





Abstract
Double-strand break (DSB) induced gene conversion in Saccharomyces cerevisiae during meiosis and MAT switching is mediated primarily by mismatch repair of heteroduplex DNA (hDNA). We used nontandem ura3 duplications containing palindromic frameshift insertion mutations near an HO nuclease recognition site to test whether mismatch repair also mediates DSB-induced mitotic gene conversion at a non-MAT locus. Palindromic insertions included in hDNA are expected to produce a stem-loop mismatch, escape repair, and segregate to produce a sectored (Ura+/−) colony. If conversion occurs by gap repair, the insertion should be removed on both strands, and converted colonies will not be sectored. For both a 14-bp palindrome, and a 37-bp near-palindrome, ~75% of recombinant colonies were sectored, indicating that most DSB-induced mitotic gene conversion involves mismatch repair of hDNA. We also investigated mismatch repair of well-repaired markers flanking an unrepaired palindrome. As seen in previous studies, these additional markers increased loop repair (likely reflecting corepair). Among sectored products, few had additional segregating markers, indicating that the lack of repair at one marker is not associated with inefficient repair at nearby markers. Clear evidence was obtained for low levels of short tract mismatch repair. As seen with full gene conversions, donor alleles in sectored products were not altered. Markers on the same side of the DSB as the palindrome were involved in hDNA less often among sectored products than nonsectored products, but markers on the opposite side of the DSB showed similar hDNA involvement among both product classes. These results can be explained in terms of corepair, and they suggest that mismatch repair on opposite sides of a DSB involves distinct repair tracts.
GENE conversion is the nonreciprocal transfer of information from a DNA duplex to a homologous duplex, a process that has been widely studied in yeast (reviewed in Petes et al. 1991). For heterozygous loci, gene conversion results in a 3:1 (or 6:2) aberrant meiotic segregation pattern, contrasting with normal 2:2 segregation. In the yeast Saccharomyces cerevisiae, several other types of aberrant segregation patterns are observed at lower frequencies, notably 5:3 patterns, in which one of the four spores yields a sectored colony. Sectoring reflects segregation of mismatches, which are formed during strand exchange, in the mitotic division immediately following meiosis. These 5:3 patterns are therefore termed postmeiotic segregation (PMS).
Two types of models have been proposed to explain the relative frequencies of gene conversion and PMS and the mechanism(s) underlying these events. One type proposes that gene conversion results from correction of mismatches in hDNA intermediates, with PMS reflecting mismatch segregation (Holliday 1964; Meselson and Radding 1975). In this view, the predominance of gene conversion over PMS reflects efficient mismatch repair. The gap repair model was proposed to explain the results of transformation experiments in which an allele containing a double-strand gap (produced in vitro) was repaired from endogenous sequences in the host genome (Orr-Weaver et al. 1981; Szostak et al. 1983). In this model, most conversion occurs in a double-strand gap, which would preclude PMS, but rare PMS events were thought to reflect the inclusion of markers in heteroduplex DNA (hDNA) adjacent to a gap during strand invasion or as a result of Holliday junction (HJ) branch migration.
In both mitotic and meiotic cells, nearly all conversion tracts are continuous (Aguilera and Klein 1989; Ahn and Livingston 1986; Borts and Haber 1987; Borts and Haber 1989; Judd and Petes 1988; Sweetser et al. 1994; Symington and Petes 1988; Willis and Klein 1987), and alleles suffering a DSB are preferentially converted (reviewed in Nickoloff and Hoekstra 1997). These observations, plus the fact that gapped substrates are repaired in vivo with information donated by a homologous duplex (Orr-Weaver et al. 1981), provided strong support for the gap repair model. However, in vivo studies of MAT conversion failed to show double-strand gap formation at double-strand breaks (DSBs) introduced into MAT by HO nuclease (White and Haber 1990), and genetic evidence suggests that markers quite close to the MAT DSB are often in hDNA (Haber et al. 1993; McGill et al. 1989; Ray et al. 1991). Furthermore, although meiotic conversion is initiated by DSBs (Cao et al. 1990; de Massy et al. 1995; Nag and Petes 1993; Sun et al. 1989; Wu and Lichten 1994), PMS increases in mismatch repair-defective strains to the same degree that gene conversion decreases, suggesting that most or all meiotic conversion results from mismatch repair of hDNA (reviewed in Petes et al. 1991). Direct evidence for hDNA mediating meiotic gene conversion was obtained in two studies (Lichten et al. 1990; Nag and Petes 1990; Nag and Petes 1993). To account for tract continuity and preferential conversion of broken alleles in an hDNA repair model, mismatch repair must involve long repair tracts (or less likely, concerted short repair tracts), and repair must be biased against mismatched bases in recipient alleles (i.e., those suffering a DSB). E. coli has systems for both long and short tract mismatch repair (Modrich 1991). The long tract repair system involves MutHLS, and yeast has MutL and MutS homologs (reviewed in Crouse 1997). Specialized short tract repair systems are unknown in S. cerevisiae, but Schizosaccharomyces pombe apparently has a system that repairs C–C mismatches (Schar and Kohli 1993). Previous studies showed that for spontaneous mitotic conversion, sectored colonies and discontinuous conversion tracts arise at frequencies of ~15% and ~3%, respectively (Ronne and Rothstein 1988; Sweetser et al. 1994). Although these results indicate that at least some mitotic conversions involve hDNA intermediates, they do not differentiate between hDNA and gap repair models for the majority of mitotic events. Indirect evidence against gap repair for general mitotic DSB-induced conversion (vs. site-specific conversion at MAT) comes from two studies involving conversion of plasmid-borne alleles interacting with chromosomal loci. In these studies, conversion tracts often extended in only one direction from a DSB (or gap) (Nelson et al. 1996; Priebe et al. 1994). This result is inconsistent with gap repair since gaps are unlikely to be formed on only one side of a DSB. In the present study we addressed this question directly by using palindromic frameshift insertion mutations adjacent to DSB sites that would produce poorly repaired loop mismatches if present in hDNA, an experiment suggested by Esposito et al. (1994). Segregation of loop mismatches produces sectored (+/−) colonies, which were seen at a frequency of ~75% among recombinant products, indicating that most DSB-induced mitotic conversion is mediated by hDNA repair. The system was then exploited to examine the fate of additional mismatches near an unrepaired loop mismatch. These experiments indicated that the additional mismatches are efficiently repaired despite their proximity to the unrepaired loop mismatch and that conversion on opposite sides of a DSB can be mediated by two independent mismatch repair tracts.
