





Abstract
The Mre11-Rad50-Nbs1 protein complex has emerged as a central player in the human cellular DNA damage response, and recent observations suggest that these proteins are at least partially responsible for the linking of DNA damage detection to DNA repair and cell cycle checkpoint functions. Mutations in scaANBS1, which encodes the apparent homolog of human nibrin in Aspergillus nidulans, inhibit growth in the presence of the antitopoisomerase I drug camptothecin. This article describes the selection and characterization of extragenic suppressors of the scaA1 mutation, with the aim of identifying other proteins that interfere with the pathway or complex in which the ScaA would normally be involved. Fifteen extragenic suppressors of the scaA1 mutation were isolated. The topoisomerase I gene can complement one of these suppressors. Synergistic interaction between the scaANBS1 and scsATOP1 genes in the presence of DNA-damaging agents was observed. Overexpression of topoisomerase I in the scaA1 mutant causes increased sensitivity to DNA-damaging agents. The scsATOP1 and the scaANBS1 gene products could functionally interact in pathways that either monitor or repair DNA double-strand breaks.
THE Mre11-Rad50-Nbs1 protein complex has emerged as a central player in the human cellular DNA damage response, and recent observations suggest that these proteins are at least partially responsible for the linking of DNA damage detection to DNA repair and cell cycle checkpoint functions (Petrini 1999). In humans, loss of the NBS1 gene is associated with the Nijmegen breakage syndrome, a rare autosomal recessive disorder that belongs to the group of inherited human chromosomal instability syndromes that includes Bloom syndrome, Fanconi’s anemia, and ataxia telangiectasia. All of these disorders are characterized by spontaneous chromosomal instability, immunodeficiency, and predisposition to cancer, but have distinct cytogenetic features and sensitivities to specific DNA-damaging agents (for reviews, see Digweed 1993; Shiloh 1997; Stewart and Elledge 2002). It has also recently been found that the NBS1 gene product, nibrin/p95, is part of the Mre11/Rad50 nuclear foci that form at sites of double-strand breaks (DSBs; Carney et al. 1998). The Rad50-Mre11-p95 complex possesses manganese-dependent single-stranded DNA endonuclease and 3′-5′ exonuclease activities (Trujillo et al. 1998). In humans and in yeast, Mre11, Rad50, and p95/Xrs2 assemble into large complexes. These proteins also appear to play a role in telomere maintenance. In addition, they are implicated in the cell’s checkpoint response to the presence of DSBs (D’Amours and Jackson 2002). In meiosis, these three proteins are required not only for resection of DSBs, but also for creation of the meiotic DSBs (Petrini 1999).
The filamentous fungus Aspergillus nidulans can normally tolerate high concentrations of the antitopoisomerase I drug camptothecin. The basic mechanism of action for camptothecin is well characterized (Froelich-Ammon and Osheroff 1995). Briefly, camptothecin generates replication-mediated DSBs, which in turn induce reversible or permanent cell cycle arrest in G2-M. Recently, we isolated camptothecin-sensitive mutants of A. nidulans, and the scaANBS1 gene, the apparent homolog of the human nibrin gene, was cloned by complementation of the camptothecin-sensitive scaA1 mutation (Bruschi et al. 2001). The scaA1 mutant does not undergo mitotic delay in the presence of DNA damage and displays abnormal nuclear morphology, suggesting that the germlings might undergo mitosis in the presence of unrepaired DNA damage, which would result in chromosome breakage, nondisjunction, or other symptoms of a catastrophic mitosis. Strains disrupted in the scaANBS1 gene showed increased sensitivity to other DNA-damaging agents, were delayed in polarization and septation, and displayed reduced viability in the presence of hydroxyurea, suggesting that the scaANBS1 is intimately involved in the DNA damage response in A. nidulans. Two-hybrid yeast assays have shown that ScaANBS1 interacts with the A. nidulans Mre11 homolog (Semighini et al. 2003).
