





Abstract
Plants regenerated from tissue culture often display somaclonal variation, that is, somatic and often meiotically heritable phenotypic variation that can result from both genetic and epigenetic modifications. To better understand the molecular basis of somaclonal variation, we have characterized four unique tissue culture-derived epialleles of the pericarp color1 (p1) gene of maize (Zea mays L.). The progenitor p1 allele, P1-wr, is composed of multiple head-to-tail tandemly arranged copies of the complete gene unit and specifies brick-red phlobaphene pigmentation in the cob glumes. The novel epialleles identified in progeny plants regenerated from tissue culture showed partial to complete loss of p1 function indicated by pink or colorless cob glumes. Loss of pigmentation was correlated with nearly complete loss of p1 steady-state transcripts. DNA gel-blot analysis and genomic bisulfite sequencing showed that silencing of the epialleles was associated with hypermethylation of a region in the second intron of P1-wr. Presence of Unstable factor for orange1 (Ufo1), an unlinked epigenetic modifier of p1, restored the cob glume pigmentation in the silenced alleles, and such reactivation was accompanied by hypomethylation of the p1 sequence. This observation confirmed that silencing of the epialleles is indeed due to epigenetic modifications and that the p1 epialleles were capable of functioning in the presence of the correct trans-acting factors. While the low-copy regions of the genome generally undergo hypomethylation during tissue culture, our study shows that the tandemly repeated genes are also prone to hypermethylation and epigenetic silencing.
SOMATICALLY and meiotically heritable mutations are common in plants regenerated from tissue culture and their progenies. Heritable variation can result from polyploidy, aneuploidy, chromosome rearrangements, changes in DNA sequence, and movement of transposable elements (Kaeppler et al. 2000).
Activity of transposable elements in tissue culture-derived progeny was first reported by Peschke et al. (1987), and transposon movement of specific element families has now been shown to be broadly important in tissue culture-induced variation (Hirochika and Otsuki 1995; Hirochika et al. 1996; Kikuchi et al. 2003; Huang et al. 2009). Consistent with activation of transposable elements are changes in their epigenetic status; reduced methylation has been reported to correspond to the active state of several transposons in tissue culture (Cheng et al. 2006; Ngezahayo et al. 2009). Although tissue culture-induced chromatin modifications have been shown to occur in many types of sequences, the extent to which these modifications affect the expression of nontransposon sequences remains unclear.
Global variation in DNA methylation in tissue culture regenerants and their progeny has been documented in species including carrot, rice, tobacco, sorghum, and barley. These studies have shown that significant alteration or resetting in the global methylation level of genomic DNA is associated with the age of the culture (Quemada et al. 1987; Müller et al. 1990), growth regulator type and concentration (LoSchiavo et al. 1989), osmotic shock (Kovařik et al. 1997), and even the tissue culture process per se (Shimron-Abarbanell and Breiman 1991; Devaux et al. 1993). Significant perturbance of DNA methylation in calli and regenerated plants was reported in sorghum in which, interestingly, hybrids were found to be resilient to such changes as compared to their parental inbred lines (Zhang et al. 2009). This body of evidence implicates physical, chemical, and genetic factors in global DNA methylation variation in vitro.
Several studies have described variation in methylation of specific DNA sequences resulting from maize tissue culture (Kaeppler and Phillips 1993; Phillips and Olhoft 1995). DNA gel-blot analysis was used to study methylation patterns using random genomic and coding DNA sequences as probes. In general, reduced DNA methylation was observed in the progeny of regenerated plants, although examples of increased DNA methylation were also reported. The observed DNA methylation changes were shown to be stably transmitted to progeny plants.
Although the literature supports a role for epigenetic modification in tissue culture-induced variation, no direct evidence of an endogenous tissue culture-induced epiallele resulting in an altered phenotype has been shown. Due to easily scorable phenotypes, the maize flavonoid biosynthetic pathway is well suited to address this question. pericarp color1 (p1) gene encodes an R2R3 Myb transcription factor and governs the biosynthesis of brick-red flavonoid pigments called phlobaphenes (Coe et al. 1988; Styles and Ceska 1989). Multiple alleles and epialleles of p1 are identified by accumulation of phlobaphenes in pericarp and cob glumes (Brink and Styles 1966; Chopra et al. 1998); for example, P1-wr specifies white pericarp and red cob glumes, while p1-ww has white pericarp and white cob glumes. p1 is particularly sensitive to epigenetic modifications, and several examples of altered expression patterns due to epigenetic suppression have been reported (Das and Messing 1994; Cocciolone et al. 2001; Sekhon et al. 2007; Robbins et al. 2008; Sekhon and Chopra 2009). Epigenetic silencing of the p1 alleles and epialleles can be relieved by the trans-acting factor Unstable factor for orange1 (Ufo1), resulting in gain of ectopic pigmentation (Chopra et al. 2003; Sekhon et al. 2007). Ufo1-induced phenotypes show poor meiotic and mitotic inheritance.
We studied the effect of tissue culture-induced modifications on P1-wr, a tandemly repeated p1 allele composed of at least six head-to-tail copies. We report that the P1-wr allele is sensitive to epigenetic silencing during the tissue culture process and that the silencing frequently leads to formation of epialleles that are stably inherited through sexual generations. Furthermore, we show that the silenced epialleles were reactivated in the presence of Ufo1, indicating that such epigenetic suppression can be reversed in the presence of appropriate modifiers.
MATERIALS AND METHODS
Plant materials:
The four novel p1 epialleles evaluated in this study were originally obtained from Catherine Mackey and Paul Jullstrom at Dekalb Plant Genetics (Groton, CT). Immature embryo explants of the genotype LH51 were grown on a modified MS media for ∼3.5 months, and regenerated plants were selfed to obtain the epialleles. Complementation analysis was performed using a p1-ww:4Co63 tester stock provided by Thomas Peterson (Iowa State University). Three independent mutants (p1-ww:DP1, p1-ww:DP6, and p1-ww:DP8) had no visible cob glume color, and one mutant (p1-ww:DP3) had pink cob glumes, indicating significantly reduced pigmentation. The noncultured wild-type P1-wr:LH51 allele was derived from sibling plants of those originally used to initiate tissue culture. The Ufo1 stock, originally identified and provided to us by Derek Styles (University of Victoria, Canada), was introgressed into inbred line 4Co63 (p1-ww c1 r-r) as described previously (Chopra et al. 2003; Sekhon et al. 2007). The 4Co63 inbred line itself does not affect p1 expression as shown by crosses of this line with a number of p1 alleles and epialleles (Sekhon et al. 2007; R. S. Sekhon and S. Chopra, unpublished data).
