 | 3.0 × 10−157 | 11.1 | 7.2 × 10−6 |
| P.s. pPSR1 Ru1B | 41.5 | 7.2 × 10−93 | 17.7 | 4.7 × 10−7 |
| B.sub. YQ JHc,d | 16.9 | 4.8 × 10−18 | 31.6 | 1.0 × 10−61 |
| B.sub. YQ JWc,d | 19.7 | 3.5 × 10−18 | 25.1 | 7.1 × 10−27 |
| B.sub. UvrXc,d | 16.8 | 7.7 × 10−11 | 16.8 | 4.3 × 10−16 |
| L.lactis ORFUc,d | 14.7 | 2.8 × 10−3 | 10.8 | 2.2 × 10−3 |
| M.genit. Dbhc,d | 12.4 | 1.4 × 10−3 | 22.2 | 8.2 × 10−25 |
| M.pneu. Dbhc,d | 14.3 | 1.9 × 10−3 | 22.8 | 8.2 × 10−25 |
| M.tuberc. Dbhc,d | 19.7 | 2.8 × 10−15 | 24.9 | 2.0 × 10−29 |
| S.sp. UmuCc,d | 31.7 | 1.0 × 10−20 | 12.9 | ND f |
| S.solf. Dbhc | 17.2 | 1.7 × 10−17 | 25.9 | 2.1 × 10−40 |
| S.cere. Rev1pc | 12.8 | ND f | 14.2 | 2.1 × 10−4 |
| S.cere. Dbh1pc,d | 14 | 0.9998 | 20.5 | 4.4 × 10−23 |
| C.eleg. Rev1hc,d,e | 12.8 | ND f | 12.8 | 6.4 × 10−7 |
| C.eleg. Dbhc,d,e | 17.5 | 6.1 × 10−6 | 30.2 | 3.2 × 10−59 |
| E. coli UmuC | E. coli DinB | |||
|---|---|---|---|---|
| Homolog | Similaritya (%) | P (N)b | Similaritya (%) | P (N)b |
| E. coli UmuC | — | — | 17.4 | 3.0 × 10−11 |
| E. coli DinBc | 17.4 | 3.0 × 10−11 | — | — |
| S.t. UmuC | 83.6 | 1.1 × 10−257 | 16.5 | 9.4 × 10−9 |
| S.t. 60-MDa SamB | 60.7 | 2.3 × 10−182 | 15.7 | 7.4 × 10−6 |
| R391 RumB | 57.3 | 3.7 × 10−174 | 17.7 | 6.5 × 10−8 |
| TP110 ImpB | 55.5 | 3.4 × 10−167 | 15.4 | 9.1 × 10−8 |
| pKM101 MucB | 53.1 | 3.0 × 10−157 | 11.1 | 7.2 × 10−6 |
| P.s. pPSR1 Ru1B | 41.5 | 7.2 × 10−93 | 17.7 | 4.7 × 10−7 |
| B.sub. YQ JHc,d | 16.9 | 4.8 × 10−18 | 31.6 | 1.0 × 10−61 |
| B.sub. YQ JWc,d | 19.7 | 3.5 × 10−18 | 25.1 | 7.1 × 10−27 |
| B.sub. UvrXc,d | 16.8 | 7.7 × 10−11 | 16.8 | 4.3 × 10−16 |
| L.lactis ORFUc,d | 14.7 | 2.8 × 10−3 | 10.8 | 2.2 × 10−3 |
| M.genit. Dbhc,d | 12.4 | 1.4 × 10−3 | 22.2 | 8.2 × 10−25 |
| M.pneu. Dbhc,d | 14.3 | 1.9 × 10−3 | 22.8 | 8.2 × 10−25 |
| M.tuberc. Dbhc,d | 19.7 | 2.8 × 10−15 | 24.9 | 2.0 × 10−29 |
| S.sp. UmuCc,d | 31.7 | 1.0 × 10−20 | 12.9 | ND f |
| S.solf. Dbhc | 17.2 | 1.7 × 10−17 | 25.9 | 2.1 × 10−40 |
| S.cere. Rev1pc | 12.8 | ND f | 14.2 | 2.1 × 10−4 |
| S.cere. Dbh1pc,d | 14 | 0.9998 | 20.5 | 4.4 × 10−23 |
| C.eleg. Rev1hc,d,e | 12.8 | ND f | 12.8 | 6.4 × 10−7 |
| C.eleg. Dbhc,d,e | 17.5 | 6.1 × 10−6 | 30.2 | 3.2 × 10−59 |
