





Abstract
Previous studies have demonstrated that the diversity of Y-linked genes is substantially lower than that of their X-linked homologs in the plant Silene latifolia. This difference has been attributed to selective sweeps, Muller's ratchet, and background selection, processes that are predicted to severely affect the evolution of the nonrecombining Y chromosome. We studied the DNA diversity of a noncoding region of the homologous genes DD44Y and DD44X, sampling S. latifolia populations from a wide geographical area and also including the closely related species S. dioica, S. diclinis, and S. heuffelii. On the Y chromosome of S. latifolia, we found substantial DNA diversity. Geographical population structure was far higher than on the X chromosome and differentiation between the species was also higher for the Y than for the X chromosome. Our findings indicate that the loss of genetic diversity on the Y chromosome in Silene occurs within local populations rather than within entire species. These results are compatible with background selection, Muller's ratchet, and local selective sweeps, but not with species-wide selective sweeps. The higher interspecific divergence of DD44Y, compared to DD44X, supports the hypothesis that Y chromosome differentiation between incipient species precedes reproductive isolation of the entire genome, forming an early stage in the process of speciation.
HETEROMORPHIC sex chromosomes have evolved independently in many animal and plant species (Bull 1983; Graves and Shetty 2001) but have apparently followed a common evolutionary pathway involving the suppression of recombination between the proto-Y and X chromosomes, followed by Y chromosome degeneration (Bull 1983). Plants of the genus Silene are especially convenient for studies of sex chromosome evolution because they allow closely related species with and without sex chromosomes to be compared. Sex chromosomes are found only in the following Silene species of the section Elisanthe: Silene latifolia, S. dioica, S. diclinis, S. heuffelii, and S. marizii. The previous assignment of the hermaphroditic species S. noctiflora to section Elisanthe was incorrect (Sandbrink et al. 1989). With the exception of S. otites, which apparently evolved dioecy independently from the Elisanthe species, the remaining plants in the genus Silene are nondioecious. Silent divergence between S. latifolia and the hermaphroditic species S. vulgaris is ∼20%, suggesting that S. latifolia sex chromosomes are not older than 10–20 million years (Filatov and Charlesworth 2002).
Previous studies of sex chromosome evolution in Silene have focused upon the comparison of DNA diversity between homologous X- and Y-linked genes (Filatov et al. 2000, 2001; Laporte et al. 2005). These studies indicated that the genetic diversity of Y-linked genes could be up to 30-fold lower than that of their X-linked homologs. The lower genetic diversity of Y-linked genes was interpreted as evidence that population genetic processes such as genetic hitchhiking and background selection were causing degeneration of the nonrecombining Y chromosome (reviewed in Charlesworth and Charlesworth 1999). The genetic hitchhiking model (Rice 1987) proposes that rare, strongly advantageous mutations can result in the fixation of associated Y chromosome haplotypes, including linked, weakly deleterious mutations. In contrast, the background selection model (Charlesworth et al. 1993, 1995) states that frequent, weakly deleterious mutations accumulate stochastically on different Y chromosomes, allowing only those chromosomes with the fewest deleterious mutations to perpetuate themselves. Both processes result in the loss of Y chromosome variability, potentially increasing the stochastic loss of chromosomes with the fewest deleterious mutations (Muller's ratchet; Charlesworth 1978; Gordo et al. 2002). The extremely low diversity of genes on the Y chromosome of S. latifolia is consistent with both selective sweeps and background selection, but this very lack of diversity has, until now, prevented discrimination between the alternative models (Laporte and Charlesworth 2002).
Population subdivision can affect Y-linked and X-linked genes in different ways, depending on differences in migration rate between seed and pollen (Laporte and Charlesworth 2002), potentially biasing comparisons of sequence diversity. Dispersal of S. latifolia seed is usually limited to a distance of <80 m from the parent population while the range of pollen dispersal by bees and moths can be many times greater than this (Richards et al. 1999). McCauley (1994, 1997) demonstrated a genetic effect of this sexual difference in dispersal, showing higher levels of fine-scale genetic structure in maternally inherited chloroplast genes than in biparentally inherited autosomal genes. The sequences analyzed in previous studies (Filatov et al. 2000, 2001; Laporte et al. 2005) came from plants collected from Western European and from recently introduced North American populations. The natural range of S. latifolia is far more extensive, stretching from Britain and Scandinavia in northwestern Europe to northern Morocco in northwestern Africa to Lake Baikal in southeastern Siberia to the Black Sea in southeastern Europe (Baker 1947). Furthermore, studies of RAPDs (Vellekoop et al. 1996), allozyme (Mastenbroek et al. 1984) and flavonoid variation (Mastenbroek and van Brederode 1986), and pollen and seed morphology all indicated that European S. latifolia populations are divided into nine geographical races (Mastenbroek and van Brederode 1986), possibly corresponding to colonization routes following the clearance of land for agriculture (Vellekoop et al. 1996). The RAPD data of Vellekoop et al. (1996) indicated that individuals from the same geographical area were more genetically similar (Jaccard's coefficient of similarity 0.50–0.82) than individuals from different geographical areas (Jaccard's coefficient of similarity 0.28–0.61). It is therefore likely that the geographically limited samples of the previous studies (Filatov et al. 2000, 2001) captured only a small proportion of the total diversity of S. latifolia Y-linked genes.
We demonstrate that widening the geographical range from which S. latifolia samples are collected reveals substantially more DNA diversity in the gene DD44Y, a Y-linked gene recently described by Moore et al. (2003). Most of these newly discovered polymorphic sites are fixed within geographical areas, a finding consistent with the background selection model for the reduction of diversity on the Y chromosome, but inconsistent with the global selective sweep model. In the case of background selection, the stochastic occurrence of mildly deleterious mutations within each population is expected to favor different haplotypes in different populations and thereby to increase population structure. On the other hand, strongly beneficial mutations have the potential to spread quickly among populations (Slatkin and Wiehe 1998), resulting in species-wide hitchhiking events that eliminate all population structure on the Y chromosome's nonrecombining region. Hitchhiking is therefore expected to increase population structure only if the spread of a beneficial mutation is restricted to local populations, due to either geographic isolation or a mutation that is beneficial only under local environmental conditions (Stephan et al. 1998; Innan and Stephan 2003).
Interspecific gene flow also has the potential to influence the relative diversity and population structure of X- and Y-linked genes. In rodent hybrid zones, Y-linked genes introgress far less readily than other genes, resulting in greater divergence of Y-linked genes between species and lower diversity within species (Vanlerberghe et al. 1986; Jaarola et al. 1997). S. latifolia occupies a geographical range overlapping those of its close relatives S. dioica, S. heuffelii, and S. diclinis. S. diclinis is cross-incompatible with S. latifolia (Prentice 1978). However, S. dioica is known to interbreed with S. latifolia under natural conditions (Baker 1948) and S. heuffelii crosses freely with S. latifolia in cultivation (Prentice 1978). We used this evidence for hybridization between S. latifolia and other dioecious Silene species as a basis to compare levels of interspecific gene flow between X- and Y-linked genes, with the aim of evaluating the importance of gene flow in creating differences in diversity and population structure between the X and Y chromosomes.