MATERIALS AND METHODS
Plasmids: Standard techniques were used to construct plasmids (Sambrook et al. 1989). Plasmid pUCUraR-HO432Δ5′Hleu is a pUC19 derivative with ura3 and LEU2 (J. W. Cho and J. A. Nickoloff, unpublished results). This plasmid contains ura3 with nine phenotypically silent RFLP markers and a 24-bp HO site at a natural NcoI site at position 432 (HO432) (Sweetser et al. 1994). This plasmid lacks the polylinker XbaI site, and the promoter proximal (5′) HindIII site of the ura3 fragment was deleted to facilitate allele rescue and simplify marker analysis. Derivatives of this plasmid were created as follows. A 14-bp palindromic frameshift mutation was created by inserting a 10-bp MluI linker (5′-CGACGCGTCG-3′) into the silent PmlI RFLP at position 409 (Pml409). Subsequent MluI digestion, fill-in, and ligation reactions converted the MluI site to a BssHII site, and created a palindromic 14-bp insertion (Bss14-409). An 8-bp NruI linker (5′-GTCGCGAC-3′) was inserted into a digested and filled-in Bss14-409 site, and a 12-bp MluI linker (5′-GCGACGCGTCGC-3′) was then inserted into the NruI site. Although the resulting insertion carried the desired MluI site, sequence analysis showed that it did not have the predicted 38-bp sequence; one bp was absent, an apparent cloning artifact. This 37-bp near-palindromic insertion is called Mlu37-409. The HO site and either Bss14-409 or Mlu37-409 were transferred to a wild-type copy of URA3 in a pUC19 derivative lacking the polylinker XbaI site by using a domain replacement procedure (Ray et al. 1994).
Yeast strains: Cells were cultured as described previously (Nickoloff et al. 1990). Strain DY3421 (J. W. Cho and J. A. Nickoloff, unpublished results) contains ura3 with a +1 frameshift mutation at position 764 (X764) (Sweetser et al. 1994) and a galactose-inducible source of HO (GalHO) integrated at lys2 using pHSS19, a kanamycin-resistant vector (Nickoloff and Reynolds 1991); this vector does not interfere with rescue of either ura3 allele, which are linked to the ampicillin-resistant pUC19 vector. DY3421 also carries a MATa-inc mutation to prevent mating-type switching when HO is induced, ade2-101, his3-200, leu2-Δ1, and trp1-Δ1 (Sweetser et al. 1994). Plasmids with ura3 containing 14- or 37-bp palindromes and LEU2 were linearized at the SmaI site in ura3 and integrated into ura3-X764 of DY3421, creating strains YW14-409 and YW37-409. Integration of a related plasmid containing a 14-bp palindrome and 8 silent RFLP markers created strain YW14-409R. Structures of the resulting duplications (Figure 1, A and B) were confirmed by Southern hybridization analysis of genomic DNA. Further characterization of rescued ura3 alleles (see below) included restriction mapping of all markers, and sequence analysis of the region containing palindromes, including the entire region amplified by PCR during domain replacement.
Recombination assays: Two-day old colonies (Ura− Leu+) of YW14-409 and YW37-409 were grown in 1.5 ml of rich medium with 2% glycerol (YPGly) for one day and then transferred to 1.5 ml of rich medium with 5% galactose (YPGal5) for 3 hours. A high concentration of galactose was used to induce HO rapidly, but a minimal induction time period was used to reduce the probability that recombinant cells would divide before plating as this would reduce the apparent sector rate. Cells were plated at a low density to reduce coplating artifacts (~100–200 per plate) on nonselective medium (YPD or LB-Ura; see below) and grown for 3 days. For colonies on YPD, Ura+ colonies and sectored Ura+/− colonies were identified
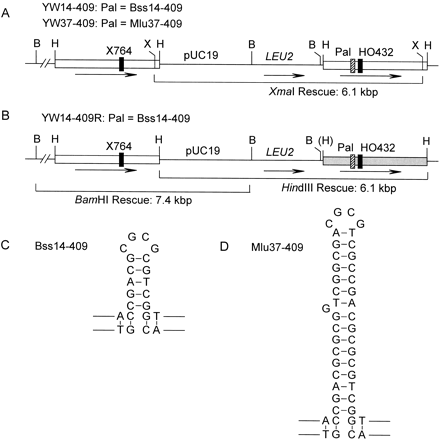
Recombination substrates. (A) Strains YW14-409 and YW37-409 have nontandem direct ura3 repeats (boxes) flanking pUC19 and LEU2 and integrated at the normal ura3 locus on chromosome V. The left allele is inactivated by the X764 frame-shift mutation and the right allele by HO432 and 14- or 37-bp palindromic insertions (Pal). Digestion with XmaI releases a 6.1-kbp fragment containing the right allele linked to pUC19 for rescue into E. coli. BamHI, HindIII, and XmaI sites are indicated by B, H, and X. (B) Strain YW14-409R is identical to YW14-409 except that the right ura3 allele has eight silent RFLP mutations indicated by the shaded box (see Figure 3). (C–D) Predicted stem-loop structures of the perfect 14-bp palindrome, and near-perfect 37-bp palindrome, as they would occur in hDNA.
by replica-plating to uracil omission medium. LB-Ura medium is identical to standard uracil omission medium except that it contains 3.4 g/liter yeast extract, 6.8 g/liter bacto-tryptone, and 3.4 g/liter NaCl. LB-Ura contains enough uracil to support growth of Ura− yeast, but allows the Ura phenotype to be determined visually in ade2 (red) mutants (Weng and Nickoloff 1997). On LB-Ura plates, three types of colonies were identified: white (Ura− parental), red (Ura+ nonsectored recombinants), and half-red/half-white (Ura+/− sectored recombinants). Leu phenotypes were subsequently determined for all recombinants, including both halves of sectored colonies. Strain YW14-409R was treated as above except that HO was induced for 6 hr. Although increasing the length of induction would tend to reduce the ratio of sectored to nonsectored products due to preplating segregation, preliminary tests indicated that the longer induction increased the total number of both sectored and nonsectored recombinants. This simplified the recovery of sectored colonies, which arose at very low frequencies in YW14-409R (see results), and did not compromise our physical analysis of sectored products.