Accession numbers for the nucleotide or amino acid sequences: E.coli UmuC, M13387; DinB, 984587; S.t. UmuC, M57431 or M35010; 60-MDa plasmid SamB, D90202; R391 RumB, U13633; TP110 ImpB, X53528; pKM101 MucB, D90147; P.s. pPSR1 Ru1B, U43696; B.sub. YQ JH, 1731071; YQ JW, 1731083; UvrX, AF014938; L.lactis. ORFU, 1039479; M.genet. Dbh, 1361895; M.pneu. Dbh, 2146696; M.tuberc. Dbh, 1781157; S.sp. UmuC, 1001815; S.solf. Dbh, 1706953; S.cere. Rev1p, M22222; Dbh1p, 927690; C.eleg. Rev1h, 599712; Dbh, 465873. Abbreviations for organism names: E.coli, Escherichia coli; S.t., Salmonella typhimurium; P.s., Pseudomonas syringae; B.sub., Bacillus subtilis; L.lactis, Lactococcus lactis; M.genit., Mycoplasma genitalium; M.pneu., Mycoplasma pneumoniae; M.tuberc, Mycobacterium tuberculosis; S.sp, Synechocystis sp. PCC6803; S.solf., Solfolobus solfataricus; S.cere., Saccharomyces cerevisiae; C.eleg, Caenorhabditis elegans.
See Table 1 notes (except that the amino acid sequence used for percent similarity and P(N) calculations in Table 2 was either the E.coli UmuC or DinB sequence).
These proteins do not appear to have adjacent UmuD-like partners.
These protein sequences have been identified via BLASTP searching of sequence databases. They have yet to be analyzed in further detail and should be considered putative homologs.
The database entries for these sequences have names different from those indicated here. The names given to them here more accurately reflect their relationships to other known proteins.
The similarity between these proteins was too weak to be identified in a BL ASTP-driven database search.
E. coli UmuC and DinB homologs
| E. coli UmuC | E. coli DinB | |||
|---|---|---|---|---|
| Homolog | Similaritya (%) | P (N)b | Similaritya (%) | P (N)b |
| E. coli UmuC | — | — | 17.4 | 3.0 × 10−11 |
| E. coli DinBc | 17.4 | 3.0 × 10−11 | — | — |
| S.t. UmuC | 83.6 | 1.1 × 10−257 | 16.5 | 9.4 × 10−9 |
| S.t. 60-MDa SamB | 60.7 | 2.3 × 10−182 | 15.7 | 7.4 × 10−6 |
| R391 RumB | 57.3 | 3.7 × 10−174 | 17.7 | 6.5 × 10−8 |
| TP110 ImpB | 55.5 | 3.4 × 10−167 | 15.4 | 9.1 × 10−8 |
| pKM101 MucB | 53.1 | 3.0 × 10−157 | 11.1 | 7.2 × 10−6 |
| P.s. pPSR1 Ru1B | 41.5 | 7.2 × 10−93 | 17.7 | 4.7 × 10−7 |
| B.sub. YQ JHc,d | 16.9 | 4.8 × 10−18 | 31.6 | 1.0 × 10−61 |
| B.sub. YQ JWc,d | 19.7 | 3.5 × 10−18 | 25.1 | 7.1 × 10−27 |
| B.sub. UvrXc,d | 16.8 | 7.7 × 10−11 | 16.8 | 4.3 × 10−16 |
| L.lactis ORFUc,d | 14.7 | 2.8 × 10−3 | 10.8 | 2.2 × 10−3 |
| M.genit. Dbhc,d | 12.4 | 1.4 × 10−3 | 22.2 | 8.2 × 10−25 |
| M.pneu. Dbhc,d | 14.3 | 1.9 × 10−3 | 22.8 | 8.2 × 10−25 |
| M.tuberc. Dbhc,d | 19.7 | 2.8 × 10−15 | 24.9 | 2.0 × 10−29 |
| S.sp. UmuCc,d | 31.7 | 1.0 × 10−20 | 12.9 | ND f |
| S.solf. Dbhc | 17.2 | 1.7 × 10−17 | 25.9 | 2.1 × 10−40 |