RNA isolation and reverse transcriptase–PCR analysis:
Cob glume tissues were isolated from 21 DAP (day after pollination) ears of mutant and wild-type plants. Tissues were immediately frozen and ground in liquid nitrogen. Total RNA extraction was conducted using Trizol reagent (Invitrogen, Carlsbad, CA) following the manufacturer's instructions. RNA was treated with DNase I (Applied Biosystems, Austin, TX). The first strand of cDNA was synthesized from 5 μg of RNA by adding 0.1 μg of oligo(dT) primer, 0.2 mm of dNTP mix, 5.0 mm of DTT, 2.5 mm of MgCl2, 1× AMV reaction buffer, 1.0 μl of RNasin, 30 units of AMV reverse transcriptase (RT), and nuclease-free water (all reagents from Promega, Madison, WI). First-strand cDNA was synthesized by incubating the mixture for 45 min at 40° followed by inactivation of AMV reverse transcriptase for 2 min at 94°. One-tenth (by volume) of the first strand of cDNA was used as template DNA for subsequent PCR amplification of two regions on cDNA sequence of P1-wr (GenBank accession U57002; Chopra et al. 1996). Primers PRTEP5-8 (5′-GGACGCACGCGCGACCAGCTGCTAACCGTG-3′) and PRTEP3-13 (5′-AAGGTAGTTGATCCACCGGAGCCGG-3′) targeted nucleotide (nt) positions 126–504 of P1-wr cDNA (coordinates based on GenBank accession EF165349; Sekhon et al. 2007) and yielded a 378-bp product. PRTEP5-8 (5′-GGACGCACGCGCGACCAGCTGCTAACCGTG-3′) and PRTEP3-12 (5′-GATCTGCCGGCTGAGGTGCGAGTTCCA-3′) gave a 543-bp product spanning nt positions 126–669 of cDNA. The coding region of Gpc1 (glyceraldehyde-3-phosphate dehydrogenase subunit C, GenBank accession X15596) of maize was amplified as the internal control using GAPCF (5′-CACTGCCACCCAGAAAACTGTTGACGGAC-3′) and GAPCR (5′-CTGTAGCCCCACTCGTTGTCGTACCAC-3′). The thermocycler program was 1 cycle of 30 sec at 94°, 40 cycles of 30 sec each at 94°, 1 cycle of 1 min at 60°, and 1 cycle of 2 min at 72°, followed by a final extension of 5 min at 72°. PCR products were visualized on agarose gels, and their identities were confirmed by sequencing.
Genomic DNA isolation and gel-blot analysis:
Leaf tissues harvested from the mutants and wild-type plant at the V7/V8 stage were immediately frozen and ground in liquid nitrogen. Genomic DNA isolation was performed using the CTAB extraction procedure (Saghai-Maroof et al. 1984). Ten micrograms of genomic DNA of the mutants and wild type was digested with 30 units of restriction enzymes following the manufacturer's instructions. All restriction enzymes were purchased from Promega, Fermentas (Glen Burnie, MD), or New England BioLabs (Ipswich, MA). Digested DNA fragments were separated by electrophoresis and transferred onto a Hybond-N+ membrane (Amersham Biosciences, Piscataway, NJ). Each membrane was prehybridized for 12 hr at 65° with 20 ml of hybridization solution containing 5× SSC, 10% Dextran sulfate, 1× Denhardt's (50× 1% Ficoll 400, 1% polyvinylpyrrolindone 360), 50 mm Tris (pH 8.0), 10 mm EDTA, 0.2% SDS, and 0.1 mg/ml of sheared salmon sperm DNA. DNA fragments used as probes were prepared from the cloned sequences of the P1-rr allele provided to us by Thomas Peterson and have been previously described (Lechelt et al. 1989; Grotewold et al. 1991). Radioactively labeled probes were prepared using oligolabeling described previously (Feinberg and Vogelstein 1983) and purified by ProbeQuant G-50 Micro Column (Amersham Biosciences, Piscataway, NJ). The purified probe DNA was denatured at 95° for 7 min and immediately added to 20 ml of preheated hybridization solution. After hybridization for at least 6 hr at 65°, membranes were washed twice in a 1× SSC and 0.1% SDS solution at room temperature for 30 min each, followed by one wash each in 0.2× SSC and 1.0% SDS and in 0.1× SSC and 1.0% SDS solutions at 65° for 15 min. Washed membranes were exposed to X-ray film (Kodak, Rochester, NY) at −80°.
Bisulfite genomic DNA sequencing:
The procedures are based mainly on Frommer et al. (1992) with minor modifications. Approximately 4 μg of genomic DNA was denatured by incubating with 3.0 m NaOH at 37° for 20 min. A total of 1.2 ml of freshly prepared sodium bisulfite solution was added directly to the denatured DNA, and the reactions were incubated in the dark for 4 hr at 55°. The reactions were desalted using a Wizard DNA purification system (Promega) and eluted in 1.0 mm of Tris (pH 8.0). Subsequently, the DNA was desulfonated with 3.0 m NaOH at 37° for up to 45 min followed by neutralization with 47 μl of 10 m ammonium acetate. Bisulfite-treated DNA was precipitated, dissolved in 1.0 mm of Tris (pH 8.0), and used for PCR amplification of regions of interest. PCR was conducted using the following conditions: 1 cycle of 30 sec at 94°, 36 cycles of 30 sec at 94°, 1 cycle of 1 min at 40°, and 1 cycle of 2 min at 72°, followed by a final extension of 5 min at 72°. The PCR products were subcloned using a pGEM-T Easy Vector (Promega), and 60 randomly selected independent subclones were subjected to the BigDye Terminator Cycle Sequencing (Applied Biosystems, Foster City, CA) according to the manufacturer's instructions using an M13 forward or reverse primer. For each genotype, five independent samples were subjected to bisulfite sequencing analysis.
To investigate the extent and pattern of DNA methylation, sequences were first aligned using Clustral X (Thompson et al. 1997), and, for each genotype, clones with similar methylation patterns and polymorphisms were grouped together. Graphical output to depict the methylation of each cytosine residue in each clone was generated through CyMATE (Hetzl et al. 2007). Overall methylation at each cytosine residue was calculated using MethTools (Grunau et al. 2000).