A. nidulans strains
| Strains | Genotypes | References |
|---|---|---|
| R21 | pabaA1 yA2 | FGSC A234 |
| GR5 | pyrG89; wA3; pyroA4 | FGSC A773 |
| sca299 | pabaA1; yA2; scaA1 | Bruschi et al. (2001) |
| sca299-16 | pyrG89 pabaA1 yA2; scaA1 | Bruschi et al. (2001) |
| scs528-180 | pyrG89 pabaA1 yA2; scsA1 | This work |
| scs528-180-10 | pyrG89 pabaA1 yA2; scaA1; scsA1 | This work |
| A781 | wA2; nimA5 | FGSC |
| GG1 | pyrG89; ΔmusN; scsA1 | This work |
| GG2 | scaA::pyr-4; scsA1 | This work |
| ASH608 | chaA1; musN227 | Steven Harris’s collection |
| ASH610 | musN227; scsA1 | Steven Harris’s collection |
| AAH16 | pabaA1 yA2; ΔmusN | Steven Harris’s collection |
| T20 | pyrG89; pyroA4; wA3; scaA::pyr-4 | Bruschi et al. (2001) |
| Strains | Genotypes | References |
|---|---|---|
| R21 | pabaA1 yA2 | FGSC A234 |
| GR5 | pyrG89; wA3; pyroA4 | FGSC A773 |
| sca299 | pabaA1; yA2; scaA1 | Bruschi et al. (2001) |
| sca299-16 | pyrG89 pabaA1 yA2; scaA1 | Bruschi et al. (2001) |
| scs528-180 | pyrG89 pabaA1 yA2; scsA1 | This work |
| scs528-180-10 | pyrG89 pabaA1 yA2; scaA1; scsA1 | This work |
| A781 | wA2; nimA5 | FGSC |
| GG1 | pyrG89; ΔmusN; scsA1 | This work |
| GG2 | scaA::pyr-4; scsA1 | This work |
| ASH608 | chaA1; musN227 | Steven Harris’s collection |
| ASH610 | musN227; scsA1 | Steven Harris’s collection |
| AAH16 | pabaA1 yA2; ΔmusN | Steven Harris’s collection |
| T20 | pyrG89; pyroA4; wA3; scaA::pyr-4 | Bruschi et al. (2001) |
FGSC, Fungal Genetics Stock Center.
A. nidulans strains
| Strains | Genotypes | References |
|---|---|---|
| R21 | pabaA1 yA2 | FGSC A234 |
| GR5 | pyrG89; wA3; pyroA4 | FGSC A773 |
| sca299 | pabaA1; yA2; scaA1 | Bruschi et al. (2001) |
| sca299-16 | pyrG89 pabaA1 yA2; scaA1 | Bruschi et al. (2001) |
| scs528-180 | pyrG89 pabaA1 yA2; scsA1 | This work |
| scs528-180-10 | pyrG89 pabaA1 yA2; scaA1; scsA1 | This work |
| A781 | wA2; nimA5 | FGSC |
| GG1 | pyrG89; ΔmusN; scsA1 | This work |
| GG2 | scaA::pyr-4; scsA1 | This work |
| ASH608 | chaA1; musN227 | Steven Harris’s collection |
| ASH610 | musN227; scsA1 | Steven Harris’s collection |
| AAH16 | pabaA1 yA2; ΔmusN | Steven Harris’s collection |
| T20 | pyrG89; pyroA4; wA3; scaA::pyr-4 | Bruschi et al. (2001) |
| Strains | Genotypes | References |
|---|---|---|
| R21 | pabaA1 yA2 | FGSC A234 |
| GR5 | pyrG89; wA3; pyroA4 | FGSC A773 |
| sca299 | pabaA1; yA2; scaA1 | Bruschi et al. (2001) |
| sca299-16 | pyrG89 pabaA1 yA2; scaA1 | Bruschi et al. (2001) |
| scs528-180 | pyrG89 pabaA1 yA2; scsA1 | This work |
| scs528-180-10 | pyrG89 pabaA1 yA2; scaA1; scsA1 | This work |
| A781 | wA2; nimA5 | FGSC |
| GG1 | pyrG89; ΔmusN; scsA1 | This work |
| GG2 | scaA::pyr-4; scsA1 | This work |
| ASH608 | chaA1; musN227 | Steven Harris’s collection |
| ASH610 | musN227; scsA1 | Steven Harris’s collection |
| AAH16 | pabaA1 yA2; ΔmusN | Steven Harris’s collection |
| T20 | pyrG89; pyroA4; wA3; scaA::pyr-4 | Bruschi et al. (2001) |
FGSC, Fungal Genetics Stock Center.
This article describes the selection and characterization of extragenic suppressors of the scaA1 mutation, with the aim of identifying other proteins that interfere with the pathway or complex in which the ScaA would normally be involved. To facilitate cloning of the suppressor genes, we searched for extragenic suppressor mutations that also confer a selectable phenotype, such as heat sensitivity (hs-). Here, we show that the topo-isomerase I gene can complement the scsA1 suppressor and demonstrate that the scaANBS1 and the scsATOP1 genes display synergistic interaction in the presence of DNA-damaging agents. Furthermore, we also show that the overexpression of the topoisomerase I increases the sensitivity of A. nidulans to DNA-damaging agents in the background of the scaA1 mutant. We suggest that the scsATOP1 and the scaANBS1 gene products functionally interact in pathways that either monitor or repair DNA DSBs.