Sectored product analysis: Conversion tracts, and segregated hybrid DNA regions in sectored products of strain YW14-409R were characterized by mapping both alleles from Ura+ and Ura− sectors. This physical analysis was performed only on sectored colonies in which both sectors were Leu+, as these were constrained to arise by gene conversion (Ura+/− sectors can also result from unequal sister chromatid exchange and other types of events, but these yield colonies with at least one Leu− sector; see results). Because physical analysis was only performed on sectored colonies, and because sectored colonies were rare in strain YW14-409R (<1 per plate) the two pairs of ura3 alleles in any sectored colony were assured of being derived from a single recombinant. Both ura3 alleles from each Ura+ and Ura− sector were independently rescued as follows. Upstream alleles of all strains were excised from genomic DNA as 7.4-kbp BamHI fragments, and downstream alleles as 6.1-kbp HindIII fragments in strain YW14-409R (Figure 1B) and as 6.1-kbp XmaI fragments in strains YW14-409 and YW37-409, each linked to pUC19 (Figure 1A). Fragments were circularized using T4 DNA ligase, and electroporated into E. coli strain HB101 as described (Gunn et al. 1995). Conversion tracts were defined in all rescued plasmids from YW14-409R by scoring all markers, including silent RFLPs, the palindrome, and the two frameshift mutations (X764 and HO432) in both donor and recipient alleles. To test whether Ura− sectors of strain YW14-409 retained HO sites, patch recombination assays were performed on solid medium as described previously (Nickoloff et al. 1990). Briefly, cells were grown as 1 cm2 patches on solid YPGly medium for 24 h, replica-plated to YPGal to induce HO nuclease and to YPD (uninduced control), incubated for 24 hr, then replica-plated to uracil-omission plates and incubated for 2 days. Strains with HO sites recombine at high frequencies after growth on YPGal and produce many Ura+ papillae; strains lacking HO sites (i.e., those in which the HO site had converted) recombine infrequently and few or no papillae arise. Statistics were performed using the Fisher exact test.
RESULTS
Experimental strategy: To determine whether general, DSB-induced gene conversion is mediated by gap or hDNA repair, we constructed strains with nontandem ura3 duplications having perfect or near perfect palindromes 23 bp from an HO site. DSBs can be delivered to HO sites in vivo, thus providing a defined recombination initiation site (Figure 1). We reasoned that if DSB-induced gene conversion involves even short double-stranded gaps, the palindrome would frequently be included in gaps extending from the nearby HO site, and Ura+ recombinants would usually be nonsectored. However, if conversion proceeds by hDNA repair, as seen in meiosis and during MAT switching (Nag and Petes 1990; Nag and Petes 1993; Petes et al. 1991; Ray et al. 1991), the palindrome should produce a stem-loop mismatch (Figure 1, C and D) that is expected to frequently escape repair (Nag and Petes 1991; Nag et al. 1989) and segregate during the next division, producing sectored Ura+/− recombinants. Since sectored colonies will only be detected if recombinants do not divide before plating, recombination was induced only briefly (3 hr in galactose medium). Cells grown in galactose divide about once per 2–3 hr (data not shown), so these conditions allow no more than one cell division during the induction period. Uninduced recombination frequencies were less than 10−4 (data not shown) while induced frequencies were ~2 × 10−1 (Table 1), confirming that essentially all recombinants from galactose cultures were induced by DSBs.
DSB-induced recombination between these ura3 direct repeats can produce a variety of products including full or half gene conversions (Ura+ or Ura+/−) in which both ura3 repeats and LEU2 are retained (Leu+ or Leu+/+) or that are associated with a deletion of one ura3 repeat and LEU2 (Leu− or Leu−/−). Deletions may reflect either crossover or single-strand annealing (SSA) events. Also expected were unequal sister chromatid exchange events (which produce a colony with a Leu+ triplication sector and a Leu− deletion sector). If recombinants are identified using selective medium, gene conversions and triplications yield identical Ura+ Leu+ phenotypes, but can be distinguished if genomic DNA is analyzed. Deletions arising by crossing-over, SSA, or unequal exchange are indistinguishable phenotypically (all are Leu−) and genotypically. The nonselective conditions used in this study allow sectored gene conversion products (Ura+/− Leu+/+) to be easily distinguished from other types of Ura+/− sectored products, such as triplication or deletion events resulting from unequal sister chromatid exchange (Ura+/− Leu−/+ and Ura+/− Leu+/−) (Figure 2).
Distribution of recombinant products: The phenotype distribution for DSB-induced recombinants of strain YW14-409 is shown in Table 1. Ura+/− Leu−/− deletion products appeared most frequently; these may arise by loop segregation during conversion events associated with a crossover or an SSA event, or from conversion of only one sister chromatid in G2 cells, although evidence presented below argues against the latter mechanism in the majority of cases. Leu+/+ gene conversion (non-deletion) products were less frequent than deletions (Leu−/−), consistent with studies using related strains (Nickoloff et al. 1989; J. W. Cho and J. A. Nickoloff, unpublished results). Leu+/− products accounted for ~40% of the total recombinants. Southern analysis of three Leu+/− products each of classes 5 and 6 (Figure 2) was consistent with these resulting from unequal sister chromatid exchange or by the mechanism proposed by Lovett et al. (1993) involving sister strand exchange of nascent strands during DNA replication
DSB-induced recombination frequencies
| Strain | na | 104 × Frequencyb | |||||||
| Full GCc | Half GC | Percent Sectored | Deletion ± GC | Deletion + Half GC | Deletion/Triplicationd | ||||
| Ura+ Leu+ | Ura+/− Leu+/+ | Ura+ Leu− | Ura+/− Leu−/− | Ura+/− Leu−/+ | Ura+/− Leu+/− | Ura+/+ Leu+/− | |||
| YW14-409 | 9,205 | 38 | 265 | 87 | 170 | 798 | 425 | 425 | 44 |
| YW37-409 | 9,789 | 90 | 427 | 83 | 150 | 682 | 256 | 341 | 19 |
| YW14-409R | 17,167 | 253 | 12 | 5 | ND | 67 | 17 | 16 | ND |
| Strain | na | 104 × Frequencyb | |||||||
| Full GCc | Half GC | Percent Sectored | Deletion ± GC | Deletion + Half GC | Deletion/Triplicationd | ||||
| Ura+ Leu+ | Ura+/− Leu+/+ | Ura+ Leu− | Ura+/− Leu−/− | Ura+/− Leu−/+ | Ura+/− Leu+/− | Ura+/+ Leu+/− | |||
| YW14-409 | 9,205 | 38 | 265 | 87 | 170 | 798 | 425 | 425 | 44 |
| YW37-409 | 9,789 | 90 | 427 | 83 | 150 | 682 | 256 | 341 | 19 |
| YW14-409R | 17,167 | 253 | 12 | 5 | ND | 67 | 17 | 16 | ND |
ND = not determined.
Number of colonies analyzed.
Frequencies of DSB-induced products with indicated phenotypes. See Figure 2 for chromosomal corresponding to each colony phenotype.