RESULTS
Identification of novel p1-ww alleles:
The four alleles analyzed in this study were identified in a group of eight regenerant-derived families with reduced cob color. Allelism analysis (data not shown) identified four independent tissue culture-derived p1-ww alleles, one c2 allele (Rhee et al. 2009), and three mutants that were not allelic to p1 or c2.
The P1-wr allele that has been previously reported is a complex locus that consists of multiple tandem copies of the complete gene unit (Chopra et al. 1998; Goettel and Messing 2009). The P1-wr:LH51 allele is consistent with this published allele on the basis of DNA gel-blot analysis (data not shown) and with sequences generated during the methylation analysis in this study. DNA gel-blot analysis using the DNA methylation-insensitive enzymes DpnI, EcoRV, HindIII, MboI, and XbaI revealed no restriction length polymorphisms between the tissue culture-induced mutants and wild type when probed with p1-specific probe fragments (data not shown). This observation indicated that, within the resolution of our analysis, insertions, deletions, or rearrangements are not likely to explain the mutant phenotypes. The suppressed phenotypes of the p1 mutant alleles show stable meiotic inheritance, and we did not note any spontaneous revertants (data not shown).
Since multiple P1-wr repeats are thought to be functional (Chopra et al. 1998, 2003; Robbins et al. 2008; Sekhon and Chopra 2009), common mutational mechanisms disrupting functionality of one of the repeats (e.g., mutagenesis with ethyl methanesulfonate) are unlikely to produce loss-of-function alleles. The unexpected frequent appearance of p1 mutant alleles (four of eight novel alleles analyzed for complementation) among this material indicates that (1) the P1-wr allele is hypersensitive to mutagenesis in culture, and (2) the mechanism of mutagenesis must affect multiple copies of the gene simultaneously.
Silenced mutants lack steady-state p1 transcripts:
To test whether loss of pigmentation in the mutant alleles is due to lack of p1 transcripts, we used RT–PCR on RNA isolated from cob glumes. Cob glumes were harvested at 21 DAP as P1-wr expression is developmentally regulated and peaks around this stage (Chopra et al. 1996). Two coding regions were targeted for amplification from three white cob mutants, one pink cob mutant, and the P1-wr:LH51 progenitor allele. As expected, products of desired sizes were amplified for all three regions in P1-wr:LH51 (Figure 1). Sequencing of the PCR products further confirmed that they belonged to the desired regions. However, no PCR product was observed in any of the mutant samples, indicating absence or undetectable levels of steady-state transcripts.
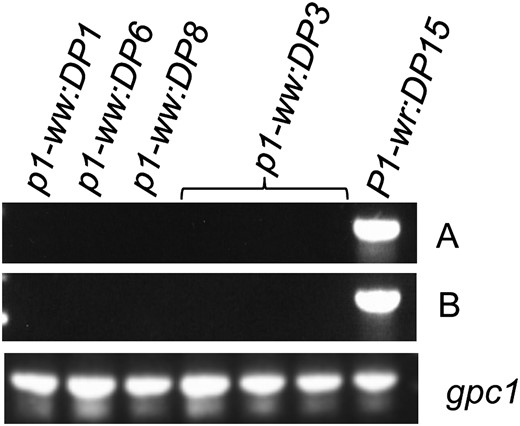
The silenced alleles lack steady-state p1 transcripts. RT–PCR was performed on total RNA extracted from cob glumes of mutant and wild-type genotypes at the 21-DAP stage. Two regions of P1-wr cDNA were amplified with p1-specific primer pairs: A, PRTEP5-8 + PRTEP3-13; B, PRTEP5-8 + PRTEP3-12 (see materials and methods for details). (Bottom) The same samples amplified with gpc1-specific primers as positive controls.
White and pink cob mutants are hypermethylated p1 epialleles:
Absence of any obvious sequence polymorphisms, and lack of steady-state transcripts in the mutant alleles led us to consider involvement of epigenetic mechanisms in P1-wr silencing. Indeed, previous reports have shown the propensity of p1 alleles toward epigenetic silencing (Das and Messing 1994; Chopra et al. 1998; Sekhon et al. 2007). To test this possibility, we used DNA gel-blot analysis to assay the methylation status of 46 HpaII (C*C*GG) and 3 SalI (GTC*GAC) restriction sites distributed across each copy of the P1-wr repeat complex. Hybridization of HpaII-digested blot with intron probe 8B produced a prominent band of ∼4.5 kb in the P1-wr:LH51 allele (Figure 2A), which is produced by restriction of sites at nt positions 600 and 4669 (Figure 2D). Absence of this band in the white cob mutants along with presence of a >10.0-kb band indicated complete methylation of the two HpaII sites. Interestingly, an intense >10.0-kb and a relatively weaker ∼4.5-kb band were present in the pink cob mutant, indicating partial methylation of the two HpaII sites in this genotype. Similarly, probe fragment 15 hybridized to a prominent band of ∼8.5 kb (Figure 2B), which was produced by restriction of HpaII sites at nt position 4669 of one P1-wr copy and at nt position 181 of the downstream copy (Figure 2D). Absence of this band and appearance of a >10.0-kb band in the white cob mutants indicated hypermethylation of the two HpaII sites. These two HpaII sites were partially hypermethylated in the pink cob mutant as indicated by presence of both an ∼8.5-kb and a >10.0-kb band. To assay the methylation of SalI sites, leaf genomic DNA was digested simultaneously with StuI, PacI, and SalI (Figure 2C). Upon hybridizing this blot with fragment 8B, a prominent 3.46-kb band was observed in P1-wr:LH51, indicating that SalI in the second intron (nt position 4034) is not methylated (Figure 2D). However, detection of an intact StuI–PacI fragment of 7.64 kb and the absence of the 3.46-kb band in the white cob and leaky mutants indicated hypermethylation of both SalI sites in the coding region (Figure 2D).