MATERIALS AND METHODS
Collection of plant material:
To capture the maximum amount of geographic variation, we collected S. latifolia samples from central, southwestern, and southeastern Europe and from Siberia as well as from Western Europe. To investigate the effects of interspecific gene flow on the relative diversity of X- and Y-linked genes, we also sequenced DD44X and DD44Y from S. diclinis, S. dioica, and S. heuffelii. S. dioica occurs throughout northern Europe, where its range broadly overlaps that of S. latifolia. S. heuffelii occurs in the northern Balkan region and the Carpathian mountains at altitudes >700–800 m, while S. diclinis is an endemic species, occupying an extremely restricted geographical range surrounding the town of Xativa in the Spanish province of Valencia (Prentice 1976).
S. latifolia leaf material was collected from four areas of Europe: (1) northwestern Europe—Britain, France, and Belgium; (2) central Europe—Denmark, Czech Republic, and western Romania; (3) southwestern Europe—Spain; and (4) southeastern Europe—Romania. These areas correspond, respectively, to Mastenbroek and van Brederode's (1986) races 1, 2, 3, 7, and 9. Additional samples were collected from Siberia, at the extreme eastern limit of S. latifolia's range. Locations and sample sizes are given in Table 1. Samples of three other dioecious Elisanthe species were also collected. S. dioica was collected from Britain, S. diclinis from Spain, and S. heuffelii from Romania. All of the dioecious Silene species form discrete patches, usually containing <100 plants, and most seed dispersal occurs within 80 m of the parent's patch (Richards et al. 1999). To eliminate any genetic structure caused by inbreeding within patches, a single male plant was sampled from each of several patches within each geographical area. Patches were sampled haphazardly but, except in the case of S. diclinis, adjacent patches were sampled only when separated by a distance >100 m. Because the geographical range of S. diclinis is now so small, samples were collected from every patch, even when patches were close together. A small quantity of leaf material was taken from each plant and frozen. DNA was extracted using Plant DNAzol according to the manufacturer's instructions.
Geographical locations of S. latifolia samples
Country, nearest town | Geographic position | Geographical racea | Sample size |
|---|---|---|---|
| Scotland, Edinburgh | 55.57 N 3.13 W | 1 | 2 |
| England, London | 51.30 N 0.10 W | 1 | 1 |
| England, Malvern | 52.07 N 2.19 W | 1 | 1 |
| England, Kidderminster | 52.23 N 2.14 W | 1 | 2 |
| England, Brighton | 50.50 N 0.08 W | 1 | 1 |
| England, Cambridge | 52.10 N 0.11 E | 1 | 1 |
| France, Biarritz | 48.43 N 3.1 E | 1 | 2 |
| Belgium, Brussels | 50.41 N 4.46 E | 1 | 1 |
| Denmark, Arhus | 56.25 N 10.48 E | 2 | 1 |
| Czech Republic, Brno | 49.12 N 16.37 E | 2 | 1 |
| Romania, Oradea | 47.03 N 21.57 E | 2 | 1 |
| Spain, Alcoy | 38.42 N 0.38 W | 7 | 6 |
| Romania, Campeni | 44.00 N 24.35 E | 9 | 8 |
| Romania, Cluj-Napoca | 46.47 N 23.36 E | 9 | 1 |
| Russia, Krasnoyarsk region 1 | 53.8 N 92.53 E | — | 1 |
| Russia, Krasnoyarsk region 2 | 56.0 N 92.51 E | — | 1 |
| Russia, Irkutsk | 52.16 N 104.20 E | — | 3 |
Country, nearest town | Geographic position | Geographical racea | Sample size |
|---|---|---|---|
| Scotland, Edinburgh | 55.57 N 3.13 W | 1 | 2 |
| England, London | 51.30 N 0.10 W | 1 | 1 |
| England, Malvern | 52.07 N 2.19 W | 1 | 1 |
| England, Kidderminster | 52.23 N 2.14 W | 1 | 2 |
| England, Brighton | 50.50 N 0.08 W | 1 | 1 |
| England, Cambridge | 52.10 N 0.11 E | 1 | 1 |
| France, Biarritz | 48.43 N 3.1 E | 1 | 2 |
| Belgium, Brussels | 50.41 N 4.46 E | 1 | 1 |
| Denmark, Arhus | 56.25 N 10.48 E | 2 | 1 |
| Czech Republic, Brno | 49.12 N 16.37 E | 2 | 1 |
| Romania, Oradea | 47.03 N 21.57 E | 2 | 1 |
| Spain, Alcoy | 38.42 N 0.38 W | 7 | 6 |
| Romania, Campeni | 44.00 N 24.35 E | 9 | 8 |
| Romania, Cluj-Napoca | 46.47 N 23.36 E | 9 | 1 |
| Russia, Krasnoyarsk region 1 | 53.8 N 92.53 E | — | 1 |
| Russia, Krasnoyarsk region 2 | 56.0 N 92.51 E | — | 1 |
| Russia, Irkutsk | 52.16 N 104.20 E | — | 3 |
According to Mastenbroek and van Brederode (1986).
Geographical locations of S. latifolia samples
Country, nearest town | Geographic position | Geographical racea | Sample size |
|---|---|---|---|
| Scotland, Edinburgh | 55.57 N 3.13 W | 1 | 2 |
| England, London | 51.30 N 0.10 W | 1 | 1 |
| England, Malvern | 52.07 N 2.19 W | 1 | 1 |
| England, Kidderminster | 52.23 N 2.14 W | 1 | 2 |
| England, Brighton | 50.50 N 0.08 W | 1 | 1 |
| England, Cambridge | 52.10 N 0.11 E | 1 | 1 |
| France, Biarritz | 48.43 N 3.1 E | 1 | 2 |
| Belgium, Brussels | 50.41 N 4.46 E | 1 | 1 |
| Denmark, Arhus | 56.25 N 10.48 E | 2 | 1 |
| Czech Republic, Brno | 49.12 N 16.37 E | 2 | 1 |
| Romania, Oradea | 47.03 N 21.57 E | 2 | 1 |
| Spain, Alcoy | 38.42 N 0.38 W | 7 | 6 |
| Romania, Campeni | 44.00 N 24.35 E | 9 | 8 |
| Romania, Cluj-Napoca | 46.47 N 23.36 E | 9 | 1 |
| Russia, Krasnoyarsk region 1 | 53.8 N 92.53 E | — | 1 |
| Russia, Krasnoyarsk region 2 | 56.0 N 92.51 E | — | 1 |
| Russia, Irkutsk | 52.16 N 104.20 E | — | 3 |
Country, nearest town | Geographic position | Geographical racea | Sample size |
|---|---|---|---|
| Scotland, Edinburgh | 55.57 N 3.13 W | 1 | 2 |
| England, London | 51.30 N 0.10 W | 1 | 1 |
| England, Malvern | 52.07 N 2.19 W | 1 | 1 |
| England, Kidderminster | 52.23 N 2.14 W | 1 | 2 |
| England, Brighton | 50.50 N 0.08 W | 1 | 1 |
| England, Cambridge | 52.10 N 0.11 E | 1 | 1 |
| France, Biarritz | 48.43 N 3.1 E | 1 | 2 |
| Belgium, Brussels | 50.41 N 4.46 E | 1 | 1 |
| Denmark, Arhus | 56.25 N 10.48 E | 2 | 1 |
| Czech Republic, Brno | 49.12 N 16.37 E | 2 | 1 |
| Romania, Oradea | 47.03 N 21.57 E | 2 | 1 |
| Spain, Alcoy | 38.42 N 0.38 W | 7 | 6 |
| Romania, Campeni | 44.00 N 24.35 E | 9 | 8 |
| Romania, Cluj-Napoca | 46.47 N 23.36 E | 9 | 1 |
| Russia, Krasnoyarsk region 1 | 53.8 N 92.53 E | — | 1 |
| Russia, Krasnoyarsk region 2 | 56.0 N 92.51 E | — | 1 |
| Russia, Irkutsk | 52.16 N 104.20 E | — | 3 |
According to Mastenbroek and van Brederode (1986).