GC = gene conversion.
May arise by unequal sister chromatid exchange or sister exchange of nascent strands during DNA replication (Lovett et al. 1993).
DSB-induced recombination frequencies
| Strain | na | 104 × Frequencyb | |||||||
| Full GCc | Half GC | Percent Sectored | Deletion ± GC | Deletion + Half GC | Deletion/Triplicationd | ||||
| Ura+ Leu+ | Ura+/− Leu+/+ | Ura+ Leu− | Ura+/− Leu−/− | Ura+/− Leu−/+ | Ura+/− Leu+/− | Ura+/+ Leu+/− | |||
| YW14-409 | 9,205 | 38 | 265 | 87 | 170 | 798 | 425 | 425 | 44 |
| YW37-409 | 9,789 | 90 | 427 | 83 | 150 | 682 | 256 | 341 | 19 |
| YW14-409R | 17,167 | 253 | 12 | 5 | ND | 67 | 17 | 16 | ND |
| Strain | na | 104 × Frequencyb | |||||||
| Full GCc | Half GC | Percent Sectored | Deletion ± GC | Deletion + Half GC | Deletion/Triplicationd | ||||
| Ura+ Leu+ | Ura+/− Leu+/+ | Ura+ Leu− | Ura+/− Leu−/− | Ura+/− Leu−/+ | Ura+/− Leu+/− | Ura+/+ Leu+/− | |||
| YW14-409 | 9,205 | 38 | 265 | 87 | 170 | 798 | 425 | 425 | 44 |
| YW37-409 | 9,789 | 90 | 427 | 83 | 150 | 682 | 256 | 341 | 19 |
| YW14-409R | 17,167 | 253 | 12 | 5 | ND | 67 | 17 | 16 | ND |
ND = not determined.
Number of colonies analyzed.
Frequencies of DSB-induced products with indicated phenotypes. See Figure 2 for chromosomal corresponding to each colony phenotype.
GC = gene conversion.
May arise by unequal sister chromatid exchange or sister exchange of nascent strands during DNA replication (Lovett et al. 1993).
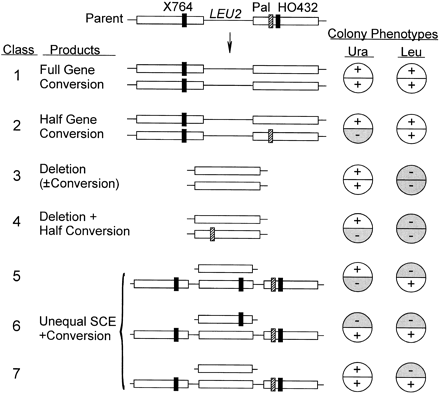
Sectored and nonsectored colony genotypes and phenotypes. The parental substrate is shown at the top as in Figure 1. Below are predicted products of reciprocal (or SSA) and nonreciprocal events shown as two segregation products following the first post-recombination division. Half-colony phenotypes corresponding to the URA3 and LEU2 genotypes for each segregation product are shown on the right. HO432 is assumed to be converted in at least one allele in all cases, whereas Pal may undergo full or half conversion. Only full gene conversions and simple deletions produce nonsectored colonies; all others involve sectoring of either URA3 or LEU2 or both. Half-gene conversions of Pal produce sectored Ura+/− colonies in which both halves are either Leu+ (no deletion) or Leu− (deletion). Unequal exchanges produce one apparent deletion product and a triplication, and can produce sectoring of URA3, LEU2, or both. Full or half gene conversion events can occur in G1 cells through intrachromosomal interactions, or in G2 cells through either intrachromosomal or sister chromatid interactions; the system does not distinguish intrachromosomal and sister chromatid conversions.
(G2 events), as Leu+ and Leu− halves had ura3 triplications and single copies of ura3, respectively (data not shown). Thus, most Leu+/− products probably reflect sister chromatid interactions, but this limited analysis may have missed products arising via deletion in only one sister chromatid in G2. This triplication frequency is considerably higher than seen in a study of a limited number of products with a related cross (Nickoloff et al. 1989). However in that study, recombinants were identified following selection. Of the three triplication classes shown in Figure 2, only classes 6 and 7 would be scored as unequal exchange if only Ura+ products were recovered due to selection, reducing this fraction to ~20% of the total. The Ura−/− Leu−/− deletion class (expected to be common, see ref. Nickoloff et al. 1989) and the fourth unequal exchange class (Ura−/− Leu+/−) were not scored. In the present study deletions and unequal exchange products were not characterized extensively because of ambiguities associated with their formation (e.g., deletions may result from reciprocal exchange or SSA). Instead, we focused on the more mechanistically constrained conversions unassociated with deletions (Leu+/+; Figure 2, classes 1 and 2) because our primary goal was to investigate the mechanism(s) of DSB-induced gene conversion. Similar product distributions were found for strains YW14-409 and YW37-409 (Table 1), indicating that the 37-base loop is also poorly recognized by the mismatch repair machinery and that loop length does not affect recombination mechanism(s).
Most DSB-induced mitotic gene conversion is mediated by hDNA repair: Among YW14-409 and YW37-409 products that remained Leu+, ~87% were sectored Ura+/−. To confirm that these arose via segregation of a palindromic mismatch, and not from conversion of only one sister chromatid in G2, we analyzed Ura− sectors from strain YW14-409. If such sectors arose from conversion of only one sister chromatid in G2 cells, they would retain the HO site and would recombine at high frequency upon growth in medium with galactose. Patch recombination assays (see materials and methods) on 17 Ura− sectors from Ura+/− Leu+/+ products showed that 4 retained an HO site. Therefore, the majority of the Ura+/− Leu+/+ sectors reflect the segregation of the palindromic frameshift mutation in hDNA. Because a significant fraction of events occur in G2 (discussed above), these data suggest that the majority G2 events involve cleavage of HO sites in both sister chromatids; products with quarter-sectors were apparent on LB-ura medium, but these were ~10-fold less frequent than half-sectors, further supporting this idea. Physical analysis of 22 Ura+/− Leu+/+ sectored products from the related strain YW14-409R (see below) indicated that none resulted from conversion of only one sister chromatid in G2 cells, further supporting the idea that the most DSB-induced mitotic gene conversion occurs via hDNA repair. Thus, ~90% of Ura+/− Leu+/+ colonies (35 of 39 from these two strains) resulted from palindrome segregation, giving a segregation rate of ~75%. The ~25% nonsectored colonies might have arisen by gap repair, residual mismatch repair and/or loop segregation prior to plating (see discussion). By ruling out gap repair for most DSB-induced gene conversions in our system, this system can be used to address specific questions about mismatch repair in recombination intermediates.