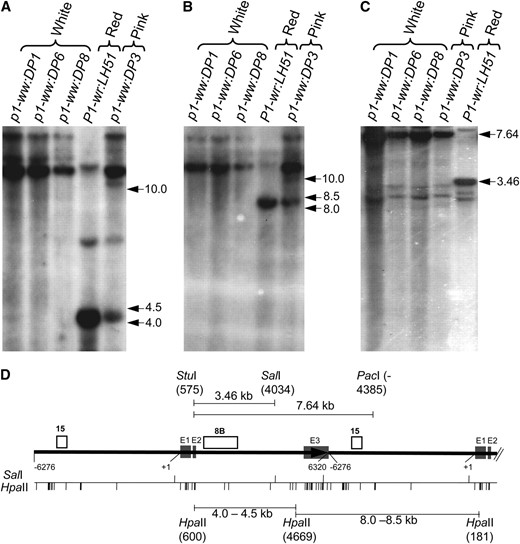
DNA gel-blot analysis showing that the silenced mutants are hypermethylated. Leaf genomic DNA of three white cob mutants, a pink cob mutant, and the red cob (wild-type) progenitor was used for the methylation assay. (A and B) Gel blot containing HpaII-digested DNA was hybridized with probe fragments 8B (A) and 15 (B). (C) DNA was simultaneously digested with a mixture of methylation-insensitive (StuI and PacI) and methylation-sensitive (SalI) enzymes, and the resulting blot was hybridized with probe 8B. Size (in kilobase pairs) and location of important hybridizing bands is to the right of each autoradiogram. (D) Gene structure and restriction map of P1-wr. Gene structure is based on Chopra et al. (1998), and the coordinates are consistent with GenBank accession EF165349 (Sekhon et al. 2007). The solid black arrow represents a complete P1-wr copy while the black solid line following the arrow represents a partial downstream copy. Shaded boxes (marked E1, E2, and E3) represent exons. Probes are shown above the gene structure diagram, and the coordinates are shown below. Also shown below the gene structure diagram are the positions of the HpaII and SalI sites. Some important restriction fragments, along with the location and coordinates of restriction enzyme sites yielding these fragments, are shown above and below the gene structure diagram.
These data together with analysis of other sites with additional probes and restriction digests (data not shown) allowed us to construct the methylation map presented in Figure 3. From the map, it is evident that methylation of a SalI site (nt position 4034) and one or both of the contiguous HpaII sites (nt positions 4069 and 4673) in the second intron was directly correlated with cob pigmentation. Thus, the silencing of the mutant alleles was associated with hypermethylation of the 3′ region of the second P1-wr intron. Furthermore, pink cob pigmentation of the p1-ww:DP3 mutant was correlated with partial methylation of certain cytosine residues, indicating that such mosaic methylation patterns can produce intermediate gene expression levels. Overall, these data showed that the white and pink cob mutants are hypermethylated P1-wr epialleles.
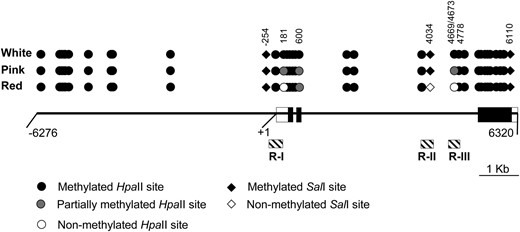
Methylation map of the HpaII and SalI sites in white cob, pink cob, and wild-type genotypes based on DNA gel-blot analysis. Solid line represents a single P1-wr copy. Boxes embedded in the solid line represent three exons; open boxes indicate UTRs. Coordinates are below the solid line; “+1” indicates the transcription start site. Hatched boxes indicate the position of three regions (R-I, R-II, and R-III) selected for bisulfite sequence analysis. Methylation status of HpaII (circles) and SalI (diamonds) sites in white cob (p1-ww:DP6) and leaky (p1-ww:DP3) mutants and their wild-type progenitor (P1-wr:LH51) is shown. Coordinates of selected sites mentioned in the text are indicated; two adjacent sites at positions 4669 and 4673 are shown as one circle.
Silencing of the p1 epialleles is accompanied by increased CG and CNG methylation of 3′ region of the second intron:
To precisely compare methylation of individual cytosine residues in white (p1-ww:DP6) and pink (p1-ww:DP3) cob epialleles relative to the progenitor P1-wr:LH51 allele, we performed genomic bisulfite sequencing on three P1-wr regions (see Figure 3 and supporting information, Table S1, for more details). The first region (R-I, 326 bp), covering 169 bp of core promoter and 157 bp of 5′ UTR, was selected because (1) this region is CG (cytosine, guanine) rich; (2) promoters are prone to methylation-induced epigenetic silencing; and (3) methylation data were unavailable for this region due to the absence of suitable restriction enzyme sites. The other two regions targeted the SalI and HpaII sites in the second intron detected by DNA gel-blot analysis. The second region (R-II, 250 bp) flanked the SalI site (nt position 4034) while the third region (R-III, 294 bp) covered the three HpaII sites, including two adjacent sites (nt position 4069 and 4073) showing hypermethylation in DNA gel-blot analysis.
Core promoter region (R-I):
In this region, methylation in CG, CNG (N = A, T, G, or C), and CHH (H = A, C, or T) contexts was limited to ∼80 bp on the 5′ end of the core promoter for all three genotypes (Figure 4A). The genotypes had very similar methylation profiles although, interestingly, the white cob (p1-ww:DP6) epiallele had slightly lower CG methylation as compared to the wild-type and pink cob genotypes. The rest of the core promoter and the 5′ UTR were devoid of methylation in all three genotypes. The overall average of methylation at 34 CG, 20 CNG, and 41 CHH sites also showed that this region was hypomethylated in all three genotypes (Table 1).