MATERIALS AND METHODS
Aspergillus strains and growth methods: All strains of A. nidulans are derived from a haploid nucleus and therefore are isogenic except for differences induced by mutagenic treatment (Pontecorvo et al. 1953). The strains used are described in Table 1. Media used were complete medium (YAG: 2% glucose, 0.5% yeast extract, 2% agar, and trace elements) and YUU, which is YAG medium supplemented with 1.2 g/liter each of uracil and uridine; YG medium has the same composition, but without agar or minimal medium (1% glucose, nitrate salts, trace elements, 2% agar, pH 6.5). Trace elements, vitamins, and nitrate salts are described in the appendix to Kafer (1977). Standard genetic techniques for A. nidulans (Kafer 1977) were used for all constructions. For the hs- scaA- mutants, the permissive temperature for growth is 28° and the restrictive temperature is 44°.
For experiments using light microscopy, strains were grown on sterile coverslips in petri dishes. Sterile glass coverslips were placed on the bottom of the petri dish and overlaid with appropriately supplemented liquid YG medium containing ∼106/ml conidia of the relevant strain. The conidia settled to the bottom of the petri dish and adhered tightly to the coverslips during germination.
Mutagenesis and isolation of revertants: Conidia from strain sca299 were mutagenized at a concentration of 1 × 106/ml in sterile distilled water using UV light as a mutagen (3.85 J/m2/sec). Approximately 70% of the conidia were killed under these conditions. Aliquots of 0.1 ml were spread on YAG plates that were incubated at 28° for 3 days. Revertants that grew were isolated and subsequently retested for growth at 28° and 44°.
Staining and microscopy: For nuclear staining of the germlings, asexual spores (conidia) were inoculated onto coverslips. After 8-12 hr incubation at 28° or 44°, coverslips with adherent germlings were transferred to fixative (3.7% formaldehyde, 50 mm sodium phosphate buffer pH 7.0, 0.2% Triton X-100) for 30 min at room temperature. Then they were briefly rinsed with PBS buffer (140 mm NaCl, 2 mm KCl, 10 mm NaHPO4, 1.8 mm KH2PO4, pH 7.4) and incubated for 5 min in a solution with 100 ng/ml of 4′,6-diamidino-2-phenylindole (DAPI; Sigma Chemical, St. Louis) and 100 ng/ml of calcofluor (fluorescent brightener, Sigma Chemical). After incubation with the dyes, germlings were washed with PBS buffer for 5 min at room temperature and then rinsed in distilled water and mounted in Citifluor. The material was photographed using a Zeiss epifluorescence microscope.
The reciprocal shift experiments to determine the stage of interphase at which the scs528-180 strain arrests were performed as described (Bergen et al. 1984). Conidia from strain scs528-180 were first incubated at 44° for 7 hr by inoculation in YG medium onto coverslips. The coverslips were either fixed (as a control) or transferred to 44° for 3 hr or to YG plus 25 mm hydroxyurea (HU) for 10 min (at 44°) and then shifted to 28° with HU for 3 hr. For the reciprocal experiments, conidia were arrested in S phase by inoculating onto coverslips in rich media containing 25 mm HU and incubated for 7 hr at 28°. The coverslips were fixed (as a control) or transferred to 28° YG medium in the absence of HU or shifted to 44° without HU. Coverslips were stained as described above and the number of nuclei per germling was determined.
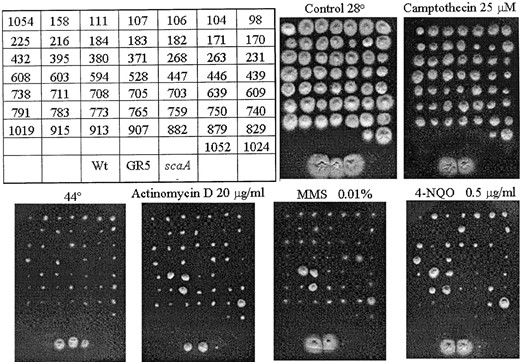
—Extragenic suppressors of the scaA1 mutation. A total of 56 extragenic suppressors with an associated heat-sensitivity defect were isolated, and 51 of them are shown here. They were grown on YUU at 28°, YUU + camptothecin (top), YUU at 44°, YUU + actinomycin D, YUU + MMS, and YUU + 4-NQO (bottom) for 72 hr. Only 15 extragenic suppressors were analyzed to determine whether the conditional hs- mutation in the mutants cosegregated with the scs mutations (see Table 2).
Molecular cloning of the scsATOP1 gene: The heat sensitivity and uridine and uracil requirements of strain sca528-180 were complemented by DNA-mediated transformation using a genomic library in the vector AMA1-pRG3, which contains the pyr-4 gene of Neurospora crassa and an autonomous replicating sequence (Ballance et al. 1983; Osherov and May 2000). Transformations were performed as described by Osmani et al. (1987). Approximately 4000 transformants were transferred to the restrictive temperature (44°) for 5 days, yielding 2 rescued transformants. Genomic DNA was prepared from these transformant colonies and used to transform electrocompetent Escherichia coli cells. Approximately 20 ampicillin-resistant colonies were analyzed for the recovery of the plasmid. More than 50% of the rescued plasmids contained identical or overlapping inserts that were then used to recapitulate the rescue of the original scsA- mutant. Complementing activity within the cloned insert was identified by randomly mutagenizing the rescuing plasmid with a transposon (GPS-1 system; New England Biolabs, Beverly, MA). Individual transposon-tagged clones were transformed into the scs mutant of interest. Plasmids that failed to rescue the mutant at the restrictive temperature were assumed to have insertions disrupting the complementing gene. These were sequenced using primers unique for the transposon ends. The DNA insert was fully sequenced using gene-specific primers and BigDye terminator cycle sequencing (Applied Biosystems, Foster City, CA).