Donor alleles are unchanged whether or not mismatches are repaired: Studies have demonstrated that spontaneous and DSB-induced gene conversion is completely nonreciprocal, i.e., donor loci are not altered (Nickoloff et al. 1989, J. W. Cho and J. A. Nickoloff, unpublished results). We were interested in whether donor loci are similarly unaltered when HO site conversions are associated with at least one segregation event. To address this question, we constructed strain YW14-409R, which is identical to YW14-409 except that the downstream ura3 allele carries eight phenotypically silent RFLP markers. These additional markers are well repaired (J. W. Cho and J. A. Nickoloff, unpublished results).
Markers were scored in both ura3 alleles from Ura+ and Ura− sectors in 22 gene conversion products. Since each sector receives one DNA strand from each half of a recombination intermediate, this analysis provided information about all four participating strands of these mitotic events (two strands each from donor and recipient alleles). The unbroken X764 allele retained its parental configuration in all 22 products. Complete nonreciprocality was also seen in nonsectored intrachromosomal gene conversion products in which 9 RFLP markers were scored (J. W. Cho and J. A. Nickoloff, unpublished results). Thus, donor alleles remain unchanged whether or not all markers in hDNA are repaired.
Additional markers reduce loop segregation: The additional markers in YW14-409R (relative to YW14-409) reduced Ura+/− sectoring from 87% to 5% among Leu+ products (Table 1). Although different HO induction times used for strains with and without silent markers preclude direct comparison of sector rates in the two types of strains, limited analysis of YW14-409R sector rates with the shorter (3 hr) induction time indicated that the silent markers reduce Ura+/− sectoring by more than 10-fold (data not shown). Increased loop repair (reduced sectoring) likely reflects the corepair of the loop and nearby well-repaired silent markers, a well-known phenomenon in yeast (reviewed in Petes et al. 1991). Such corepair can also explain the large decrease in the two measured unequal exchange classes (Ura+/− Leu−/+ and Ura+/− Leu+/−) in YW14-409R compared with YW14-409 and YW37-409, as corepair would shift these to the nonsectored Ura+/+ Leu+/− class.
Mismatches adjacent to an unrepaired palindromic loop mismatch are repaired efficiently: The sectored products of strain YW14-409R can be used to examine mismatch repair of markers near an unrepaired palindromic loop mismatch. As mentioned above, the X764 donor allele was unchanged in both Ura+ and Ura− sectors in all 22 products and therefore remained Ura−. Conversion and segregation patterns in the recipient alleles (which suffered DSBs) are summarized in Figure 3. As expected, recipient alleles of Ura+ sectors lost both frameshift mutations (the HO site and the palindrome). The HO site was also absent in Ura− sectors in all 22 recipient alleles. Thus, the HO site was completely converted in all 22 products. We also expected that the palindrome would be present in recipient alleles in Ura− sectors because sectoring was likely to result from loop segregation, and this was true for 21 of 22 products. In the single exception, both the loop and HO site converted, but hDNA extended to X764, which escaped repair (Figure 3, type 9).
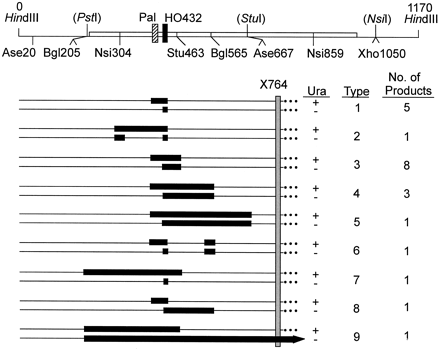
Conversion/segregation patterns among sectored Ura+/− Leu+/+ (half-gene conversion) products of YW14-409R. (Top) Map of the right ura3 allele. Wild-type restriction sites are shown above the line, silent RFLPs below. Sites in parentheses are absent. (Bottom) Structures of conversion products in the two sectors are shown by pairs of lines with converted markers indicated by black bars. For example, in the type 1 products, HO432 was fully converted, Pal half-converted (yielding the Ura+ and Ura− products as indicated), and none of the silent RFLPs converted. In the type 9 product, the lower allele also half-converted Nsi859 and Xho1050 (not shown). The relative position of the X764 frameshift in the donor allele is indicated by the shaded bar; full conversions of X764 are not detected as these retain the parental Ura−/− phenotype.
Among these products, hybrid DNA was usually more extensive than the minimum required to produce a sectored colony. This minimum is represented by the five examples of product type 1, in which only the region between the HO site and the palindrome insertion may have been included in hybrid DNA. The silent RFLPs are efficiently repaired in standard conversion assays (J. W. Cho and J. A. Nickoloff, unpublished results). Among the 17 products known to have arisen from intermediates with silent RFLPs in hDNA (types 2–9), 14 showed no evidence of silent marker segregation (types 2–6), indicating that these markers are efficiently repaired even when the nearby loop escapes repair. These 17 products had at least 32 silent markers in hDNA, 22 of which were repaired. This 69% repair efficiency is likely an underestimate as repair that restores a marker is not detected. Short tract repair was evident in two products. The type 6 product had a discontinuity due to restoration of Stu463 and conversion of Bgl565. The discontinuity in the Ura− sector of the type 2 product resulted from segregation of the loop rather than repair. However, the conversion of the Nsi304 site in this product adjacent to the unrepaired loop is further evidence for short tract repair.
Of the three products in which silent markers escaped repair (types 7–9), two showed a complete lack of mismatch repair. Note that conversion of the HO site does not involve mismatch repair; instead, the nonhomologous ends produce unpaired single-stranded tails upon invasion, and these are thought to be removed by Rad1/10p endonuclease (Fishman-Lobell and Haber 1992). Thus, the HO site, unlike other markers in homology, is converted by a mechanism different from either gap or hDNA repair.