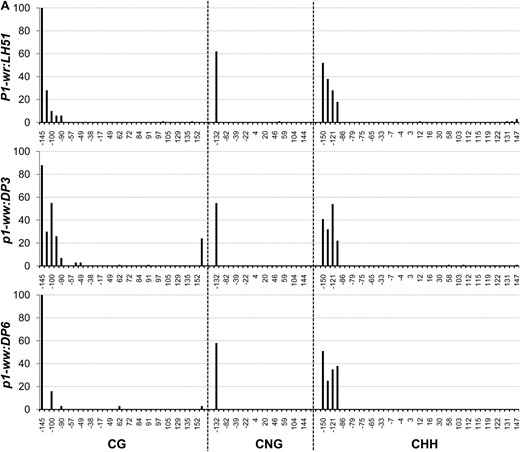
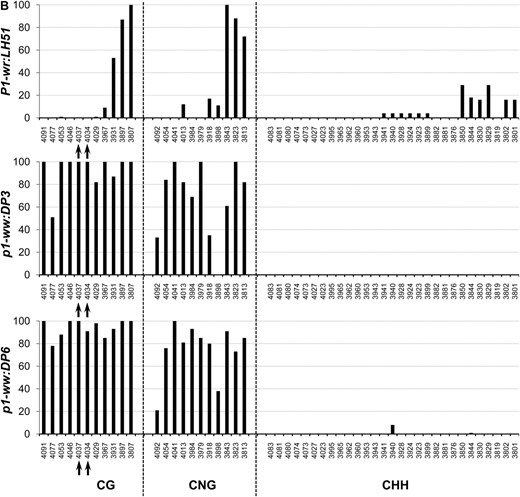
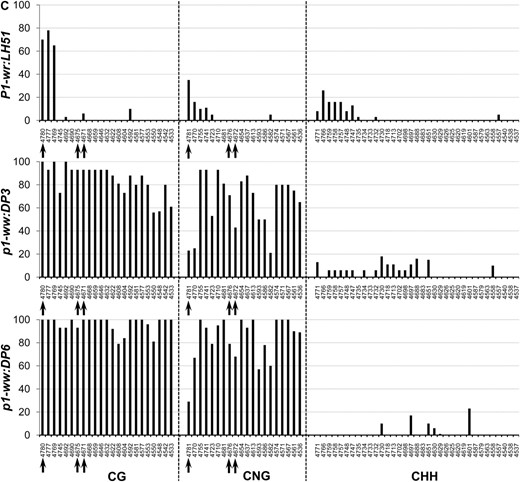
Genomic bisulfite data showing the methylation profile of three P1-wr regions in wild-type (P1-wr:LH51), pink cob (p1-ww:DP3), and white cob (p1-ww:DP6) genotypes. The regions include (A) a 326-bp sense-strand spanning promoter and 5′ UTR (R-I), (B) a 294-bp antisense strand flanking the SalI site in the second intron (R-II), and (C) a 250-bp antisense sequence covering three HpaII sites in the second intron (R-III) (see Figure 3 and Table S1 for more details). For each region, the percentage methylation of individual sites in CG, CNG (N is any nucleotide), and CHH (H is A, C, or T) contexts is shown on the y-axis while the coordinates of the sites are shown on the x-axis. The CGG sites were counted as CG sites and thus are not included in the CNG category. The percentage methylation was calculated by dividing the methylated clones for a cytosine residue by the total number of clones. Positions of the SalI (GTC*GAC) and HpaII (C*C*GG) sites mentioned in the text are indicated by arrows in B and C, respectively.
Methylation in three P1-wr regions in wild type and the leaky and white cob epialleles
R-I | R-II | R-III | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Genotype | CG (34) | CNG (20) | CHH (41) | CG (11) | CNG (11) | CHH (29) | CG (23) | CNG (20) | CHH (35) |
| P1-wr:LH51 (red cob) | 4 | 3 | 3 | 23 | 27 | 5 | 10 | 4 | 3 |
| p1-ww:DP3 (pink cob) | 7 | 3 | 4 | 93 | 68 | 0 | 85 | 66 | 5 |
| p1-ww:DP6 (white cob) | 4 | 3 | 4 | 94 | 75 | 0 | 96 | 84 | 2 |
R-I | R-II | R-III | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Genotype | CG (34) | CNG (20) | CHH (41) | CG (11) | CNG (11) | CHH (29) | CG (23) | CNG (20) | CHH (35) |
| P1-wr:LH51 (red cob) | 4 | 3 | 3 | 23 | 27 | 5 | 10 | 4 | 3 |
| p1-ww:DP3 (pink cob) | 7 | 3 | 4 | 93 | 68 | 0 | 85 | 66 | 5 |
| p1-ww:DP6 (white cob) | 4 | 3 | 4 | 94 | 75 | 0 | 96 | 84 | 2 |
For each genotype and region, the average methylation (percentage) was calculated by dividing the total number of methylated cytosines in each sequence context with total cytosines in all the clones. For each region, the total number of cytosines in a particular context is given in parentheses.
Methylation in three P1-wr regions in wild type and the leaky and white cob epialleles
R-I | R-II | R-III | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Genotype | CG (34) | CNG (20) | CHH (41) | CG (11) | CNG (11) | CHH (29) | CG (23) | CNG (20) | CHH (35) |
| P1-wr:LH51 (red cob) | 4 | 3 | 3 | 23 | 27 | 5 | 10 | 4 | 3 |
| p1-ww:DP3 (pink cob) | 7 | 3 | 4 | 93 | 68 | 0 | 85 | 66 | 5 |
| p1-ww:DP6 (white cob) | 4 | 3 | 4 | 94 | 75 | 0 | 96 | 84 | 2 |
R-I | R-II | R-III | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Genotype | CG (34) | CNG (20) | CHH (41) | CG (11) | CNG (11) | CHH (29) | CG (23) | CNG (20) | CHH (35) |
| P1-wr:LH51 (red cob) | 4 | 3 | 3 | 23 | 27 | 5 | 10 | 4 | 3 |
| p1-ww:DP3 (pink cob) | 7 | 3 | 4 | 93 | 68 | 0 | 85 | 66 | 5 |
| p1-ww:DP6 (white cob) | 4 | 3 | 4 | 94 | 75 | 0 | 96 | 84 | 2 |
For each genotype and region, the average methylation (percentage) was calculated by dividing the total number of methylated cytosines in each sequence context with total cytosines in all the clones. For each region, the total number of cytosines in a particular context is given in parentheses.
Region flanking SalI site in intron 2 (R-II):
In the P1-wr:LH51 allele, hypermethylation was confined to the 50-bp sequence in the 3′ end of this region where high levels of CG and CNG and relatively lower levels of CHH methylation were observed (Figure 4B). In contrast, the pink and white cob epialleles had higher levels of CG and CNG methylation throughout this region. The pink cob epiallele (p1-ww:DP3) had slightly lower levels of CG and CNG methylation as compared to highly silenced p1-ww:DP6 epiallele. In agreement with the gel-blot data, two cytosines in the recognition sequence (GTC*GAC) of the SalI site were fully methylated in the mutants whereas they were not methylated in the wild type (see the two CG sites marked by arrows in Figure 4B). Notably, the CHH methylation was not detected in either of the epialleles, indicating that P1-wr silencing was accompanied by loss of CHH methylation in this region. Average methylation over 10 CG, 11 CNG, and 29 CHH sites was higher in the two epialleles as compared to the wild-type P1-wr allele (Table 1).