RNA isolation: A total of 1 × 108 conidia/ml were used to inoculate liquid cultures that were incubated in a reciprocal shaker at 37° for either 8 or 12 hr. Mycelia were aseptically transferred to a fresh YG medium in the absence or presence of 0.025% of methyl methanesulfonate (MMS) for 3 hr. A. nidulans mycelia were harvested by filtration through a Whatman filter no. 1, washed thoroughly with sterile water, quickly frozen in liquid nitrogen, and disrupted by grinding, and total RNA was extracted with Trizol (Life Technologies). RNAse-free DNAse treatment was done as previously described by Semighini et al. (2002).
RT-PCR reactions: All the PCR and RT-PCR reactions were performed using an ABI Prism 7700 sequence detection system (Perkin-Elmer Applied Biosystems, Norwalk, CT). Taq-Man EZ RT-PCR and PCR kits (Applied Biosystems) were used for RT-PCR and PCR reactions, respectively. The reactions and calculations were performed according to Semighini et al. (2002). The following primers and probes (Applied Biosystems) were used in this work: for β-tubulin (tubC), tubCfw, 5′-CGGAAACTGGCCGTCAATAT-3′; tubCrv, 5′-GGGCAAACCCGACCATAAA-3′; tubCprobe, 6FAM-5′-TCCCTTCCCGCG GTTGCATTT-TAMRA; for uvsCRAD51, uvsCfw, 5′-TGCTCGACGAAGCGAACTTA-3′; uvsCrv, 5′-CACCTCCCAGAAGCGTA TCC-3′; uvsCprobe, TET-5′-CAATCACAACGGGCTCCAAGCAGC-3′-TAMRA; and for top1, topo1fw, 5′-CAACTTCCGAGTC GAGCCTC-3′; topo1rv, 5′-GCGACCTGTCTTAGGGTGCT; topo1probe, 5′-TET-5′-TTCCCTTTTCCGCGGTCGTGG-3′-TAMRA (6FAM, 6-carboxyfluorescein; TET, 6-carboxy-4,7,2′,7′-tetrachlorofluorescein; and TAMRA, 6-carboxy-N,N,N′,N′-tetramethylrhodamine).
RESULTS
Isolation of cptR revertants of the camptothecin-sensitive scaA1 mutation: Revertants of scaA1 camptothecin sensitivity were isolated as follows. Conidia of the scaA1 mutant were mutagenized using UV light and spread on complete medium plates with 25 μm camptothecin and incubated at 28° for 3 days. The camptothecin-resistant (cptR) revertants that grew were isolated and retested for growth on camptothecin at 28°. A total of 1064 revertants were selected for their ability to grow in the presence of 25 μm camptothecin, a concentration at which scaA1 exhibits little growth. We screened these revertants for suppressor mutations that also confer a selectable phenotype, such as heat sensitivity. The rationale was to facilitate the direct cloning of the suppressor genes by DNA-mediated transformation and complementation of their heat sensitivity. Fifty-six of the revertants were hs- (Figure 1); these revertants were first tested to determine whether the mutations were caused by back mutations in scaANBS1 or by extragenic suppressor mutations in another gene. Each revertant was crossed with a scaA+ strain, GR5. The appearance of camptothecin-sensitive microcolonies among the progeny of a cross plated on YAG + camptothecin at 28° indicated that the camptothecin resistance of the original strain was caused by a second-site extragenic suppressor mutation. Reversion was caused by a second-site suppressor mutation in all 56 hs- extragenic revertants (data not shown); these suppressors were designated scs (for supressor of camptothecin sensitivity) mutations.