Differential involvement of markers in hDNA on opposite sides of the DSB: Previously, we performed plasmid × chromosome and intrachromosomal crosses with identical ura3 alleles as in YW14-409R except they lacked the palindrome. Among Ura+ products from these crosses, markers 5′ of the DSB converted significantly more often than equidistant 3′ markers (Sweetser et al. 1994, J. W. Cho and J. A. Nickoloff, unpublished results). We demonstrated that this asymmetry reflected a selection bias against Ura− products that arise when tracts extending 3′ of the DSB reach the X764 frameshift mutation (Weng et al. 1996). The sectored colonies described here were selected on the basis of segregation of palindromic insertions, and 21 of 22 were of this type. Thus, selection pressure was imposed for hDNA forming 5′ from the DSB. It is therefore striking that among sectored products, the Nsi304 marker 5′ of the DSB converted at less than half the rate of the equidistant 3′ marker, Bgl565 (Figure 4). Although a formal possibility, it is unlikely that these reduced values reflect increased restoration repair for these markers among the sectored YW14-409R products. Another possibility is that hDNA formation is blocked by the palindrome, and in this case, markers 5′ of the palindrome would be expected to exist in hDNA at reduced levels whether the loop mismatch was repaired or not. To test this idea, we generated a tract spectrum from 17 YW14-409R products that converted the palindrome (nonsectored Ura+/+ Leu+/+ products). As shown in Figure 4, conversion rates of 5′ markers were higher than 3′ markers among nonsectored products. The tract spectrum of nonsectored YW14-409R products is essentially the same as conversion spectra obtained with these same alleles, but lacking the palindrome, in both plasmid × chromosome and intrachromosomal crosses (Sweetser et al. 1994, J. W. Cho and J. A. Nickoloff, unpublished results). As this and prior studies indicate that most DSB-induced conversion is mediated by hDNA repair, the tract spectrum of the nonsectored products would indicate that the palindrome does not block hDNA formation. Instead, these results suggest that the 5′ markers occur in hDNA less often in intermediates that produce sectored products. In contrast, the 3′ markers either converted at similar or slightly higher rates in sectored recombinants compared with nonsectored recombinants (Figure 4). We argue below that these results indicate that conversion on opposite sides of a DSB involves distinct mismatch repair processing events.
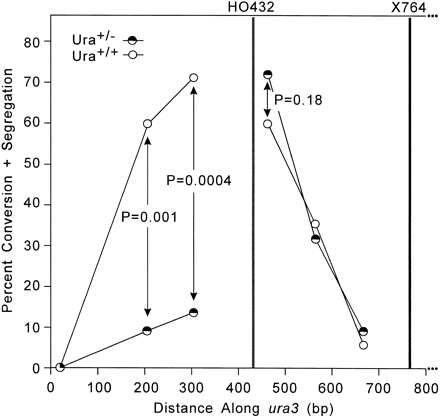
Percent conversion + segregation of silent RFLP markers among sectored and nonsectored products of YW14-409R. Marker positions in ura3 are plotted on the x-axis. Data for the sectored products are from the 22 tract structures shown in Figure 3 (half-filled circles). Data for the nonsectored products (open circles) are from 17 tract structures (not shown). P values calculated by using the Fisher exact test.
DISCUSSION
Meiotic conversion occurs at relatively high frequencies (a few percent or more), which allows recombinants to be identified without selection. This facilitates the analysis of all four strands involved in a single meiotic recombination event. In contrast, spontaneous conversion in mitotic cells occurs about 1000-fold less often than in meiotic cells, and selection strategies are typically required to identify events that produce functional alleles. Selection can conceal certain types of recombinant events. For example, selection for Ura+ products can make some unequal sister chromatid exchange events appear as deletions. By using HO nuclease to stimulate mitotic recombination at high frequencies, selection can be avoided, and information can be gained for all four strands participating in a single mitotic event. In meiosis, there are eight strands that can interact, but only rarely are more than four involved. The haploid mitotic system described here shows some similarity to the meiotic situation, but with different topological features. In G1 cells there is only one copy of each chromosome and only two copies of the alleles under study; thus interactions involve both available alleles. In G2 cells, most interactions are still likely to involve only two of the four copies, similar to meiosis. However, in meiotic cells all alleles are unlinked whereas in haploid mitotic cells, interacting partners are always linked in G1 cells (intrachromosomal events), while in G2 cells they may be linked or unlinked (sister chromatids). In mitosis, interactions typically occur between sisters rather than homologs (Kadyk and Hartwell 1992). Our data suggest an apparent rate of 40% for interactions between sisters in mitosis. However, Esposito (1978) described how events initiated in G1 could give rise to products that would appear to have occurred in G2. If the events we observe are truly G2 events, the 40% value is a minimum estimate of sister interactions as we cannot distinguish nonreciprocal interactions between alleles on sister chromatids from intrachromosomal interactions.
Most DSB-induced mitotic gene conversion is mediated by hDNA mismatch repair: DSB-induced conversion during meiosis and MAT switching involves hDNA repair and our study confirms this for general mitotic DSB-induced conversion. Broken alleles are preferentially converted and conversion tracts are usually continuous (reviewed in ref. Nickoloff and Hoekstra 1997), and these features can be explained by an end-directed, excision-based mismatch repair mechanism analogous to that mediated by MutHLS in E. coli (Modrich 1991), as proposed for both MAT and meiotic conversions in yeast (Detloff et al. 1992; Haber et al. 1993). It is not known whether spontaneous mitotic recombination results from DSBs, other types of endogenous damage, or normal DNA dynamics. Similar gene conversion:crossover ratios were found for spontaneous and DSB-induced events in two studies (Nickoloff et al. 1986; Ray et al. 1988). These results are consistent with the idea, but do not prove, that spontaneous events are initiated by DSBs.
In the absence of other markers, we observed about 25% conversion of the palindrome. Despite our efforts to prevent cell division of recombinants prior to plating, it is possible that some of the apparent palindrome conversions seen here were in fact segregation events. However, we believe that this is not the case because similar conversion rates are seen in meiosis with palindromes, and with point mutations in mismatch repair-deficient pms, msh, or mlh mutants (Crouse 1997), and meiotic analyses are not susceptible to this type of plating artifact. Loop conversion may reflect low-level mismatch repair, either by the MLH/MSH pathway or by an unknown pathway. Msh2p-Msh6p binds to 12 and 14 base palindromic mismatches (Alani 1996). Genetic evidence is also consistent with Msh binding to palindromic mismatches in vivo (Manivasakam et al. 1996; Moore et al. 1988; Nag et al. 1989). It has been suggested that palindromic stem-loop structures may be recognized at both the unpaired bases in the loop and at the base of the stem (Alani et al. 1995). Msh2p-Msh6p binding to single-base mismatches is reduced by ATP, but this effect is not seen for palindromic mismatches. This differential response to ATP might explain why palindromic loop mismatches are bound by Msh proteins, but not well repaired (Alani 1996). Residual loop repair might reflect an incomplete response of Msh2p-Msh6p to ATP in vivo. In addition, within the recently sequenced yeast genome are two previously unknown genes with homology to MLH and MSH genes (Crouse 1997). These, or other unknown genes, may be responsible for residual repair of palindromic loops. DSBs within HO site insertions produce unpaired single-stranded tails upon invasion of a homologous sequence, and Rad1/10p is thought to cleave at homology/heterology borders (Fishman-Lobell and Haber 1992). It is possible that Rad1/10p also cleaves at palindromic loop mismatches and thus contributes to palindrome conversion. Kirkpatrick and Petes (1997) recently showed that Rad1p is involved in repair of nonpalindromic loop mismatches and it will be of interest to learn if Rad1p also cleaves near palindromic loop mismatches.