Region covering HpaII sites in intron 2 (R-III):
The P1-wr:LH51 allele had high levels of CG and relatively lower levels of CNG and CHH methylation in the 60-bp sequence in the 5′ end of this region while the rest of the cytosines were hypomethylated (Figure 4C). Both silenced epialleles had dramatically higher levels of CG and CNG methylation spanning the whole region. The pink cob (p1-ww:DP3) epiallele had relatively lower levels of CG and CNG methylation as compared to its white cob counterpart. The bisulfite analysis confirmed our gel-blot methylation data of the three HpaII sites present in this region. Cytosines in the recognition sequence (C*C*GG) of the two adjacent HpaII sites (positions 4069/4073 in Figure 3) were hypomethylated in the wild-type allele (see CG sites at 4671 and 4675 and CNG sites at 4672 and 4676, marked by arrows in Figure 4C). These sites were highly methylated in the white cob epiallele while intermediate methylation levels of these sites were observed in the pink cob epiallele. Cytosine residues in the third HpaII site (position 4778 in Figure 3) were methylated in all three genotypes (see arrow marked CG and CNG sites at positions 4780 and 4781, respectively, in Figure 4C). The CHH methylation was very low in both epialleles although the pink cob epiallele had a slightly higher number of methylated CHH sites scattered across the region. Overall, methylation at 23 CG and 20 CNG sites revealed that the wild-type allele was hypomethylated, the white cob epiallele was hypermethylated, and the pink cob epialleles had intermediate methylation (Table 1).
Methylation patterns in the pink cob allele:
The pink cob allele (p1-ww:DP3) was particularly interesting in that there was a weak phenotype and partial methylation as assessed by DNA gel-blot analysis and bisulfite sequencing. The intermediate methylation level of the pink cob allele could be due either to stochastic partial methylation of random sites across sequences or to hypermethylation of specific contiguous strands. Detailed analysis indicated that the latter explanation was correct and that a proportion of the clones had stretches of non-methylated cytosine residues (Figure 5, Figure S1). For example, in some clones (group I, Figure 5), a contiguous stretch of cytosines was non-methylated in the 3′ end of R-III (indicated by the yellow shading) while many of the cytosines, particularly in the CHH context, were methylated in the 5′ end (marked by the red shading). Groups II and III had lesser numbers of non-methylated cytosine residues in the 3′ end. Interestingly, clones belonging to group IV had a contrasting pattern in which the hypomethylated stretch was in the 5′ end of the region. Partial methylation of the two adjacent HpaII sites (sites 4669/4673 in Figure 3) in the pink cob (p1-ww:DP3) was also confirmed through bisulfite analysis. These sites were non-methylated in group I (gray-shaded column in Figure 5) but methylated in the rest of the clones, thereby producing partial HpaII restriction digest. None of the clones from the white cob allele had contiguous non-methylated cytosine residues, although there was some stochastic variation in the methylation of specific sites (Figure 5). Most of the cytosines in R-III of P1-wr:LH51 were non-methylated. From these data, it appears that partial functionality of p1-ww:DP3 is contributed by relatively longer non-methylated stretches that could have a regulatory role in p1 expression.
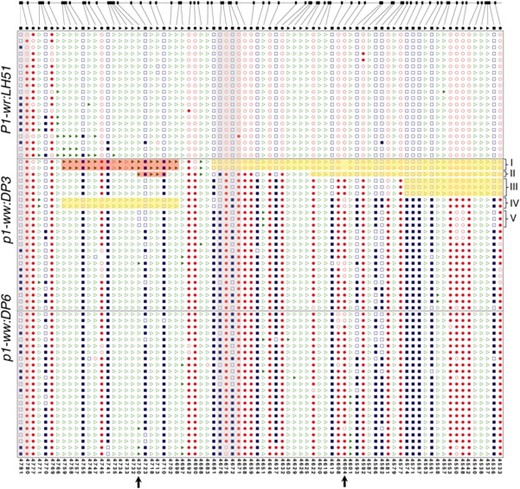
Mosaic methylation patterns of the pink cob epiallele in the R-III region of the second intron. Shown are the methylation patterns of cytosine residues in representative individual clones that were sequenced after bisulfite treatment. The clones were selected to represent the frequency of methylation patterns observed in all the sequenced clones. Location of individual CG (ovals), CNG (squares), and CHH (triangles) sites in R-III in the reference P1-wr sequence is at the top. In sequenced clones, open and solid shapes represent non-methylated and methylated sites, respectively. Some discrepancies between certain sites in the reference sequence and the corresponding sites in the cloned sequences indicate sequence polymorphisms. Coordinates of individual cytosine residues are indicated below. Five groups of p1-ww:DP3 clones showing interesting mosaic methylation patterns and/or sequence polymorphisms are indicated on the right. Red- and yellow-shaded boxes indicate hyper- and hypomethylated cytosine residues, respectively. Gray-shaded columns mark the three HpaII sites that are covered in R-III; two adjacent sites showing partial methylation in the pink cob (p1-ww:DP3) allele based on DNA gel-blot analysis are at nt positions 4671 and 4675 (on the antisense strand) in the middle of the region. Arrows at the x-axis mark two polymorphisms mentioned in the text.
Silenced P1-wr epialleles are reactivated in the presence of Ufo1:
Ufo1 is a trans-acting epigenetic modifier that reactivates epigenetically silenced p1 alleles and induces ectopic pericarp pigmentation in the wild-type P1-wr allele (Chopra et al. 2003; Sekhon et al. 2007; Sekhon and Chopra 2009). To further confirm that the loss of cob pigmentation is due to epigenetic silencing and not a sequence polymorphism(s), we crossed the silenced epialleles with inbred 4Co63 carrying the Ufo1 mutation (see materials and methods for details). Indeed, the presence of Ufo1 led to restoration of cob pigmentation in the F1 progenies of crosses involving a leaky (p1-ww:DP3) and white cob (p1-ww:DP8) epiallele (Figure 6, A and B, respectively). Interestingly, the Ufo1-induced cob pigmentation was darker in the leaky epiallele as compared to its white cob counterpart. Since Ufo1 shows poor penetrance (Chopra et al. 2003; Sekhon and Chopra 2009), only a subset of F1 progeny plants showed a gain of cob pigmentation. A 4Co63 p1-ww stock (without the Ufo1 mutation) was crossed to the p1-ww in this study and, in the absence of Ufo1, did not increase cob pigmentation (data not shown).