Amplification and sequencing:
DD44X and DD44Y were amplified from the genomic DNA of male plants (sequences for all PCR and sequencing primers are provided in supplementary material at http://www.genetics.org/supplemental/). DD44X was amplified as a single fragment using the primers DD44XY4.1F and DD44XY2.1R. The same pair of primers was used to amplify DD44 from S. vulgaris, a hermaphroditic species without sex chromosomes in which DD44 occurs as a single-copy gene on one pair of autosomes. The longer DD44Y gene was amplified in four fragments using the primer pairs DD44XY4F:DD44Y4R, DD44XY3.2F:DD44Y3R5, DD44Y3F5:DD44Y3R10, and DD44Y3F9:DD44XY2.1R. Amplifications were performed under the following PCR conditions: initial denaturation at 94° for 2 min followed by 30 cycles of 94° for 10 sec, 63° for 30 sec, and 68° for 3 min (DD44X) or 8 min (DD44Y). Expand high fidelity DNA polymerase (Roche Diagnostics) was used for DD44X while Expand long template DNA polymerase (Roche Diagnostics) was used for DD44Y. Because males possess only a single X and a single Y chromosome, DD44X and DD44Y PCR products amplified from male genomic DNA contained no heterozygous loci and so could be directly sequenced.
DD44Y phylogeny:
Y-linked genes do not recombine and can therefore be used to study the evolutionary history of individual haplotypes within a species. A neighbor-joining tree of S. latifolia DD44Y sequences was constructed with the program MEGA 2.1 (Kumar et al. 1993) using the Kimura two-parameter model (Kimura 1980). The tree included S. latifolia DD44Y sequences from each geographical area and also included out-group sequences from S. dioica, S. heuffelii, and S. diclinis. The support for each node was tested by bootstrapping with 1000 replicates.
Geographical structure:
To test for geographical population structure within S. latifolia, FST values were calculated for DD44X and DD44Y between samples from each geographical area. The program Proseq 3.0 (Filatov 2002) was used to calculate 95% confidence intervals for each FST value by bootstrapping across polymorphic loci with 100 replicates. Proseq was also used to test the significance of these FST values using permutation tests with 1000 replicates according to the methods of Hudson et al. (1992).
Comparisons of nucleotide diversity:
The program Proseq was used to calculate two measures of nucleotide diversity [π (Nei 1987) and Watterson's (1975) θw] for DD44X and DD44Y overall, within each species, and within each S. latifolia population.
The sequence diversity of DD44X was compared with that of DD44Y using the HKA test (Hudson et al. 1987), implemented in DNAsp. A single DD44 sequence from the hermaphroditic species S. vulgaris was used as an out-group. The split of S. latifolia (and the other Elisanthe species) from S. vulgaris preceded the differentiation of the sex chromosomes and so the divergence of DD44X and DD44Y from this single out-group could be used to calibrate the diversity of the two genes, allowing for potential differences between them in mutation rate.
Population genetic structure is a potential cause of bias in tests based on coalescent theory (Hudson et al. 1987). However, Wakeley (1999) demonstrated that the genealogy of a subdivided population could be divided into a brief, recent “scattering phase” preceded by a long “collecting phase.” If a single individual is sampled from each population, then the scattering phase is bypassed and the genealogy of the sample should approximate a coalescent, provided that the number of samples is sufficiently great. Since we had evidence of significant subdivision among geographical areas, we repeated each HKA test using only a single individual from each geographical area. This strategy was used so that the scattering phase of the S. latifolia genealogy would be bypassed even if scattering had occurred over a very wide geographical area (i.e., if there had been a recent, rapid population expansion) (Wakeley 1999).
Interspecific gene flow:
To test for differentiation of DD44X and DD44Y among the four Elisanthe species, interspecific FST values were calculated for both genes. The program Proseq (Filatov 2002) was used to test the significance of these FST values using permutation tests with 1000 replicates according to the methods of Hudson et al. (1992). Reduction in Y chromosome genetic diversity due to selective sweeps or background selection can inflate the divergence of Y-linked genes between populations, biasing FST values (Laporte and Charlesworth 2002). For this reason, an additional test of interspecific gene flow between S. latifolia and S. dioica was performed using the Markov chain Monte Carlo approach of Nielsen and Wakeley (2001) implemented in the program MDIV. This model jointly estimates parameters for population size, migration rate, and time of divergence from the most recent common ancestor within a likelihood framework. Simulations were performed using a Markov chain of 5,000,000 cycles with a burn-in time of 500,000 cycles. Maximum values for both the scaled migration rate (M) and the divergence time (T) were set at 10. The hypothesis that S. latifolia and S. dioica evolved in genetic isolation following their divergence from a common ancestor was tested using a likelihood-ratio test in which the maximum likelihood when M = 0 was compared with the maximum likelihood when M > 0 (see Nielsen and Wakeley 2001). Likelihood-ratio tests were also used to test for differences in M between DD44X and DD44Y.
Site-frequency distribution:
Because hitchhiking events eradicate diversity from a gene, polymorphic sites must have arisen only from mutations that occurred since the last hitchhiking event. If a hitchhiking event has occurred recently, a bias of the site-frequency spectrum toward rare alleles is therefore predicted (Braverman et al. 1995). We tested the DD44X and DD44Y sequences of each Silene population for such bias of the site-frequency distribution using Tajima's test (Tajima 1989).
RESULTS
Phylogeny of DD44Y:
A neighbor-joining tree of DD44Y (Figure 1) supported Mastenbroek and van Brederode's (1996) division of S. latifolia into separate geographical races. In agreement with earlier RAPD, allozyme, biochemical, and morphological data, the DD44Y tree indicated early divergence between southeastern and western European populations of S. latifolia while also allowing populations from southwestern Europe to be differentiated from those of northwestern Europe. Together with the samples from Siberia, the central European samples form a monophyletic group. This suggests that central European S. latifolia constitute a separate race rather than a contact zone between eastern and western races as was suggested by Mastenbroek and van Brederode (1986). The close similarity of the Siberian and central European samples raises the possibility that S. latifolia spread to Siberia from central Europe as Siberian land was cleared for cultivation.
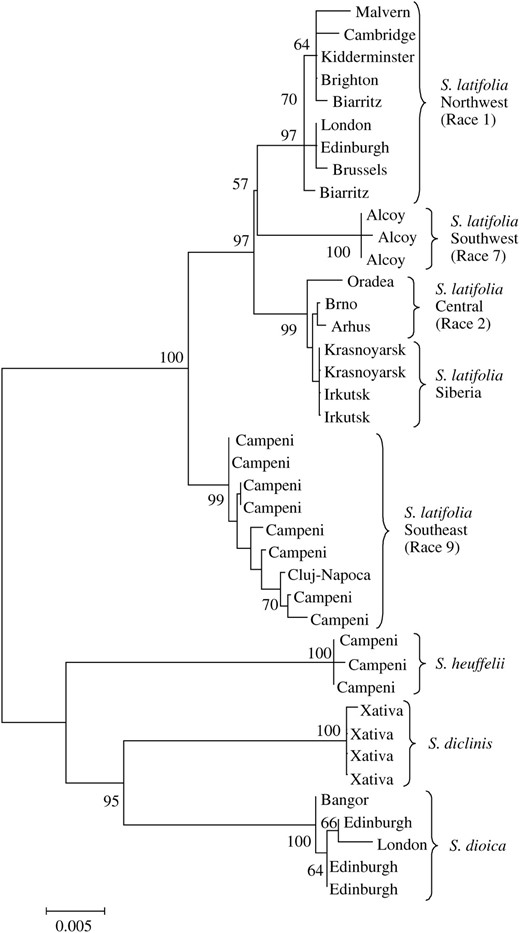
Neighbor-joining tree of Silene DD44Y sequences, showing bootstrap support for each node. Silene species and S. latifolia geographical races are clearly differentiated.