| S.cere. Rev1pc | 12.8 | ND f | 14.2 | 2.1 × 10−4 |
| S.cere. Dbh1pc,d | 14 | 0.9998 | 20.5 | 4.4 × 10−23 |
| C.eleg. Rev1hc,d,e | 12.8 | ND f | 12.8 | 6.4 × 10−7 |
| C.eleg. Dbhc,d,e | 17.5 | 6.1 × 10−6 | 30.2 | 3.2 × 10−59 |
| E. coli UmuC | E. coli DinB | |||
|---|---|---|---|---|
| Homolog | Similaritya (%) | P (N)b | Similaritya (%) | P (N)b |
| E. coli UmuC | — | — | 17.4 | 3.0 × 10−11 |
| E. coli DinBc | 17.4 | 3.0 × 10−11 | — | — |
| S.t. UmuC | 83.6 | 1.1 × 10−257 | 16.5 | 9.4 × 10−9 |
| S.t. 60-MDa SamB | 60.7 | 2.3 × 10−182 | 15.7 | 7.4 × 10−6 |
| R391 RumB | 57.3 | 3.7 × 10−174 | 17.7 | 6.5 × 10−8 |
| TP110 ImpB | 55.5 | 3.4 × 10−167 | 15.4 | 9.1 × 10−8 |
| pKM101 MucB | 53.1 | 3.0 × 10−157 | 11.1 | 7.2 × 10−6 |
| P.s. pPSR1 Ru1B | 41.5 | 7.2 × 10−93 | 17.7 | 4.7 × 10−7 |
| B.sub. YQ JHc,d | 16.9 | 4.8 × 10−18 | 31.6 | 1.0 × 10−61 |
| B.sub. YQ JWc,d | 19.7 | 3.5 × 10−18 | 25.1 | 7.1 × 10−27 |
| B.sub. UvrXc,d | 16.8 | 7.7 × 10−11 | 16.8 | 4.3 × 10−16 |
| L.lactis ORFUc,d | 14.7 | 2.8 × 10−3 | 10.8 | 2.2 × 10−3 |
| M.genit. Dbhc,d | 12.4 | 1.4 × 10−3 | 22.2 | 8.2 × 10−25 |
| M.pneu. Dbhc,d | 14.3 | 1.9 × 10−3 | 22.8 | 8.2 × 10−25 |
| M.tuberc. Dbhc,d | 19.7 | 2.8 × 10−15 | 24.9 | 2.0 × 10−29 |
| S.sp. UmuCc,d | 31.7 | 1.0 × 10−20 | 12.9 | ND f |
| S.solf. Dbhc | 17.2 | 1.7 × 10−17 | 25.9 | 2.1 × 10−40 |
| S.cere. Rev1pc | 12.8 | ND f | 14.2 | 2.1 × 10−4 |
| S.cere. Dbh1pc,d | 14 | 0.9998 | 20.5 | 4.4 × 10−23 |
| C.eleg. Rev1hc,d,e | 12.8 | ND f | 12.8 | 6.4 × 10−7 |
| C.eleg. Dbhc,d,e | 17.5 | 6.1 × 10−6 | 30.2 | 3.2 × 10−59 |
Accession numbers for the nucleotide or amino acid sequences: E.coli UmuC, M13387; DinB, 984587; S.t. UmuC, M57431 or M35010; 60-MDa plasmid SamB, D90202; R391 RumB, U13633; TP110 ImpB, X53528; pKM101 MucB, D90147; P.s. pPSR1 Ru1B, U43696; B.sub. YQ JH, 1731071; YQ JW, 1731083; UvrX, AF014938; L.lactis. ORFU, 1039479; M.genet. Dbh, 1361895; M.pneu. Dbh, 2146696; M.tuberc. Dbh, 1781157; S.sp. UmuC, 1001815; S.solf. Dbh, 1706953; S.cere. Rev1p, M22222; Dbh1p, 927690; C.eleg. Rev1h, 599712; Dbh, 465873. Abbreviations for organism names: E.coli, Escherichia coli; S.t., Salmonella typhimurium; P.s., Pseudomonas syringae; B.sub., Bacillus subtilis; L.lactis, Lactococcus lactis; M.genit., Mycoplasma genitalium; M.pneu., Mycoplasma pneumoniae; M.tuberc, Mycobacterium tuberculosis; S.sp, Synechocystis sp. PCC6803; S.solf., Solfolobus solfataricus; S.cere., Saccharomyces cerevisiae; C.eleg, Caenorhabditis elegans.