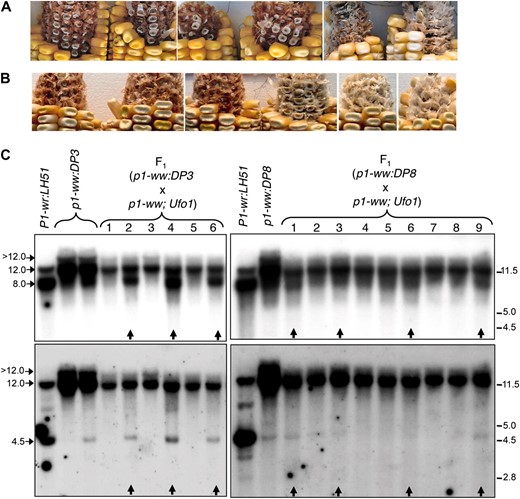
Reactivation of the silenced P1-wr epialleles in the presence of Ufo1. (A and B) Ufo1-induced gain of cob glume pigmentation in F1 progenies of a cross of p1-ww; Ufo1 with p1-ww:DP3 (A) and p1-ww:DP8 (B) plants. Cob glume and kernel phenotypes of six representative ears of each cross are shown. (C) DNA gel blots showing methylation modifications of the P1-wr epialleles in the presence of Ufo1. Gel blots carrying HpaII-digested leaf genomic DNA of F1 progeny plants of (p1-ww:DP3 × p1-ww; Ufo1) and (p1-ww:DP8 × p1-ww; Ufo1) were sequentially hybridized with probe fragments 15 (top panels) and 8B (bottom panels). Sizes and positions of bands hybridizing with the two probes are shown on the left, and molecular weight markers in kilobase pairs are shown on the right. Arrows at the bottom of the panels indicate the plants that showed gain of cob pigmentation in the presence of Ufo1.
Ufo1-induced upregulation of p1 is associated with a decrease in DNA methylation of the P1-wr multi-copy complex (Chopra et al. 2003; Sekhon et al. 2007; Sekhon and Chopra 2009). To test whether such interaction extends to the epialleles reported in this study, leaf genomic DNA of the F1 progeny plants was digested with HpaII, and the gel blots were probed with fragments 15 and 8B. In P1-wr:LH51, the wild-type progenitor of the silenced epialleles, probe 15 produced an intense ∼8.0-kb and a weaker ∼12.0-kb band (Figure 6C). Hypermethylation of the leaky (p1-ww:DP3) and white cob (p1-ww:DP8) epialleles was characterized by reversal of the intensities of ∼8.0- and ∼12.0-kb bands and the appearance of a >12.0-kb band. Hypomethylation of the F1 plants showing Ufo1-induced gain of cob pigmentation was evident from the reappearance of an ∼8.0-kb band and reduced intensities of ∼12.0- and >12.0-kb bands (lanes marked with vertical arrows in Figure 6C). Similarly, probe 8B hybridized to two prominent bands of ∼12.0 and 4.5 kb in P1-wr:LH51. Hypermethylation of the two epialleles was indicated by the absence (in p1-ww:DP3) or the lower intensity (in p1-ww:DP8) of the 4.5-kb band, the increased intensity of the 12.0-kb band, and the appearance of a >12.0-kb band. Finally, the gain of cob pigmentation in F1 plants was accompanied by hypomethylation as indicated by the reappearance of 4.5-kb band and the lower intensity of 12.0- and >12.0-kb bands. In summary, these data demonstrate that the silenced epialleles can be reactivated by Ufo1 and show loss of methylation at the P1-wr repeat complex. Interestingly, it appears that the to Ufo1-induced hypomethylation and phenotypic modification.
DISCUSSION
Tissue culture-induced epigenetic changes lead to generation of epialleles:
This study provides evidence that epigenetic modifications underlie heritable phenotypic variation observed in progenies of plants regenerated from tissue culture. While many studies have reported tissue culture-induced epigenetic modifications, this study reports a direct link between novel epialleles and phenotype. The results of our study are generally consistent with the model proposed by Phillips and Colleagues (1990), which implicate DNA methylation modifications as an underlying molecular mechanism leading to cytogenetic and phenotypic changes in culture. While both hyper- and hypomethylation have been reported in tissue culture-derived plants, hypomethylation is the predominant epigenetic modification observed in single- or low-copy sequences (Kaeppler and Phillips 1993; Olhoft 1996; Kaeppler et al. 2000). In contrast, a higher rate of epimutations in P1-wr upon tissue culture suggest that tandemly duplicated genes have increased propensity to hypermethylation and epigenetic silencing. Given that approximately one-third of the maize genes are tandemly duplicated (Messing et al. 2004), such epigenetic reprogramming may have a significant impact on the transcriptome of the tissue culture-derived transgenic cultivars.
Possible mechanisms underlying tissue culture-induced silencing of P1-wr:
Since our analysis was completed on progenies of plants regenerated from tissue culture where the epigenetic silencing was already established, the exact nature of the mechanisms responsible for inducing the silencing could not be studied. However, some specific observations allow us to speculate about the mechanisms responsible for establishment of silencing that are discussed below.
Antisense or aberrant RNA-mediated silencing:
Coordinated de novo methylation of an intronic region in multiple P1-wr gene copies spanning a over >100-kb region suggests a mechanism capable of targeting multiple genomic regions based on sequence homology. In plants, an RNA-directed DNA methylation pathway is a predominant mechanism for de novo DNA methylation (Chan et al. 2005) whereby the cytosines in the region homologous to the small interfering RNA (siRNA) molecules are methylated in all (CG, CNG, and CHH) contexts (Pelissier et al. 1999). Thus, a potential model to explain the formation of the p1 epialleles is via a transiently expressed antisense or aberrant RNA homologous to the differentially methylated region. Given the general global hypomethylation during tissue culture (Kaeppler et al. 2000), such aberrant RNA could be produced from demethylation of a quiescent cryptic promoter adjacent to a p1 or a p1-homologous sequence. Consistent with such a model, appearances of several epialleles in an Arabidopsis cell suspension culture was associated with de novo DNA and H3K9me2 hypermethylation and a concurrent appearance of novel 24-nt siRNA molecules (Martienssen et al. 2008; Tanurdzic et al. 2008). Loss of DECREASE IN DNA METHYLATION 1 in Arabidopsis, while causing global hypomethylation mechanistically similar to that observed in tissue culture, can also induce CG, CNG, and CHH hypermethylation in genic regions via siRNA-mediated chromatin remodeling (Saze and Kakutani 2007). It should be noted, however, that the hypermethylation observed in the P1-wr epialleles is in the CG and CNG contexts. Lack of CHH methylation might suggest that such an RNA-mediated mechanism occurred transiently in tissue culture and that the CG and CNG hypermethylation reflects only the maintenance of the silenced state. Our experiments to date have not searched for such an inducing RNA. Crosses of our epialleles to wild-type P-wr stocks indicate that the alleles in their current state are not paramutagenic, which is also consistent with the notion that they are currently in a maintenance, rather than an inducing, condition.