Comparisons of nucleotide diversity:
Within each of the S. latifolia samples collected from the northwestern, central, and southeastern areas of Europe, the nucleotide diversity (measured as π and θw) of DD44X was >15 times higher than that of DD44Y (Table 2, Figure 2). HKA tests using an S. vulgaris out-group indicated that these differences in diversity were statistically significant, despite the low number of samples collected from each geographical area (Table 2). A difference in diversity of similar magnitude between a homologous pair of X- and Y-linked genes was obtained by Filatov et al. (2000) between the genes SlX1 and SlY1 in samples collected mainly from northwestern Europe. In contrast, the diversity of DD44X in the southwestern area was not significantly higher than that of DD44Y. This may simply reflect the very low size of this sample. However, a significantly negative value of Tajima's D (D = −1.50, P < 0.05) indicated that the site-frequency spectrum for DD44X in the southwestern S. latifolia sample was biased toward rare polymorphisms, suggesting that there may have been a recent selective sweep or population expansion in this area. Large differences (>20-fold) in diversity between DD44X and DD44Y were also found in S. dioica, S. heuffelii, and S. diclinis (Table 2, Figure 2). Each of these species was collected from only a single geographical area.
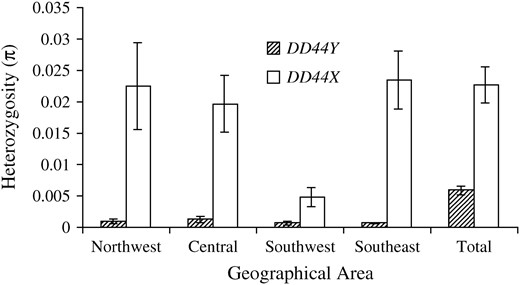
Genetic diversity (π) of DD44X and DD44Y in S. latifolia from northwestern, central, southwestern, and southeastern Europe.
DD44X and DD44Y sequences from S. latifolia, S. dioica, S. heuffelii, and S. diclinis
N | Length (bp) | Segregating sites | Nucleotide diversity (π) | Nucleotide diversity (θw) | HKA test | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Species | DD44X | DD44Y | DD44X | DD44Y | DD44X | DD44Y | DD44X | DD44Y | DD44X | DD44Y | χ2 | P |
| S. latifolia NW | 7 | 8 | 1964 | 5313 | 94 | 18 | 0.0225 | 0.0010 | 0.0195 | 0.0013 | 10.01 | <0.001 |
| S. latifolia C | 6 | 7 | 1131 | 4349 | 49 | 17 | 0.0197 | 0.0013 | 0.0190 | 0.0016 | 7.579 | <0.001 |
| S. latifolia SW | 6 | 3 | 2954 | 7096 | 23 | 2 | 0.0033 | 0.0007 | 0.0043 | 0.0007 | 1.230 | NS |
| S. latifolia SE | 9 | 9 | 2480 | 6495 | 176 | 11 | 0.0234 | 0.0007 | 0.0261 | 0.0006 | 20.50 | <0.001 |
| S. latifolia | 28 | 27 | 401 | 2690 | 40 | 56 | 0.0227 | 0.0059 | 0.0256 | 0.0032 | 4.28 | NS |
| S. dioica | 7 | 5 | 2985 | 6298 | 152 | 7 | 0.0259 | 0.0006 | 0.0239 | 0.0007 | 16.97 | <0.001 |
| S. diclinis | 6 | 4 | 2932 | 7436 | 40 | 1 | 0.0047 | 0.0002 | 0.0060 | 0.0002 | 4.09 | <0.05 |
| S. heuffelii | 4 | 3 | 2225 | 7768 | 96 | 4 | 0.0237 | 0.0002 | 0.0235 | 0.0002 | 12.14 | <0.001 |
N | Length (bp) | Segregating sites | Nucleotide diversity (π) | Nucleotide diversity (θw) | HKA test | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Species | DD44X | DD44Y | DD44X | DD44Y | DD44X | DD44Y | DD44X | DD44Y | DD44X | DD44Y | χ2 | P |
| S. latifolia NW | 7 | 8 | 1964 | 5313 | 94 | 18 | 0.0225 | 0.0010 | 0.0195 | 0.0013 | 10.01 | <0.001 |
| S. latifolia C | 6 | 7 | 1131 | 4349 | 49 | 17 | 0.0197 | 0.0013 | 0.0190 | 0.0016 | 7.579 | <0.001 |
| S. latifolia SW | 6 | 3 | 2954 | 7096 | 23 | 2 | 0.0033 | 0.0007 | 0.0043 | 0.0007 | 1.230 | NS |
| S. latifolia SE | 9 | 9 | 2480 | 6495 | 176 | 11 | 0.0234 | 0.0007 | 0.0261 | 0.0006 | 20.50 | <0.001 |
| S. latifolia | 28 | 27 | 401 | 2690 | 40 | 56 | 0.0227 | 0.0059 | 0.0256 | 0.0032 | 4.28 | NS |
| S. dioica | 7 | 5 | 2985 | 6298 | 152 | 7 | 0.0259 | 0.0006 | 0.0239 | 0.0007 | 16.97 | <0.001 |
| S. diclinis | 6 | 4 | 2932 | 7436 | 40 | 1 | 0.0047 | 0.0002 | 0.0060 | 0.0002 | 4.09 | <0.05 |
| S. heuffelii | 4 | 3 | 2225 | 7768 | 96 | 4 | 0.0237 | 0.0002 | 0.0235 | 0.0002 | 12.14 | <0.001 |
S. latifolia samples are divided among the geographical areas of northwestern Europe (NW), central Europe (C), southwestern Europe (SW), and southeastern Europe (SE). Differences between regions and species in the number of base pairs analyzed are due to large, polymorphic indels. HKA tests compare the noncoding DNA diversity of DD44X with that of DD44Y. The diversity of DD44X was calibrated with that of DD44Y using the divergence of the two genes from the out-group S. vulgaris.