See Table 1 notes (except that the amino acid sequence used for percent similarity and P(N) calculations in Table 2 was either the E.coli UmuC or DinB sequence).
These proteins do not appear to have adjacent UmuD-like partners.
These protein sequences have been identified via BLASTP searching of sequence databases. They have yet to be analyzed in further detail and should be considered putative homologs.
The database entries for these sequences have names different from those indicated here. The names given to them here more accurately reflect their relationships to other known proteins.
The similarity between these proteins was too weak to be identified in a BL ASTP-driven database search.
other eukaryotes, have evolved a translesion synthesis system that is dependent on a specialized DNA polymerase as opposed to the prokaryotic system, which modulates the function of a high-fidelity, replicative DNA polymerase.
A role for umuD+C+ in cell-cycle regulation: Recent experiments performed in our laboratory have shed new light on the functions of the umuD+C+ gene products and on how an E. coli cell responds to DNA damage. These findings evolved out of a more detailed analysis of the phenomenon of umuD+C+-mediated cold sensitivity for growth, i.e., cells that constitutively express umuD+C + from a multicopy plasmid can grow at 42° but cannot grow at 30°; Marsh and Walker 1985. Our results have revealed that cold sensitivity for growth and SOS mutagenesis are genetically separable functions of the umuD+C+ gene products (Opperman et al. 1996). The growth defect at 30° can be very extreme. For example, transformation of a pBR322-based plasmid carrying the umuD+C+ operon under its native promoter into a lexA (Def) strain was 40,000-fold more efficient when the transformants were grown at 42° than at 30°. A similar effect was seen in the absence of SOS induction when umuD+C+ was expressed from a plasmid under control of an exogenous PBAD promoter. UmuD or uncleavable UmuD mutants in combination with UmuC conferred a greater degree of cold sensitivity than UmuD′ with UmuC (Opperman et al. 1996). These observations were exceptionally interesting because intact UmuD had previously been viewed as a molecule whose primary roles were confined to (1) being an inactive precursor to UmuD′and (2) helping to shut off SOS mutagenesis as the cell recovered from DNA damage by trapping UmuD′ in heterodimers (Figure 2). Cold sensitivity for growth was also independent of recA+, which further differentiated the activity genetically from the traditional role of umuD+C + in SOS mutagenesis.