Analysis of progeny from crosses of extragenic suppressor strains to wild type
| Wild type × suppressor | CPTR hs+a | CPTR hs- | CPTS hs+b | CPTS hs- |
|---|---|---|---|---|
| GR5 × scs528 | 96 | 16 | 25 | 0 |
| GR5 × scs603 | 110 | 13 | 32 | 0 |
| GR5 × scs639 | 92 | 20 | 45 | 0 |
| GR5 × scs705 | 93 | 19 | 44 | 0 |
| GR5 × scs706 | 68 | 54 | 27 | 0 |
| GR5 × scs708 | 74 | 67 | 15 | 0 |
| GR5 × scs711 | 60 | 49 | 47 | 0 |
| GR5 × scs738 | 90 | 27 | 39 | 0 |
| GR5 × scs752 | 54 | 68 | 33 | 0 |
| GR5 × scs773 | 81 | 48 | 27 | 0 |
| GR5 × scs792 | 50 | 78 | 28 | 0 |
| GR5 × scs829 | 63 | 60 | 32 | 0 |
| GR5 × scs1019 | 87 | 28 | 41 | 0 |
| GR5 × scs1024 | 54 | 63 | 39 | 0 |
| GR5 × scs1054 | 95 | 17 | 43 | 0 |
| Wild type × suppressor | CPTR hs+a | CPTR hs- | CPTS hs+b | CPTS hs- |
|---|---|---|---|---|
| GR5 × scs528 | 96 | 16 | 25 | 0 |
| GR5 × scs603 | 110 | 13 | 32 | 0 |
| GR5 × scs639 | 92 | 20 | 45 | 0 |
| GR5 × scs705 | 93 | 19 | 44 | 0 |
| GR5 × scs706 | 68 | 54 | 27 | 0 |
| GR5 × scs708 | 74 | 67 | 15 | 0 |
| GR5 × scs711 | 60 | 49 | 47 | 0 |
| GR5 × scs738 | 90 | 27 | 39 | 0 |
| GR5 × scs752 | 54 | 68 | 33 | 0 |
| GR5 × scs773 | 81 | 48 | 27 | 0 |
| GR5 × scs792 | 50 | 78 | 28 | 0 |
| GR5 × scs829 | 63 | 60 | 32 | 0 |
| GR5 × scs1019 | 87 | 28 | 41 | 0 |
| GR5 × scs1024 | 54 | 63 | 39 | 0 |
| GR5 × scs1054 | 95 | 17 | 43 | 0 |
CPTR, camptothecin resistant; hs-, heat sensitive.
CPTS, camptothecin sensitive; hs+, heat resistant.
Analysis of progeny from crosses of extragenic suppressor strains to wild type
| Wild type × suppressor | CPTR hs+a | CPTR hs- | CPTS hs+b | CPTS hs- |
|---|---|---|---|---|
| GR5 × scs528 | 96 | 16 | 25 | 0 |
| GR5 × scs603 | 110 | 13 | 32 | 0 |
| GR5 × scs639 | 92 | 20 | 45 | 0 |
| GR5 × scs705 | 93 | 19 | 44 | 0 |
| GR5 × scs706 | 68 | 54 | 27 | 0 |
| GR5 × scs708 | 74 | 67 | 15 | 0 |
| GR5 × scs711 | 60 | 49 | 47 | 0 |
| GR5 × scs738 | 90 | 27 | 39 | 0 |
| GR5 × scs752 | 54 | 68 | 33 | 0 |
| GR5 × scs773 | 81 | 48 | 27 | 0 |
| GR5 × scs792 | 50 | 78 | 28 | 0 |
| GR5 × scs829 | 63 | 60 | 32 | 0 |
| GR5 × scs1019 | 87 | 28 | 41 | 0 |
| GR5 × scs1024 | 54 | 63 | 39 | 0 |
| GR5 × scs1054 | 95 | 17 | 43 | 0 |
| Wild type × suppressor | CPTR hs+a | CPTR hs- | CPTS hs+b | CPTS hs- |
|---|---|---|---|---|
| GR5 × scs528 | 96 | 16 | 25 | 0 |
| GR5 × scs603 | 110 | 13 | 32 | 0 |
| GR5 × scs639 | 92 | 20 | 45 | 0 |
| GR5 × scs705 | 93 | 19 | 44 | 0 |
| GR5 × scs706 | 68 | 54 | 27 | 0 |
| GR5 × scs708 | 74 | 67 | 15 | 0 |
| GR5 × scs711 | 60 | 49 | 47 | 0 |
| GR5 × scs738 | 90 | 27 | 39 | 0 |
| GR5 × scs752 | 54 | 68 | 33 | 0 |
| GR5 × scs773 | 81 | 48 | 27 | 0 |
| GR5 × scs792 | 50 | 78 | 28 | 0 |
| GR5 × scs829 | 63 | 60 | 32 | 0 |
| GR5 × scs1019 | 87 | 28 | 41 | 0 |
| GR5 × scs1024 | 54 | 63 | 39 | 0 |
| GR5 × scs1054 | 95 | 17 | 43 | 0 |
CPTR, camptothecin resistant; hs-, heat sensitive.
CPTS, camptothecin sensitive; hs+, heat resistant.
The segregants from the crosses between 15 revertants and strain GR5 were also analyzed to determine whether the conditional hs- mutation in the revertants cosegregated with the scs mutations. The presence of cptR, hs- progeny signified a lack of cosegregation between the scs mutation and the conditional mutation. However, the conditional phenotypes cosegregated with the extragenic suppressor mutations in all 15 mutants (Table 2). This shows that these extragenic suppressor mutations cause the associated conditional phenotypes.