Repeats have enhanced susceptibility to silencing in tissue culture:
Another possible model for p1 epiallele formation is that the repeated structure of the allele itself makes P1-wr a target for silencing. It has been shown that repeat structures serve as a signal to initiate epigenetic silencing in many cases (Ye and Signer 1996; Irelan and Selker 1997; Selker 1999; Chandler and Stam 2004), and for all cases cited, DNA–DNA pairing between repeat sequences was proposed to trigger DNA methylation. Chopra et al. (1998) proposed that somatic pairing of P1-wr multicopy repeats may be controlled by tissue-specific alteration in chromatin structure (Ceccarelli and Cionini 1993), which in turn affects the accessibility of the P1-wr gene copies to nuclear trans-acting factors. Higher propensity of P1-wr to epigenetic silencing is also evident from characterization of P1-wr*, another epiallele of P1-wr that originated during development of recombinant inbred lines (Sekhon et al. 2007).
Role of epigenetic modifications of introns in regulation of gene expression:
A relatively straightforward explanation for the inactivation of P1-wr is that the differentially methylated region of the second intron contains sequences important for P1-wr transcription. Findings from our study are consistent with a previous report (Sekhon et al. 2007) that also implicates hypermethylation of sequences in the second intron in epigenetic silencing of P1-wr*, a spontaneous epiallele of P1-wr. Furthermore, both P1-wr* and the epialleles examined in our study can be reactivated in the presence of Ufo1, and such derepression is associated with hypomethylation of the intron 2 region. These findings suggest that the differentially methylated region of intron 2 may include an important uncharacterized positive cis-regulatory DNA element in the P1-wr repeats. Once the labile DNA region is subject to epigenetic silencing, transcription initiation will be incomplete and gene transcription, and thereby function, will be reduced. Alternatively, it is also possible that methylation of intron regions hinders the transcript elongation, thereby suppressing gene expression.
In mammals, there are several reports documenting the role of the epigenetic state of introns in gene regulation. Downregulation of EGR2, a tumor suppressor gene, was associated with hypermethylation of a putative enhancer in the intron region (Unoki and Nakamura 2003). Similarly, DNA methylation of intronic regions has been proposed to affect the expression of the aldolase B gene in rats and the Igf2r gene in mice (Daimon et al. 1986; Wutz et al. 1997; Murrell et al. 2001). In plants, however, the role of the epigenetic state of introns in gene regulation is less clear. Genome-wide methylation studies have shown that 20–30% methylation (Zhang et al. 2006; Vaughn et al. 2007; Zilberman et al. 2007), suggesting that such methylation has little or no effect on gene transcription. A closer look at the bisulfite sequencing data, however, revealed that such hypermethylation is almost exclusively found in the CG context. Such conspicuous absence of non-CG methylation indicated that it could hinder gene expression and that the plant introns are kept free of non-CG methylation by a yet-unknown mechanism. Indeed, increased CNG methylation of the BONSAI gene body in Arabidopsis plants lacking INCREASE IN BONSAI METHYLATION1, a putative H3 lysine 9 demethylase, resulted in loss of gene expression (Miura et al. 2009). This is consistent with our observation that silencing the p1 epialleles is associated with high levels of CNG methylation in the second intron. In conclusion, our study provides further evidence supporting the role of epigenetic modifications of introns in the regulation of gene expression.
Intermediate-expression phenotypes of tissue culture-induced epialleles correlated with mosaic DNA methylation patterns:
The appearance of stable and meiotically heritable epialleles with intermediate (pink cob) phenotypes indicates that intermediate alleles can be formed during the induction process. In a previous study, attenuated expression of cell culture-derived epialleles of an invertedly repeated transgene in tobacco was attributed to methylation mosaicism at the cellular level (Krizova et al. 2009). Our study, however, provides evidence that such intermediate expression might also be due to mosaic methylation patterns of individual gene copies. This observation suggests that multiple copies contribute to P1-wr expression and that stable silencing of some of the copies is responsible for intermediate phenotypes. Nonetheless, such tissue culture-induced epigenetic variation in general and the appearance of allelic variants with intermediate phenotypes in particular provide a new source of genetic variability for improved germplasm.
Footnotes
Supporting information is available online at http://www.genetics.org/cgi/content/full/genetics.110.117929/DC1.
Present address: The Institute of Life Science and Technology, Sungkyunkwan University, Natural Sciences Campus, Suwon 440-746, Korea.
Footnotes
Communicating editor: E. J. Richards
Acknowledgements
We thank Nives Kovacevic for her help in DNA methylation analyses. Part of this work was supported by National Science Foundation award MCB-0619330 and Hatch 4152 award to S.C.
References
Brink, R. A., and E. D. Styles,
Chopra, S., P. Athma and T. Peterson,
Coe, E. H., M. G. Neuffer and D. A. Hoisingto,
Devaux, P., A. Kilian and A. Kleinhofs,
Frommer, M., L. E. McDonald, D. S. Millar, C. M. Collis, F. Watt et al.,
Grotewold, E., P. Athma and T. Peterson,
Hirochika, H., K. Sugimoto, Y. Otsuki, H. Tsugawa and M. Kanda,
Kaeppler, S. M., and R. L. Phillips,
Kaeppler, S. M., H. F. Kaeppler and Y. Rhee,
Kovařik, A., B. Koukalová, M. Bezděk and Z. Opatrný,
Messing, J., A. K. Bharti, W. M. Karlowski, H. Gundlach, H. R. Kim et al.,
Olhoft, P.,
Phillips, R. L., and P. Olhoft,
Phillips, R. L., S. M. Kaeppler and V. M. Peschke,
Rhee, Y., H. Lin, R. Buell, K. Childs and S. Kaeppler,
Saghai-Maroof, M. A., K. M. Soliman, R. A. Jorgensen and R. W. Allard,
Styles, E. D., and O. Ceska,
Tanurdzic, M., M. W. Vaughn, H. Jiang, T. J. Lee, R. K. Slotkin et al.,
Ye, F., and E. R. Signer,
Zhang, M., C. Xu, H. Yan, N. Zhao, D. von Wettstein et al.,

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}