These studies also revealed that UmuD and UmuC could inhibit cell division, i.e., inhibit septation via a novel pathway, causing the production of cellular filaments at 30° in lexA(Def) strains when expressed from the single chromosomal copy of the umuD+C+ operon (Opperman et al. 1996). Filamentation was independent of the previously known DNA damage-induced inhibitors of cell division, sulA+ and sfiC+. However, another unidentified LexA-regulated function was required. Interestingly, the SOS-regulated dinH locus appears to be ftsK+, a gene whose function is required late in cell division (Begg et al. 1995; Lewis et al. 1992). It is not clear how a gene required for cell division could also be involved in UmuDC-mediated inhibition of septation, but a closer look at ftsK's possible involvement is called for. This filamentation could be suppressed by the introduction of a plasmid overexpressing the FtsQ, FtsA, and FtsZ proteins, suggesting that UmuD and UmuC may act upstream of these Fts proteins to inhibit septation. Because it is the target of the cell division inhibitor SulA, FtsZ, which acts early in cell division by forming a polymeric ring at the site of septation (Bi and Lutkenhaus 1991; Lutkenhaus and Addinall 1997), is the likely target of this filamentation-promoting activity of UmuD and UmuC. The overexpression of the Fts proteins did not suppress cold sensitivity for growth resulting from the overexpression of UmuD and UmuC. Along with the requirement of SOS induction for filamentation but not cold sensitivity for growth, this indicates that cold-sensitive growth and filamentation are genetically separable and involve different cellular targets of the UmuD and UmuC proteins. A later report described the analysis of an sfi-independent filamentation pathway that is regulated in a lexA+- and recA+-dependent manner and that requires DNA damage (Hill et al. 1997). It seems likely that this pathway for filamentation is the same as the umuD+C +-dependent pathway described by Opperman and his colleagues (1996). In both studies, 4′,6-diamino-2-phenyl-indole (DAPI) staining revealed abnormal nucleoid morphologies in some filaments. Thus, umuD+C+-induced filamentation may also disrupt chromosome segregation to some degree. Whether this potential segregation defect is due to a direct function of the Umu proteins or is a side effect resulting from the disruption of the normal bacterial cell cycle remains to be determined.
The cold sensitivity for growth, filamentation, and inhibition of DNA replication phenotypes observed when umuD+C+ is overexpressed indicated the possibility of a novel function(s) of the umuD+C+ operon when expressed at physiologically relevant levels. The first hint of what this function might be was provided by previous work in our laboratory in which an allele of umuC, umuC125, was isolated; when overexpressed with umuD+ on a pBR322-based plasmid, this allele did not cause cold sensitivity for growth even though it was active in SOS mutagenesis. Interestingly, the umuC125 mutation also made the cells more sensitive to UV irradiation than strains expressing umuC+ (Marsh et al. 1991). If SOS mutagenesis was the only way in which the umuD+C+ gene products participated in the protection of the bacterial cell from DNA damage, then why was a cell expressing umuD+C125 more sensitive to UV irradiation than one expressing umuD+C +?
The answer to that question is suggested by various clues indicating that the umuD+C+ gene products are involved in regulating aspects of the cell cycle after DNA damage. Recent observations indicate that UmuC and the intact UmuD protein are involved in the inhibition of DNA replication after the cell's DNA is damaged. The RecA*-mediated processing of UmuD to UmuD′ results in the lifting of this inhibition. It is attractive to view the cleavage of UmuD to UmuD′ as a timing mechanism. Once the umuD+C + operon is induced, the gene products will act to inhibit DNA replication, thereby allowing error-free repair of the DNA to occur. If the damage is extremely extensive and cannot be repaired promptly, the DNA damage signal will persist long enough to allow RecA* to promote the cleavage of UmuD to UmuD′, stimulating translesion synthesis and allowing the cell to continue its cell cycle as best it can (T. Opperman, S. Murli and G. C. Walker, unpublished results). Supporting this model is the fact that the RecA*-mediated cleavage of UmuD is much less efficient than that of other coprotease substrates such as LexA and MucA (Burckhardt et al. 1988; Woodgate and Ennis 1991; Hauser et al. 1992), which suggests that the reduced cleavage efficiency may have evolved specifically to enhance this timing function.