Mismatch repair in the vicinity of an unrepaired loop mismatch: The analysis of tract structures in sectored products showed that nearly 80% had hDNA extending across one or more silent markers, and more than 70% had hDNA on both sides of the DSB. These are minimum values because restoration repair effectively hides hDNA. Studies are in progress in mismatch repair mutants to more accurately identify the extent of hDNA. The amount of “hidden” hDNA may be significant because the majority of silent markers were repaired, despite their proximity to an unrepaired loop mismatch. We did observe segregation of silent markers at a (maximum) rate of 31%, which is about 10-fold higher (P = 0.001) than that seen in gene conversion products of a related intrachromosomal cross lacking the palindrome (J. W. Cho and J. A. Nickoloff, unpublished results).
A marker might escape repair due to a total or partial lack of repair. Partial repair could reflect the termination of a long repair tract prior to reaching a marker, or short tract repair acting individually on some, but not all markers. Most conversion tracts are continuous (Petes et al. 1991), consistent with excision-based, long tract repair, as in E. coli, a view that is strengthened by the identification of yeast homologs of E. coli MutL and MutS (Crouse 1997). E. coli MutH induces nicks in hemi-methylated DNA to target a strand for repair. Yeast DNA is not methylated, but for DSB-induced conversions, the DNA ends could serve the same purpose as MutH-induced nicks (Detloff et al. 1992; Haber et al. 1993). We found three of 22 products in which silent markers escaped repair. Two showed no evidence of repair (types 7 and 8; Figure 3) and a third showed complete repair on one side of the DSB and no repair on the other (type 9). Although it is possible that the type 9 product resulted from termination of a single mismatch repair tract, we believe that it reflects independent repair processing on opposite sides of the DSB. Further evidence for this idea is discussed below.
Evidence for rare, short mismatch repair tracts: Discontinuous tracts, which arise at low frequencies in both mitotic and meiotic cells, can result from either short tract repair or partial repair. Although short tract repair systems are known in bacteria (Lieb 1991), S. pombe (Schar and Kohli 1993), and higher eukaryotes (Gallinari et al. 1997; Neddermann and Jiricny 1994), previous evidence for short tract repair in yeast has been indirect, limited to observations of discontinuous conversion tracts. In meiosis, short tract and partial repair can be distinguished since all strands can be followed. However, this distinction cannot be made in mitotic studies that employ selection to identify recombinants. Using a nonselective assay, we found two products with discontinuous tracts (Figure 3, types 2 and 6), both of which resulted from repair. In the type 2 product, repair of the Nsi304 marker did not include flanking Bgl205 and loop markers, each only 100 bp away. In the type 6 product adjacent markers were repaired in opposite directions. These products provide direct evidence of short tract repair processing in S. cerevisiae, albeit at a low level.
Independent mismatch repair processing on opposites sides of a DSB: Despite selecting for events that extended 5′ of the DSB (toward the palindrome), silent markers 5′ of the DSB were converted (or segregated) less often than equidistant markers 3′ of the DSB. These results are opposite of previous results of related plasmid × chromosome and intrachromosomal crosses that lacked the palindrome (Sweetser et al. 1994; J. W. Cho and J. A. Nickoloff, unpublished results). This conversion bias toward 5′ markers reflects selection against longer 3′ tracts, as such tracts transfer the donor X764 frameshift mutation to the recipient allele (Weng et al. 1996). It is unlikely that a relatively short palindrome could block branch migration in view of E. coli data showing that heterologies greater than 1 kb are incorporated into hDNA in vitro (Bianchi and Radding 1983) and in vivo (Holbeck and Smith 1992; Lichten and Fox 1984). Although multiple mismatches were found to inhibit pairing (Negritto et al. 1997; Worth et al. 1994), these studies involved 4- to 20-fold more markers than the current study. We found that among products that converted the palindrome, 5′ marker conversion rates were even higher (though not significantly; P = 0.07) than rates seen in a cross lacking the palindrome (J. W. Cho and J. A. Nickoloff, unpublished results), thus ruling out the possibility that the palindrome prevents hDNA formation at these 5′ markers. We cannot rule out the possibility that there is increased restoration repair of 5′ markers among products that segregated the palindrome. However, there is no obvious mechanism that would produce such an effect. The simplest explanation for the observed differential marker conversion derives from the observation that poorly repaired mismatches are repaired efficiently when near well-repaired mismatches (Petes et al. 1991), reflecting corepair. Such corepair indicates that poorly repaired mismatches are not refractory to repair per se, but that they are unable to initiate repair. For markers 5′ of the DSB (on the same side as the palindrome), the reduced conversion rates in sectored products, and the normal rates in nonsectored products can be explained in terms of corepair. In this view, if hDNA on the 5′ side of the DSB is limited and includes only the palindrome, this marker is likely to escape repair and produce a sectored colony. If hDNA is more extensive, other 5′ markers will be included in hDNA, and these will stimulate their own repair and lead to corepair of the palindrome, producing a nonsectored product. In contrast, conversion rates for markers 3′ of the DSB were similar among sectored and nonsectored products, indicating that the presence of these markers in hDNA did not stimulate corepair of the palindrome. These results therefore suggest that conversion on opposite sides of the DSB involves independent mismatch repair tracts, and rule out models involving a single mismatch repair tract.
As discussed above, our data and those of others are inconsistent with DSBs stimulating conversion in a double-strand gap. Sun et al. (1991) proposed a modified DSB repair model in which only 5′ ends are degraded, producing long 3′ extensions. In this model, both ends invade an unbroken, homologous duplex (donor allele) and prime repair synthesis (Figure 5, A–C). Resolution of the two HJs gives two products that each have hDNA (Figure 5D). However, Gilbertson and Stahl (1996) showed that hDNA rarely forms in unbroken alleles, prompting the model shown in Figure 5, E–F in which only one HJ is resolved enzymatically. The resolution of one HJ produces an intermediate with three single-strand nicks (Figure 5E; filled triangles). Branch migration shown by the rightward arrow resolves the second junction when it reaches two of the nicks, yielding two duplexes, only one of which has hDNA. The nicks adjacent to hDNA (open triangles) are potential entry points for end-directed mismatch repair (Figure 5F), as suggested for meiotic conversion and MAT conversion (Detloff et al. 1992; Haber et al. 1993). Our data are consistent with hDNA forming only in broken alleles, as are data indicating that broken alleles rarely donate information to unbroken alleles (reviewed in Nickoloff and Hoekstra 1997).