In addition to camptothecin, the sca299 strain also displays sensitivity to other genotoxic agents, such as actinomycin D, MMS, and 4-nitroquinoline oxide (4-NQO; Bruschi et al. 2001). Thus, the revertants were tested for resistance to these DNA-damaging agents (Figure 1). The revertants scs708, -711, -773, and 829 showed actinomycin, MMS, and 4-NQO resistance. In these mutants, the heat-sensitivity phenotype cosegregated with extragenic suppressor mutations (Table 1) and the resistance to the DNA-damaging agents (data not shown). In addition, several revertants (such as scs104, -183, and -608) also showed 4-NQO resistance (Figure 1).
To determine whether the mutations were allelic, each of the original scaA1, scs strains was crossed to each of the other extragenic suppressor strains. First, each scaA1 scs mutant was crossed to GR5 (wA1, pyrG89, pyroA1) to generate a pyrG89 scaA1 scs recombinant with auxotrophic and color markers complementary to and suitable for crossing with the original scaA1, scs strains. Fifteen individual strains carrying an scs mutation were then crossed with each other. However, since none of the strains crossed, it was not possible to define linkage groups among these 15 scs mutants. As mentioned above, the revertants could be backcrossed to the wild-type strain GR5. Accordingly, these results strongly suggest that the presence of two mutated copies of the scaA1 allele causes sexual sterility in A. nidulans. Instead of making further attempts to establish the linkage groups in these mutants by using either diploid formation or heterokaryon complementation, we decided to clone the scs genes that complement some of these mutants.
The topoisomerase I gene (scsATOP1) complements the extragenic suppressor scs528: As an initial step in the characterization of the scs mutants, we cloned the scsA gene by complementing the heat sensitivity and the pyrG- deficiency of the scs528-180 (scaA+ scsA-) mutant with a plasmid from the AMA1 genomic library (Figure 2A). Two transformants were identified that were no longer heat sensitive for growth. We then used transposon-mediated insertional inactivation of the rescued plasmid to map the complementing gene (see materials and methods). Sequences derived from transposon-inactivated genes were used to test for homology with known genes in the data banks. The two transformants have the same complementing DNA fragment that harbors the gene encoding A. nidulans topoisomerase I. When we compared the DNA sequence of the wild-type scsATOP1 to the A. nidulans topoisomerase I sequence deposited in the database (Van Dross et al. 1997), differences were noted at the following positions (downstream of the predicted translational start site): 880, AGG [amino acid (aa) 227, from lysine to arginine]; 892, GAT (aa 231, from glycine to aspartate); 1204, TTC (aa 335, from serine to phenylalanine); 1787, GAT (aa 529, from aspartate to aspartate); 2247, CGT (aa 666, from valine to arginine); 2256, CAC (aa 668, from aspartate to histidine); and 2613, GCT (aa 764, from alanine to alanine). From these differences, two are conservative substitutions while five are nonconservative substitutions. These differences could represent polymorphisms in the A. nidulans topoisomerase I gene.
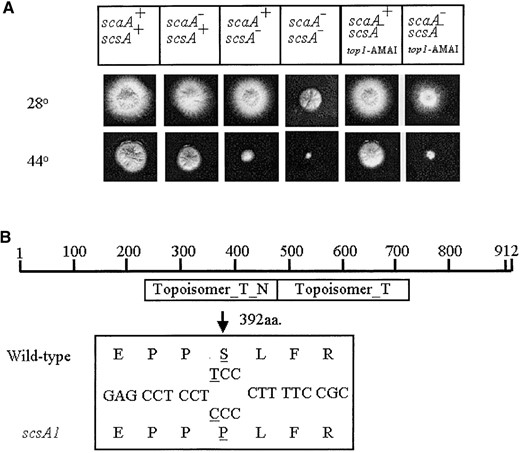

—Growth phenotypes of scaA1 scsA1 transformed with the pRG3-AMA1-top1. (A) Strains were grown for 72 hr at 28° or 44° in YAG medium. (B) Diagram of the scsATOP1 gene; Topoisomer_T_N indicates the domain that is responsible for DNA binding, whereas the Topoisomer_T denotes the domain that is responsible for the catalytic activity. The bottom box shows a small region of the wild type and mutated topoisomerase I with the respective nucleotide and predicted protein sequences (the single nucleotide and the corresponding amino acid changes are underlined). (C) Conidia from the wild type and the scsA- strains were germinated at 28° and 44° for 10 hr. The germlings were fixed and stained with DAPI.