The process by which UmuD and UmuC influence DNA synthesis after DNA damage is related to the observation that following sublethal doses of UV, DNA replication rates in E. coli decrease and then, shortly afterwards, resume to normal levels. This resumption has been termed “induced replisome reactivation” (IRR; Khidir et al. 1985) or “replication restart” (Echols and Goodman 1991). The recovery of DNA synthesis has been shown to require RecA and postirradiation synthesis of another protein (Khidir et al. 1985). Interestingly, the resumption of replication was found to be independent of umuD+C + in a recA+ strain but was dependent on umuD+C+ in a partial loss-of-function recA718 background (Witkin et al. 1987). The question of what causes this inhibition was not answered, although it is independent of RecA and does occur in a mutant lexA3 strain that is incapable of SOS induction (Khidir et al. 1985). The postirradiation inhibition could be caused merely by the accumulation of lesions in the chromosome that inhibits the progress of the replication machinery. Alternatively (or in addition), there could be an active inhibition of DNA replication in response to the detection of DNA damage in the cell. Such a system is termed a DNA-damage checkpoint and is ubiquitous in eukaryotic cells (reviewed in Elledge 1996). The possible presence or absence of a DNA-damage checkpoint in E.coli has been discussed at length elsewhere (Bridges 1995).
The increased UV sensitivity of mutant strains that completely lack umuD+C + function (Woodgate 1992) had previously been attributed exclusively to the loss of the translesion synthesis activity of UmuD′ and UmuC. However, in a lexA(Def) recA430 background (in which the mutant RecA protein is incapable of mediating the cleavage of UmuD to UmuD′ and SOS mutagenesis is not observed) a ΔumuDC mutation still increases UV sensitivity (T. Opperman, S. Murli and G. C. Walker, unpublished results). This experiment clearly demonstrates a role for the UmuD and UmuC proteins in the protection of the cell from DNA damage, which is distinct from their role in SOS mutagenesis. Further investigation of this additional function revealed that, while the inhibition and recovery of DNA synthesis after UV irradiation occurs normally in a ΔumuDC strain, increasing the dosage of umuD+C +(but not of umuD′C+) using a pBR322-based plasmid delayed the resumption of DNA synthesis relative to the pBR322-containing, ΔumuDC control strain. With a plasmid carrying an operon encoding a mutant UmuD molecule that is refractive to RecA*-mediated cleavage [umuD(SA60)C+; Nohmi et al. 1988], recovery of DNA synthesis was not observed within the time scale of the experiment (60 min). In contrast, with the plasmid-borne umuD+C125 operon, which exhibits increased UV sensitivity and does not cause cold sensitivity when overexpressed, inhibition of DNA synthesis did not occur after UV irradiation (T. Opperman, S. Murli and G. C. Walker, unpublished results).
Analysis of these data must take into account the previously mentioned work on the inhibition and resumption of DNA synthesis after UV irradiation, which showed the following: (1) a strain unable to induce the SOS response (lexA3) exhibited inhibition of DNA synthesis after UV irradiation but not the subsequent resumption that is seen in a wild-type strain (Khidir et al. 1985); (2) in a recA+ strain, umuDC mutations do not affect either the inhibition or resumption of DNA synthesis (Khidir et al. 1985; Witkin et al. 1987); and (3) umuD+C+ is required for resumption of DNA synthesis but not for inhibition in a partial loss-of-function recA718 strain (Witkin et al. 1987). One interpretation of our findings in light of these observations is that, when present, the umuD+C + gene products exert an additional layer of control over DNA synthesis, perhaps by enhancing either an active inhibitory factor or the passive ability of accumulated lesions in the DNA to impede the DNA polymerase. The inhibitory activity of UmuD and UmuC proteins is relieved by the RecA*-mediated cleavage of UmuD to UmuD′, which could explain, in part, the requirement of RecA for resumption of DNA synthesis. In this way, the uncleavable umuD(SA60) protein, in combination with UmuC, would inhibit replication in an unregulated manner. In contrast, the umuC125 protein appears to be capable of rendering the DNA polymerase insensitive to inhibition after DNA damage occurs. Whether this is due to a loss of the wild-type UmuC's inhibitory function when combined with UmuD, or to a gain of a replication-promoting function akin to its role with UmuD′ in translesion synthesis, is not clear.