Although casual inspection of the recombination intermediate in Figure 5F might suggest the possibility of independent mismatch repair processing on either side of the DSB, this structure is better viewed as having a single region of hDNA; it is therefore likely that mismatch repair initiated on either side of the DSB would process the entire hDNA region in a single repair tract, which is not consistent with our data. Furthermore, because donor information occurs on complementary strands on opposite sides of the DSB, long repair tracts (as seen by Detloff and Petes 1992) would produce unidirectional tracts. Although such tracts were predominant in DSB-induced plasmid × chromosome
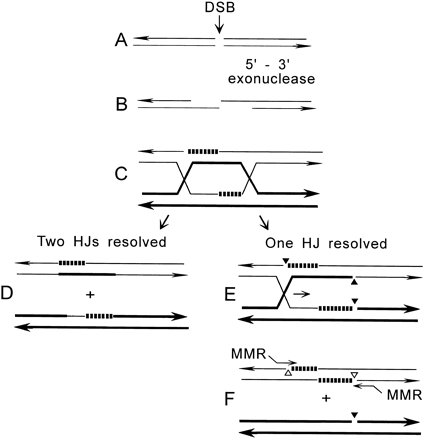
Two versions of the DSB repair model. An unbroken homolog is shown by thick lines, repair synthesis by dashed lines, single-strand nicks by triangles, and end-directed mismatch repair of hDNA by “MMR” initiating at open triangles. (Left) The original DSB repair model (Szostak et al. 1983) as modified by Sun et al. (1991), in which both HJs are resolved. (Right) A model proposed by Gilbertson and Stahl (1996) to account for the lack of hDNA in unbroken alleles. In both models, mismatch repair occurs after HJs are resolved. HJs may be resolved in two senses to yield crossover products or noncrossover products; here only the latter are shown. See text for further details.
conversion with a resolution level of 20–30 bp (Sweetser et al. 1994), these same alleles in an intrachromosomal cross yielded uni- and bidirectional tracts at similar frequencies (J. W. Cho and J. A. Nickoloff, unpublished results). Unidirectional tracts were also predominant in the meiotic study by Gilbertson and Stahl (1996), although here the level of resolution was lower (≥130 bp). Thus, the model shown in Figure 5, E–F does not fully explain the available evidence.
In Figure 6 are shown two versions of a DSB repair model in which two physically separated regions of hDNA are formed on either side of the DSB; consequently, these regions will be processed by independent mismatch repair tracts. The first (Figure 6, A–E) is a noncrossover version of a model proposed by Gilbertson and Stahl (see Figure 6 in Gilbertson and Stahl 1996), and is similar to that diagrammed in Figure 5, E–F, except that mismatch repair initiates at a nick produced by enzymatic resolution of one of the two HJs (Figure 6B). After this first hDNA region is repaired, branch migration resolves the remaining HJ (Figure 6C) producing an intermediate with a second hDNA region that is subject to an independent, nick-directed
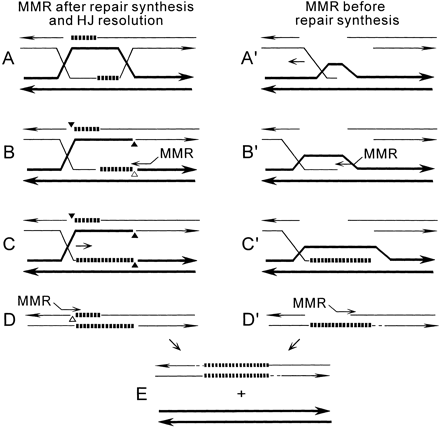
Independent mismatch repair processing on opposite sides of a DSB. Symbols as in Figure 5. Only noncrossover products are shown; see text for details.
mismatch repair tract (Figure 6D). Note that this model predicts that repair synthesis (Figure 6A) occurs before mismatch repair (Figure 6, B and D); however, the newly synthesized DNA is completely removed and then replaced during mismatch repair. The second model (Figure 6, A′–D′) is a single-end invasion model (Belmaaza and Chartrand 1994; Nelson et al. 1996), which is essentially the same as synthesis-dependent strand annealing (Nassif et al. 1994). This model suggests that hDNA formed upon initial strand invasion, and/or branch migration, signals mismatch repair (Figure 6B′) before repair synthesis (Figure 6C′). This produces a strand that can anneal to the non-invading strand, yielding a second hDNA region that is subject to a second mismatch repair event (Figure 6D′). As described for Figure 5, E–F, the models in Figure 6 predict that no hDNA will form in the unbroken allele.
The model shown in Figure 6, A′–D′ suggests that only one end invades a homolog; however, these same steps can be invoked in a symmetric fashion in a two-ended invasion model that also predicts independent mismatch repair tracts on opposite sides of the DSB, and yields the same products shown in Figure 6E (not shown). In these dual mismatch repair tract models, products are shown with bidirectional tracts. However, unidirectional tracts can arise if hDNA either does not form on one side of the DSB (i.e., no branch migration), or by appropriate mismatch repair processing. In a cross that lacked the palindrome, 42% of intrachromosomal conversion tracts were bidirectional (J. W. Cho and J. A. Nickoloff, unpublished results). In contrast, among products in which the palindrome segregated, there were significantly more bidirectional tracts (73%; P = 0.01), possibly reflecting more frequent two-ended invasions. If sectored products only appear when hDNA on the 5′ side of the DSB is limited and includes only the palindrome, as argued above, such intermediates may be stabilized by a second invasion on the 3′ side of the DSB. This idea is consistent with the significantly higher level of involvement of the 3′ Stu463 marker among sectored products (73%) than seen previously among conversion products from a cross lacking the palindrome (47%; P < 0.03). Conversion of Stu463 was slightly higher among sectored than nonsectored products of YW14-409R (Figure 4). If such effects are found to be general, it would suggest that successful recombination requires intermediates to be stabilized by a minimum length of hybrid DNA.
Acknowledgement
We thank Jennifer W. Cho and Doug Sweetser for technical assistance, and Richard Kolodner, Fred Winston, Gerry Smith, and Stephanie Ruby for helpful comments. This research was supported by grant CA55302 to J.A.N. from the National Cancer Institute, National Institutes of Health.
Footnotes
Communicating editor: S. Jinks-Robertson
LITERATURE CITED

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}