DD44X and DD44Y sequences from S. latifolia, S. dioica, S. heuffelii, and S. diclinis
N | Length (bp) | Segregating sites | Nucleotide diversity (π) | Nucleotide diversity (θw) | HKA test | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Species | DD44X | DD44Y | DD44X | DD44Y | DD44X | DD44Y | DD44X | DD44Y | DD44X | DD44Y | χ2 | P |
| S. latifolia NW | 7 | 8 | 1964 | 5313 | 94 | 18 | 0.0225 | 0.0010 | 0.0195 | 0.0013 | 10.01 | <0.001 |
| S. latifolia C | 6 | 7 | 1131 | 4349 | 49 | 17 | 0.0197 | 0.0013 | 0.0190 | 0.0016 | 7.579 | <0.001 |
| S. latifolia SW | 6 | 3 | 2954 | 7096 | 23 | 2 | 0.0033 | 0.0007 | 0.0043 | 0.0007 | 1.230 | NS |
| S. latifolia SE | 9 | 9 | 2480 | 6495 | 176 | 11 | 0.0234 | 0.0007 | 0.0261 | 0.0006 | 20.50 | <0.001 |
| S. latifolia | 28 | 27 | 401 | 2690 | 40 | 56 | 0.0227 | 0.0059 | 0.0256 | 0.0032 | 4.28 | NS |
| S. dioica | 7 | 5 | 2985 | 6298 | 152 | 7 | 0.0259 | 0.0006 | 0.0239 | 0.0007 | 16.97 | <0.001 |
| S. diclinis | 6 | 4 | 2932 | 7436 | 40 | 1 | 0.0047 | 0.0002 | 0.0060 | 0.0002 | 4.09 | <0.05 |
| S. heuffelii | 4 | 3 | 2225 | 7768 | 96 | 4 | 0.0237 | 0.0002 | 0.0235 | 0.0002 | 12.14 | <0.001 |
N | Length (bp) | Segregating sites | Nucleotide diversity (π) | Nucleotide diversity (θw) | HKA test | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Species | DD44X | DD44Y | DD44X | DD44Y | DD44X | DD44Y | DD44X | DD44Y | DD44X | DD44Y | χ2 | P |
| S. latifolia NW | 7 | 8 | 1964 | 5313 | 94 | 18 | 0.0225 | 0.0010 | 0.0195 | 0.0013 | 10.01 | <0.001 |
| S. latifolia C | 6 | 7 | 1131 | 4349 | 49 | 17 | 0.0197 | 0.0013 | 0.0190 | 0.0016 | 7.579 | <0.001 |
| S. latifolia SW | 6 | 3 | 2954 | 7096 | 23 | 2 | 0.0033 | 0.0007 | 0.0043 | 0.0007 | 1.230 | NS |
| S. latifolia SE | 9 | 9 | 2480 | 6495 | 176 | 11 | 0.0234 | 0.0007 | 0.0261 | 0.0006 | 20.50 | <0.001 |
| S. latifolia | 28 | 27 | 401 | 2690 | 40 | 56 | 0.0227 | 0.0059 | 0.0256 | 0.0032 | 4.28 | NS |
| S. dioica | 7 | 5 | 2985 | 6298 | 152 | 7 | 0.0259 | 0.0006 | 0.0239 | 0.0007 | 16.97 | <0.001 |
| S. diclinis | 6 | 4 | 2932 | 7436 | 40 | 1 | 0.0047 | 0.0002 | 0.0060 | 0.0002 | 4.09 | <0.05 |
| S. heuffelii | 4 | 3 | 2225 | 7768 | 96 | 4 | 0.0237 | 0.0002 | 0.0235 | 0.0002 | 12.14 | <0.001 |
S. latifolia samples are divided among the geographical areas of northwestern Europe (NW), central Europe (C), southwestern Europe (SW), and southeastern Europe (SE). Differences between regions and species in the number of base pairs analyzed are due to large, polymorphic indels. HKA tests compare the noncoding DNA diversity of DD44X with that of DD44Y. The diversity of DD44X was calibrated with that of DD44Y using the divergence of the two genes from the out-group S. vulgaris.
When S. latifolia samples were pooled from all geographical areas, an HKA test indicated that DD44X was not significantly more diverse than DD44Y (χ2 = 4.280, NS). This result was maintained when the HKA test was repeated using a single S. latifolia individual from each of the four geographical areas from which the samples were collected (χ2 = 3.642, NS). Overall, the diversity of DD44X was only 3.85 times greater than that of DD44Y (Table 2, Figure 2), far less than the 20-fold difference reported between SlX1 and SlY1 (Filatov et al. 2000) and only slightly higher than the threefold difference in diversity predicted due to the difference in ploidy between X- and Y-linked genes (Caballero 1995). Given the high population structure of DD44Y (see below) and the fact that only a subset of four of the nine geographical races was sampled, it is possible that we failed to capture the full diversity of DD44Y and so even this relatively modest difference is likely to be an overestimate.
Differentiation among species:
Permutation tests of FST among the four Elisanthe species indicated significant differentiation of both DD44X (FST = 0.30, P < 0.001) and DD44Y (FST = 0.93, P < 0.001) among species. While the FST values of DD44Y were uniformly high, the significant differentiation among species shown by DD44X could be attributed almost entirely to divergence between S. diclinis and the other three species (Table 3). Unlike the other Elisanthe species, S. diclinis is reproductively isolated from S. latifolia (Prentice 1978). S. diclinis is also geographically isolated from S. dioica and S. heuffelii and has an extremely low population size. Any of these attributes may have contributed to the relatively high value of FST for DD44X between S. diclinis and the other Elisanthe species.
Ninety-five-percent confidence intervals for pairwise genetic differentiation (FST) between Elisanthe species
S. latifolia | S. dioica | S. heuffelii | ||||
|---|---|---|---|---|---|---|
| Species | DD44X | DD44Y | DD44X | DD44Y | DD44X | DD44Y |
| S. dioica | 0.05 < FST < 0.17 | 0.82 < FST < 0.91 | — | — | — | — |
| S. heuffelii | 0.00 < FST < 0.18 | 0.85 < FST < 0.92 | 0.12 < FST < 0.25 | 0.96 < FST < 0.99 | — | — |
| S. diclinis | 0.28 < FST < 0.60 | 0.84 < FST < 0.91 | 0.37 < FST < 0.48 | 0.96 < FST < 0.99 | 0.28 < FST < 0.44 | 0.97 < FST < 1.00 |
S. latifolia | S. dioica | S. heuffelii | ||||
|---|---|---|---|---|---|---|
| Species | DD44X | DD44Y | DD44X | DD44Y | DD44X | DD44Y |
| S. dioica | 0.05 < FST < 0.17 | 0.82 < FST < 0.91 | — | — | — | — |
| S. heuffelii | 0.00 < FST < 0.18 | 0.85 < FST < 0.92 | 0.12 < FST < 0.25 | 0.96 < FST < 0.99 | — | — |
| S. diclinis | 0.28 < FST < 0.60 | 0.84 < FST < 0.91 | 0.37 < FST < 0.48 | 0.96 < FST < 0.99 | 0.28 < FST < 0.44 | 0.97 < FST < 1.00 |
Confidence intervals were calculated by bootstrapping across polymorphic loci.
Ninety-five-percent confidence intervals for pairwise genetic differentiation (FST) between Elisanthe species
S. latifolia | S. dioica | S. heuffelii | ||||
|---|---|---|---|---|---|---|
| Species | DD44X | DD44Y | DD44X | DD44Y | DD44X | DD44Y |
| S. dioica | 0.05 < FST < 0.17 | 0.82 < FST < 0.91 | — | — | — | — |
| S. heuffelii | 0.00 < FST < 0.18 | 0.85 < FST < 0.92 | 0.12 < FST < 0.25 | 0.96 < FST < 0.99 | — | — |
| S. diclinis | 0.28 < FST < 0.60 | 0.84 < FST < 0.91 | 0.37 < FST < 0.48 | 0.96 < FST < 0.99 | 0.28 < FST < 0.44 | 0.97 < FST < 1.00 |
S. latifolia | S. dioica | S. heuffelii | ||||
|---|---|---|---|---|---|---|
| Species | DD44X | DD44Y | DD44X | DD44Y | DD44X | DD44Y |
| S. dioica | 0.05 < FST < 0.17 | 0.82 < FST < 0.91 | — | — | — | — |
| S. heuffelii | 0.00 < FST < 0.18 | 0.85 < FST < 0.92 | 0.12 < FST < 0.25 | 0.96 < FST < 0.99 | — | — |
| S. diclinis | 0.28 < FST < 0.60 | 0.84 < FST < 0.91 | 0.37 < FST < 0.48 | 0.96 < FST < 0.99 | 0.28 < FST < 0.44 | 0.97 < FST < 1.00 |
Confidence intervals were calculated by bootstrapping across polymorphic loci.