Additional observations indicate that the umuD+C+ gene products can act to increase the resistance of stationary-phase cells to DNA damage by inhibiting the transition of the cells from stationary phase to exponential growth. This presumably allows time for the accumulated lesions to be repaired before cells begin rapid DNA synthesis and growth. This growth-phase arrest appears to occur via a umuD+C +-dependent inhibition of an activity of Fis (S. Murli, T. Opperman and G. C. Walker, unpublished results), a protein that is maximally expressed in the first two generations after cells emerge from stationary phase and is involved in a variety of processes, including growth-phase and transcriptional regulation as well as site-specific recombination (Ball et al. 1992; Finkel and Johnson 1992; Osuna et al. 1995). The finding that the SOS response is induced in stationary-phase cells supports the hypothesis that DNA damage incurred while a cell is quiescent must be repaired prior to the resumption of growth (Taddei et al. 1995). Further work will be required to elucidate the roles of UmuD and UmuC in all of these processes, but it is clear that they are capable of protecting the cell from DNA damage in a manner that is genetically, and perhaps biochemically, separable from the more familiar roles of UmuD′ and UmuC in SOS mutagenesis.
Evolution of the umu-like operons: With evidence accumulating that supports a DNA-damage checkpoint role for the umuD+C+ gene products, an entirely new light is cast on the question of the evolution of the umuD+C+ operon and its many homologs (Tables 1 and 2). Previous discussions of the evolution of the umu-like operons focused on the difficulty of rationalizing the apparent selective disadvantage of an inducible mutagenesis system, considering the fact that the vast majority of mutagenic events will be unfavorable, with the widespread occurrence of highly homologous operons in a variety of prokaryotic (and eukaryotic, in the case of REV1) organisms. Usually, it has been concluded that the translesion synthesis activity that these operons promote provides the cell with an additional means of surviving a serious challenge by DNA-damaging agents, with the resulting mutations being the price of survival (Woodgate and Sedgwick 1992; Friedberg et al. 1995). However, it has also been argued that induced mutagenesis could help cells to survive periods of extreme environmental stress by acting as a mechanism of inducible evolution (Radman et al. 1978; Echols 1981). With the recent experiments indicating a role for the umuD+C+operon in the regulation of various aspects of the E. coli cell cycle following DNA damage, additional evolutionary pressures that might have brought the umu-like operons into existence and then maintained them can be considered. A DNA-damage checkpoint is an advantageous response for a cell to have. As a result, the umuD+C+ operon could be selected for because it provides the cell with both a checkpoint activity and an activity, i.e., translesion synthesis, that allows the cell to continue DNA synthesis and the cell cycle when the DNA damage is too severe for rapid repair. This perspective also provides a possible explanation for why S. typhimurium is poorly mutable in spite of the presence of two umu-like operons, umuD+C+ and samA+ST B+, as well as for the poor mutability of a variety of other, less well-characterized bacterial strains that appear to have umu-like operons (Sedgwick et al. 1991). If the primary function of these operons is the regulation of the cell cycle in response to DNA damage, the translesion synthesis activity may have been acquired subsequently in some operons but not in others. This could explain the range of mutability that has been observed among organisms that contain umu-like operons.
Study of the molecular basis of UV and chemical mutagenesis in the model system E. coli has not only provided insights into the process of mutagenesis but also into the regulation of gene expression in response to DNA damage (the SOS response), into a novel mechanism of post-translational regulation (the RecA*-mediated autodigestion of LexA and UmuD), and into the regulation of the cell cycle in response to DNA damage (the role of umuD+C+ in filamentation, inhibition of DNA replication, and growth-phase transition). Future investigation into the molecular mechanisms of translesion synthesis and into the roles of RecA, UmuD, UmuD′, UmuC, and DNA Pol III in that process will undoubtedly reveal much about the mechanism by which mutations become fixed in an organism's genome. These inquiries may also illuminate more general features of both DNA replication and the cellular response to DNA damage.
Acknowledgement
The authors thank Dr. Timothy Opperman for his critical reading and discussion of the manuscript. We also thank Drs. Ann Ferentz, Gerhard Wagner, Kendall Knight and Haruo Ohmori for sharing unpublished data. The other members of the Walker laboratory are also acknowledged for their helpful comments and assistance. This work was supported by Public Health Service grant CA-21615, awarded by the National Cancer Institute.
LITERATURE CITED

{kind=link}
{kind=link}
{kind=link}