Because the heat-resistant scs528 transformants described above could reflect complementation by either wild-type scsA sequences or an extragenic suppressor of scsA, we used a 4-kb DNA fragment containing the scsATOP1 gene to target integration of the pyr-4+ selectable marker into an scsA- strain. Integration of the circular scsA DNA presumably would occur by a single crossover event at the scsA locus, thus replacing the mutant allele with a wild-type copy of scsA. Many pyr-4+ transformants were heat resistant, and two were crossed to a wild-type strain to determine whether integration was at the scsA locus or at another (suppressor) locus. If the gene replacement occurred at the scsA locus, a cross back to a wild-type strain should produce only heat-resistant progeny. On the other hand, if the gene represented an extragenic suppressor, which had integrated at its own locus in the genome, then a cross back to wild type should still segregate progeny with the original heat-sensitive mutation. In both strains, we found no heat-sensitive progeny from a total of 300 segregants. We therefore conclude that we have complemented the heat-sensitive phenotype with the wild-type copy of the scsA gene and not with a suppressor.
The genomic sequence of the scsA gene from the wild type and suppressor scsA- strains was determined. A single base (T-to-C) transition was identified in the scaA- mutant 1374 bases downstream of the predicted translational start site. This transition changes the predicted serine residue (aa 392) into a proline residue. Since this mutation is located at the topoisomerase I DNA-binding domain (Figure 2B), it could potentially impair DNA binding, causing a decrease in the formation of the ternary DNA-topoisomerase I-camptothecin complex. This could help to explain the camptothecin resistance in the scaA1 scsA1 mutant strain.
We also cloned the genes that complement the hs- scs603, -708, and -711. Gene complementation and additional two-step gene replacement experiments showed that scs528 and -603 suppressors correspond to the same locus, scsA, while scs708 and -711 are alleles of a second locus designated scsB. Besides the camptothecin sensitivity, the latter two suppressors can suppress the actinomycin D, MMS, and 4-NQO sensitivity caused by the scaA1 mutation. Cloning of the genes that complement these suppressors resulted in the identification of peptide release factor (eRF). The A. nidulans eRF gene has been recently deposited in the National Center for Biotechnology Information. Peptide release factor is responsible for recognizing the stop codon and for transmitting the signal from the mRNA stop codon occupying the ribosomal A site to the peptidyl transferase center, where it is thought to trigger the hydrolysis of peptidyl-tRNA (Kisselev and Buckingham 2000). As shown by Bruschi et al. (2001), the genomic sequence of the scaA gene from the mutant scaA- is a single base (T-to-G) transversion that was identified in the scaA- mutant 749 bases downstream of the predicted translational start site of scaA. This transversion changes the predicted leucine residue (aa 249) into a stop codon. This alteration should result in a truncated protein less than half the length of the wild-type protein. Thus, the mechanism of suppression by the peptide release factor could be failure to recognize the stop codon because of informational suppression. To our knowledge, this is the first time such a mechanism of informational suppression by peptide release factor has been described in eukaryotes.
Double reciprocal shift assay of the strain scaA1 scsA1
| Initial conditions | Shift | ||||
|---|---|---|---|---|---|
| Treatmenta | Temperature | Time (hr) | Temperature | Time (hr) | % with two nucleib |
| Downshifts | |||||
| A | 44° | 7 | No shift | 0 | 0 |
| B | 44° | 7 | 28° | 3 | 66 |
| C | 44° | 7 | 28°+ 25 mm HUc | 3 | 54 |
| Upshifts | |||||
| D | 28° | 7 | No shift | 0 | 0 |
| E | 28°+ 25 mm HU | 7 | 28° | 3 | 64 |
| F | 28°+ 25 mm HU | 7 | 44° | 3 | 0 |
| Initial conditions | Shift | ||||
|---|---|---|---|---|---|
| Treatmenta | Temperature | Time (hr) | Temperature | Time (hr) | % with two nucleib |
| Downshifts | |||||
| A | 44° | 7 | No shift | 0 | 0 |
| B | 44° | 7 | 28° | 3 | 66 |
| C | 44° | 7 | 28°+ 25 mm HUc | 3 | 54 |
| Upshifts | |||||
| D | 28° | 7 | No shift | 0 | 0 |
| E | 28°+ 25 mm HU | 7 | 28° | 3 | 64 |
| F | 28°+ 25 mm HU | 7 | 44° | 3 | 0 |
One hundred germlings were counted for each experiment and the percentage with one nuclear division was determined.
Percentage of cells completing at least one nuclear division.
Before transferring the germlings to treatment C (28°+ HU), the germlings were incubated in the presence of 25 mm HU for 10 min at 44°.