When S. diclinis was excluded from the analysis, differentiation of DD44X between the remaining three species (S. latifolia, S. dioica, and S. heuffelii) was considerably lower than that of DD44Y (see Table 3). Ninety-five-percent confidence intervals of FST for DD44X and DD44Y did not overlap in any pairwise comparison other than those involving S. diclinis, indicating that the divergence of DD44Y among S. latifolia, S. dioica, and S. heuffelii is significantly higher than that of DD44X (Table 3). This difference could have been produced by higher interspecific gene flow of DD44X, creating new, shared polymorphic sites and destroying fixed differences through introgression and recombination. Alternatively, the difference in FST might have been produced by more rapid loss of ancestral polymorphic sites from DD44Y populations due to their smaller size, decreasing the number of polymorphisms shared among species and increasing the number of fixed differences.
Nielsen and Wakeley's (2001) Markov chain Monte Carlo approach supported the hypothesis that the lower differentiation of DD44X was due to higher gene flow among species. When the divergence of S. latifolia from S. dioica was analyzed, DD44Y best fit a model with low migration (
When the divergence of S. latifolia from S. diclinis was analyzed, both DD44X and DD44Y best fit models with low migration (DD44X:
Geographical structure:
Permutation tests of FST among S. latifolia samples collected from northwestern, central, southwestern, and southeastern areas indicated significant geographical structure in both DD44X (FST = 0.42, P < 0.0001) and DD44Y (FST = 0.87, P < 0.0001). However, while the FST values of DD44Y were uniformly high, the significant geographical structure shown by DD44X could be attributed almost entirely to the highly divergent southwestern sample (Table 4). Ninety-five-percent confidence intervals of FST for DD44X and DD44Y did not overlap in any pairwise comparison other than in those involving the southwestern sample, indicating that in other areas the population structure of DD44Y is significantly higher than that of DD44X (Table 4). The southwestern sample's high values of FST for DD44X appear to be due to a dramatic reduction of genetic diversity (Table 2). Tajima's test indicates that this may be due to a recent selective sweep affecting DD44X (D = −1.50, P < 0.05).
Ninety-five-percent confidence intervals for pairwise genetic differentiation (FST) between S. latifolia samples from different geographical areas
Northwestern Europe | Central Europe | Southwestern Europe | ||||
|---|---|---|---|---|---|---|
| FST | DD44X | DD44Y | DD44X | DD44Y | DD44X | DD44Y |
| Central Europe | 0.03 < FST < 0.22 | 0.64 < FST < 0.87 | — | — | — | — |
| Southwestern Europe | 0.33 < FST < 0.45 | 0.81 < FST < 0.93 | 0.65 < FST < 0.83 | 0.78 < FST < 0.95 | — | — |
| Southeastern Europe | 0.12 < FST < 0.22 | 0.80 < FST < 0.92 | 0.10 < FST < 0.21 | 0.74 < FST < 0.89 | 0.60 < FST < 0.74 | 0.83 < FST < 0.97 |
Northwestern Europe | Central Europe | Southwestern Europe | ||||
|---|---|---|---|---|---|---|
| FST | DD44X | DD44Y | DD44X | DD44Y | DD44X | DD44Y |
| Central Europe | 0.03 < FST < 0.22 | 0.64 < FST < 0.87 | — | — | — | — |
| Southwestern Europe | 0.33 < FST < 0.45 | 0.81 < FST < 0.93 | 0.65 < FST < 0.83 | 0.78 < FST < 0.95 | — | — |
| Southeastern Europe | 0.12 < FST < 0.22 | 0.80 < FST < 0.92 | 0.10 < FST < 0.21 | 0.74 < FST < 0.89 | 0.60 < FST < 0.74 | 0.83 < FST < 0.97 |
Confidence intervals were calculated by bootstrapping across polymorphic loci.
Ninety-five-percent confidence intervals for pairwise genetic differentiation (FST) between S. latifolia samples from different geographical areas
Northwestern Europe | Central Europe | Southwestern Europe | ||||
|---|---|---|---|---|---|---|
| FST | DD44X | DD44Y | DD44X | DD44Y | DD44X | DD44Y |
| Central Europe | 0.03 < FST < 0.22 | 0.64 < FST < 0.87 | — | — | — | — |
| Southwestern Europe | 0.33 < FST < 0.45 | 0.81 < FST < 0.93 | 0.65 < FST < 0.83 | 0.78 < FST < 0.95 | — | — |
| Southeastern Europe | 0.12 < FST < 0.22 | 0.80 < FST < 0.92 | 0.10 < FST < 0.21 | 0.74 < FST < 0.89 | 0.60 < FST < 0.74 | 0.83 < FST < 0.97 |
Northwestern Europe | Central Europe | Southwestern Europe | ||||
|---|---|---|---|---|---|---|
| FST | DD44X | DD44Y | DD44X | DD44Y | DD44X | DD44Y |
| Central Europe | 0.03 < FST < 0.22 | 0.64 < FST < 0.87 | — | — | — | — |
| Southwestern Europe | 0.33 < FST < 0.45 | 0.81 < FST < 0.93 | 0.65 < FST < 0.83 | 0.78 < FST < 0.95 | — | — |
| Southeastern Europe | 0.12 < FST < 0.22 | 0.80 < FST < 0.92 | 0.10 < FST < 0.21 | 0.74 < FST < 0.89 | 0.60 < FST < 0.74 | 0.83 < FST < 0.97 |
Confidence intervals were calculated by bootstrapping across polymorphic loci.
Among the S. latifolia samples from northwestern, central, and southeastern Europe, differentiation of DD44Y was extremely high while differentiation was much lower for DD44X. Nielsen and Wakeley's (2001) Markov chain Monte Carlo approach supported the hypothesis that the lower differentiation of DD44X was due to higher gene flow among populations (Table 5). In all cases, except divergence between the northwestern and southwestern samples, the data for DD44Y best fit a model with lower migration and a greater time since the most recent common ancestor rather than the corresponding model of best fit for DD44X. Likelihood-ratio tests comparing maximum likelihood when M = 0 with that when M > 0 failed to reject the null hypothesis of isolation since divergence for DD44Y in all pairwise comparisons among geographical samples (Table 5). For DD44X the pattern was more complex. Between northwestern and southwestern, between the northwestern and southeastern, and between central and southeastern, the null hypothesis of isolation since divergence was rejected, suggesting that substantial flow of DD44X, but not of DD44Y, has occurred between these areas. The southwestern S. latifolia sample is unusual in that both DD44X and DD44Y best fit models with low migration and long times of divergence from other areas.
Estimated pairwise scaled divergence times (
Northwestern Europe | Central Europe | Southwestern Europe | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DD44X | DD44Y | DD44X | DD44Y | DD44X | DD44Y | |||||||
\({\hat{T}}\) , \({\hat{M}}\) | \({\hat{T}}\) | \({\hat{M}}\) | \({\hat{T}}\) | \({\hat{M}}\) | \({\hat{T}}\) | \({\hat{M}}\) | \({\hat{T}}\) | \({\hat{M}}\) | \({\hat{T}}\) | \({\hat{M}}\) | \({\hat{T}}\) | \({\hat{M}}\) |
| Central Europe | 0.56 | 0.60 | 1.38 | 0.1 | — | — | — | — | — | — | — | — |
| Southwestern Europe | 2.02 | 0.36ab | 1.76 | 0.1 | 2.28 | 0.0 | 2.30 | 0.0 | — | — | — | — |
| Southeastern Europe | 0.34 | 1.22ab | 2.78 | 0.0 | 0.16 | 1.46ab | 2.08 | 0.0 | 0.92 | 0.34 | 3.6 | 0.0 |
Northwestern Europe | Central Europe | Southwestern Europe | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DD44X | DD44Y | DD44X | DD44Y | DD44X | DD44Y | |||||||
\({\hat{T}}\) , \({\hat{M}}\) | \({\hat{T}}\) | \({\hat{M}}\) | \({\hat{T}}\) | \({\hat{M}}\) | \({\hat{T}}\) | \({\hat{M}}\) | \({\hat{T}}\) | \({\hat{M}}\) | \({\hat{T}}\) | \({\hat{M}}\) | \({\hat{T}}\) | \({\hat{M}}\) |
| Central Europe | 0.56 | 0.60 | 1.38 | 0.1 | — | — | — | — | — | — | — | — |
| Southwestern Europe | 2.02 | 0.36ab | 1.76 | 0.1 | 2.28 | 0.0 | 2.30 | 0.0 | — | — | — | — |
| Southeastern Europe | 0.34 | 1.22ab | 2.78 | 0.0 | 0.16 | 1.46ab | 2.08 | 0.0 | 0.92 | 0.34 | 3.6 | 0.0 |
Log-likelihood test of
Log-likelihood test of
Estimated pairwise scaled divergence times (
Northwestern Europe | Central Europe | Southwestern Europe | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DD44X | DD44Y | DD44X | DD44Y | DD44X | DD44Y | |||||||
\({\hat{T}}\) , \({\hat{M}}\) | \({\hat{T}}\) | \({\hat{M}}\) | \({\hat{T}}\) | \({\hat{M}}\) | \({\hat{T}}\) | \({\hat{M}}\) | \({\hat{T}}\) | \({\hat{M}}\) | \({\hat{T}}\) | \({\hat{M}}\) | \({\hat{T}}\) | \({\hat{M}}\) |
| Central Europe | 0.56 | 0.60 | 1.38 | 0.1 | — | — | — | — | — | — | — | — |
| Southwestern Europe | 2.02 | 0.36ab | 1.76 | 0.1 | 2.28 | 0.0 | 2.30 | 0.0 | — | — | — | — |
| Southeastern Europe | 0.34 | 1.22ab | 2.78 | 0.0 | 0.16 | 1.46ab | 2.08 | 0.0 | 0.92 | 0.34 | 3.6 | 0.0 |
Northwestern Europe | Central Europe | Southwestern Europe | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DD44X | DD44Y | DD44X | DD44Y | DD44X | DD44Y | |||||||
\({\hat{T}}\) , \({\hat{M}}\) | \({\hat{T}}\) | \({\hat{M}}\) | \({\hat{T}}\) | \({\hat{M}}\) | \({\hat{T}}\) | \({\hat{M}}\) | \({\hat{T}}\) | \({\hat{M}}\) | \({\hat{T}}\) | \({\hat{M}}\) | \({\hat{T}}\) | \({\hat{M}}\) |
| Central Europe | 0.56 | 0.60 | 1.38 | 0.1 | — | — | — | — | — | — | — | — |
| Southwestern Europe | 2.02 | 0.36ab | 1.76 | 0.1 | 2.28 | 0.0 | 2.30 | 0.0 | — | — | — | — |
| Southeastern Europe | 0.34 | 1.22ab | 2.78 | 0.0 | 0.16 | 1.46ab | 2.08 | 0.0 | 0.92 | 0.34 | 3.6 | 0.0 |
Log-likelihood test of
Log-likelihood test of
Introgression and diversity:
Given the evidence for higher interspecific gene flow of DD44X than of DD44Y, it is possible that the difference in diversity between the two genes was due to the isolation of DD44Y rather than to a loss of diversity on the Y chromosome through background selection. In that case, we would expect the higher diversity of DD44X to be explicable through the acquisition of alleles from other species. Assuming that gene flow between two species is bidirectional, its main effect upon diversity is to convert fixed differences into shared polymorphisms.
The maximum contribution that could have been made by introgression to the diversity of DD44X in S. latifolia would have occurred if all of the polymorphic sites that S. latifolia shared with DD44X in other Silene species were generated by introgression. If, given this extreme assumption, the diversity of DD44X is still higher than that of DD44Y, then we can reject the hypothesis that the difference in diversity between the two genes is due to introgression. Within geographical races, a significant difference in diversity between DD44X and DD44Y persisted in S. latifolia even when all polymorphic sites shared with other species were removed (HKA tests, northwest: χ2 = 5.345, P < 0.05; southeast: χ2 = 11.583, P < 0.001). This enabled us to reject the hypothesis that the large differences in diversity between DD44X and DD44Y at the population level are entirely due to different levels of introgression following secondary contact between Silene species. However, the diversity of DD44X may still have been increased by higher gene flow between geographical races. When all polymorphic sites shared with other geographical races were removed from the British sample, an HKA test indicated that the diversity of DD44X was no longer significantly greater than that of DD44Y (χ2 = 1.39, NS).
DISCUSSION
The population structure of the Y-linked gene DD44Y is extremely high in S. latifolia, far higher than that of the homologous X-linked gene DD44X. The distribution of variation in DD44Y revealed major subdivisions coinciding with the “geographical races” of Mastenbroek and van Brederode (1986) and supporting the hypothesis that S. latifolia colonized Europe from several different glacial refugia in North Africa and Asia (Vellekoop et al. 1996). Owing to its greater population structure, the diversity of DD44Y within each geographical area was far lower than that of DD44X. However, when the genetic diversity of the two genes was compared at a species-wide level, the difference in diversity was not significant. Relatively high population structure on the Y chromosome is inconsistent with the species-wide selective sweeps of Y chromosome haplotypes predicted by the hitchhiking model of Rice (1987) and could have been maintained during the spread of beneficial mutations only if their spread was circumscribed by barriers to gene flow or by habitat specificity. On the other hand, high Y chromosome population structure is consistent with the background selection model of Charlesworth et al. (1995), since in this case recurrent selection of the least deleterious haplotypes can occur separately within each population.
An alternative to background selection is the hypothesis that Y-linked genes have undergone less gene flow than their X-linked homologs. The possibility that X-linked genes could achieve higher levels of gene flow than their Y-linked homologs in S. latifolia is suggested by the tendency for interspecific crosses between species within section Elisanthe to produce female-biased sex ratios (Taylor 1994). Given that few hybrid males are produced, the capacity of Y-linked genes to introgress is reduced relative to their X-linked homologs. Taylor (1994) also discovered evidence linking the female-biased sex ratios of crosses between S. latifolia and S. dioica to the presence of X-linked sex-ratio distorters and Y-linked suppressors of distortion. When two different species are crossed, an X-linked sex-ratio distorter is decoupled from its Y-linked restorer and a female-biased sex ratio results. Evidence that both distorters and restorers are polymorphic within S. latifolia (Taylor 1999) suggests that intraspecific gene flow among populations may also result in female-biased sex ratios. Sex-ratio distortion may therefore disfavor Y-linked genes when crosses occur between parents from different S. latifolia populations.
Evidence that the intraspecific flow of Y-linked genes among populations can be more restricted than that of autosomal genes has been provided by studies of the rodents Mus musculus and Microtus agrestis (Vanlerberghe et al. 1986; Jaarola et al. 1997). In these cases the lack of Y chromosome introgression has been attributed to reproductive incompatibility resulting from either chromosomal rearrangements on the Y chromosome (Jaarola et al. 1997) or the accumulation of incompatibility alleles in the Y chromosome's nonrecombining region (Tucker et al. 1992; Boissinot and Boursot 1997). In mammals, chromosomal rearrangements occur infrequently on the X chromosome (Wakefield and Graves 1996; Murphy et al. 1999), an observation that Ohno (1967) attributed to the disruption that rearrangements would cause to X-inactivation. If rearrangements cause reproductive incompatibility, then mammalian X-linked genes are therefore expected to experience fewer barriers to introgression than either Y-linked or autosomal genes (Wakefield and Graves 1996). Forsdyke (2000) speculated that this process could form the first stage of speciation, eventually leading to bidirectional sexual incompatibility due to further rearrangements on autosomes. In contrast, fixation of positively selected recessive and partially recessive alleles is expected to occur more rapidly on both X and Y chromosomes than on autosomes, due to the exposure to selection of recessive alleles on the sex chromosomes in hemizygous males (Charlesworth et al. 1987). Because epistasis between sex-linked and autosomal loci produces a disproportionate contribution of the sex chromosomes to hybrid sterility or hybrid lethality in the heterogametic (male) sex, this genic hypothesis predicts restricted gene flow of both X and Y chromosomes.
Initially, we investigated the hypothesis that the higher population structure of DD44Y was due to reduced gene flow by comparing the introgression of DD44X and DD44Y between S. latifolia and the closely related species S. dioica, S. heuffelii, and S. diclinis. While the sequences of DD44Y segregated clearly among the four different species, those of DD44X did not, indicating the possibility of introgression of DD44X among S. latifolia, S. dioica, and S. heuffelii. The Monte Carlo simulation models of Nielsen and Wakeley (2001) supported introgression of DD44X, but not of DD44Y, indicating that our DD44X data best fit models in which gene flow occurred following speciation while our DD44Y data best fit models in which there had been no gene flow since speciation. The only species showing no evidence of introgression according to the simulations was S. diclinis, a finding that adds weight to our conclusions, since S. diclinis is also the only one of these species that is reproductively isolated from the others (Prentice 1978).
Given the number of polymorphic sites shared among species, interspecific introgression could not account for the magnitude of differences in diversity between DD44X and DD44Y within either the British or the Transylvanian S. latifolia populations. However, they could be explained by the observation that DD44Y appears to have undergone less gene flow among the geographical races of S. latifolia than DD44X. Isolation followed by secondary contact and gene flow between the western and eastern S. latifolia races is consistent with the hypothesis that S. latifolia colonized Western and Eastern Europe from isolated refugia in North Africa and western Asia and that these separate waves of invasion were brought into contact by the clearance of land for agriculture (Vellekoop et al. 1996). Because our results suggest that the flow of Y-linked S. latifolia genes is more restricted than that of X-linked or autosomal genes, they agree more closely with the chromosomal than with the genic hypothesis. However, Ohno's (1967) chromosomal hypothesis invokes the X-inactivation mechanism of dosage compensation to explain the rarity of rearrangements on the mammalian X chromosome. Given that the relative frequency of rearrangements of the S. latifolia X and Y chromosomes has not been measured and that the method of dosage compensation used by S. latifolia is unknown, it is therefore not yet possible to ascertain whether Ohno's (1967) hypothesis is applicable in this case.
In conclusion, our findings confirm theoretical predictions that genes on the Y chromosome should be more affected by population structure than X-linked or autosomal genes (Laporte and Charlesworth 2002) and indicate that the dramatic losses of genetic diversity associated with suppression of recombination on the Y chromosome in Silene occur at the level of local populations rather than in entire species. Our findings also support the hypothesis that Y chromosome differentiation among incipient species precedes reproductive isolation of the entire genome, forming an early stage in the process of speciation (Forsdyke 2000).
Footnotes
Footnotes
Communicating editor: V. Michel
Acknowledgement
We thank Irina Goia of Babes Bolyai University, Romania, for assisting with the collection and identification of specimens from Romania. We also thank Deborah Charlesworth for her help and advice during the preparation of this article. Finally, we thank Michel Veuille and two anonymous reviewers for their insightful comments. This work was supported by a grant to D.A.F. from the Biotechnology and Biological Sciences Research Council.
References
Baker, H. G.,
Baker, H. G.,
Boissinot, S., and P. Boursot,
Braverman, J. M., R. R. Hudson, N. L. Kaplan, C. H. Langley and W. Stephan,
Caballero, A.,
Charlesworth, B.,
Charlesworth, B., and D. Charlesworth,
Charlesworth, B., J. A. Coyne and N. H. Barton,
Charlesworth, B., M. T. Morgan and D. Charlesworth,
Charlesworth, D., B. Charlesworth and M. T. Morgan,
Filatov, D. A.,
Filatov, D. A., and D. Charlesworth,
Filatov, D. A., V. Laporte, C. Vitte and D. Charlesworth,
Filatov, D. A., F. Moneger, I. Negrutiu and D. Charlesworth,
Forsdyke, D. R.,
Gordo, I., A. Navarro and B. Charlesworth,
Graves, J. A. M., and S. Shetty,
Hudson, R. R., M. Kreitman and M. Aguade,
Hudson, R. R., D. D. Boos and N. L. Kaplan,
Innan, H., and W. Stephan,
Jaarola, M., H. Tegelstrom and K. Fredga,
Kimura, M.,
Kumar, S., K. Tamura and M. Nei,
Laporte, V., and B. Charlesworth,
Laporte, V., D. A. Filatov, E. Kamau and D. Charlesworth,
Mastenbroek, O., and J. van Brederode,
Mastenbroek, O., P. Hogeweg, J. Heringa, G. J. Niemann and J. van Nigtevecht,
McCauley, D. E.,
McCauley, D. E.,
Moore, R. C., O. Kozyreva, S. Lebel-Hardenack, J. Siroky, R. Hobza et al.,
Murphy, W. J., S. Sun, Z. Q. Chen, J. Pecon-Slattery and S. J. O'Brien,
Nielsen, R., and J. Wakeley,
Prentice, H. C.,
Rice, W. R.,
Richards, C. M., S. Church and D. E. McCauley,
Sandbrink, J. M., L. J. N. M. Geurts, T. W. J. Gadella and J. van Brederode,
Slatkin, M., and T. Wiehe,
Stephan, W., L. Xing, D. A. Kirby and J. M. Braverman,
Tajima, F.,
Taylor, D. R.,
Taylor, D. R.,
Tucker, P. K., R. D. Sage, J. Warner, A. C. Wilson and E. M. Eicher,
Vanlerberghe, F., B. Dod, P. Boursot, M. Bellis and F. Bonhomme,
Vellekoop, P., J. B. Buntjer, J. W. Maas and J. van Brederode,
Wakefield, M. J., and J. A. M. Graves,
Watterson, G. A.,

{kind=link}
{kind=link}