Double reciprocal shift assay of the strain scaA1 scsA1
| Initial conditions | Shift | ||||
|---|---|---|---|---|---|
| Treatmenta | Temperature | Time (hr) | Temperature | Time (hr) | % with two nucleib |
| Downshifts | |||||
| A | 44° | 7 | No shift | 0 | 0 |
| B | 44° | 7 | 28° | 3 | 66 |
| C | 44° | 7 | 28°+ 25 mm HUc | 3 | 54 |
| Upshifts | |||||
| D | 28° | 7 | No shift | 0 | 0 |
| E | 28°+ 25 mm HU | 7 | 28° | 3 | 64 |
| F | 28°+ 25 mm HU | 7 | 44° | 3 | 0 |
| Initial conditions | Shift | ||||
|---|---|---|---|---|---|
| Treatmenta | Temperature | Time (hr) | Temperature | Time (hr) | % with two nucleib |
| Downshifts | |||||
| A | 44° | 7 | No shift | 0 | 0 |
| B | 44° | 7 | 28° | 3 | 66 |
| C | 44° | 7 | 28°+ 25 mm HUc | 3 | 54 |
| Upshifts | |||||
| D | 28° | 7 | No shift | 0 | 0 |
| E | 28°+ 25 mm HU | 7 | 28° | 3 | 64 |
| F | 28°+ 25 mm HU | 7 | 44° | 3 | 0 |
One hundred germlings were counted for each experiment and the percentage with one nuclear division was determined.
Percentage of cells completing at least one nuclear division.
Before transferring the germlings to treatment C (28°+ HU), the germlings were incubated in the presence of 25 mm HU for 10 min at 44°.
The scaANBS1 and the scsATOP1 genes showed synergistic interaction in the presence of DNA-damaging agents: When scsA1 hyphae were incubated at 44° for 12 hr and stained with DAPI, nuclei arrested in interphase were observed (Figure 2C, right). In contrast, wild-type germlings normally undergo one or two nuclear divisions under these conditions (Figure 2C, left). The scsA1 block is reversible by shifting to 28° until ∼12 hr of incubation at 44°. To determine at which stage of interphase arrest occurs, reciprocal shift assays were performed (Table 3). Conidia were inoculated into YG medium at 44° and incubated 7 hr at 44° and then shifted to YG medium at 28° in either the absence or the presence of 25 mm HU for 3 hr. After shift to 28°, ∼60% of nuclei underwent mitotic division, regardless of the presence of HU (Table 3). This suggests that the cells exit from G1- or G2-phase arrest at 44°, traverse mitosis and/or G1 phase, and arrest in S phase in the presence of HU. As a control, the reciprocal experiment was performed, in which conidia were inoculated in YG medium at 28° in either the absence or the presence of 25 mm HU for 7 hr and then shifted to YG medium at 28° or 44° for 3 hr. After shift to 28°, ∼60% of the nuclei of the treatment without HU underwent mitotic division (Table 3). However, no germlings completed one nuclear division in the treatment F (28°+ HU; Table 3). These results indicate that the scsA1 mutant is blocked in G2 at the restrictive temperature.
The scsA1 mutation was isolated by screening for suppressors of scaA1 that could grow in the presence of camptothecin. As mentioned above, the scsA- mutation does not confer resistance to other DNA-damaging agents, such as actinomycin D, MMS, and 4-NQO. We also verified that scsA1 mutants could grow in the presence of hydroxyurea and bleomycin to the same extent as the wild-type strain. However, the concentrations of MMS and 4-NQO in Figure 1 were too high to reveal possible interactions between the scaA- and scsA- mutations. By decreasing the concentrations of MMS and 4-NQO, we noted that the scaANBS1 and the scsATOP1 genes showed synergistic interaction in the presence of DNA-damaging agents (Figure 3). In addition, the double mutant is also more sensitive to bleomycin than is the suppressor alone (scaA+ scsA-) or the scaA1 mutant.
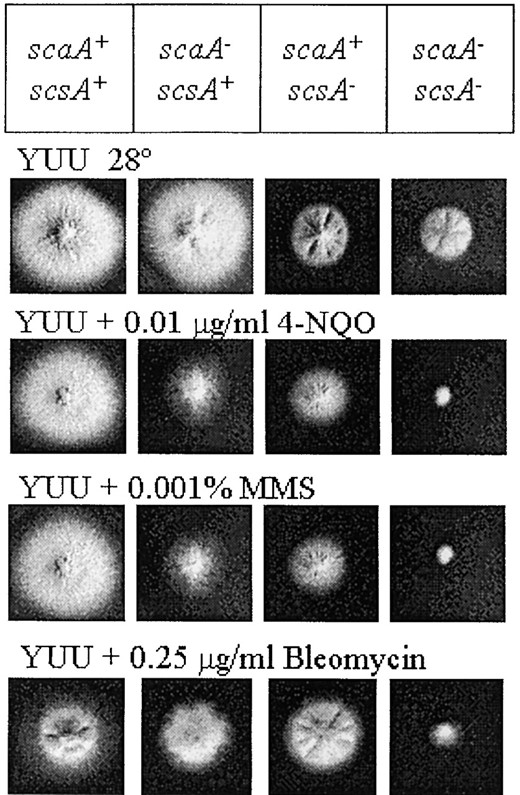
—Growth phenotypes of the scaA1 scsA1 double mutant in the presence of different DNA-damaging agents. Strains wild type, sca299 (scaA1), scs528-180 (scsA1), and scs528-180-10 (scaA1 scsA1) were grown for 72 hr at 28° in YUU, YUU + 4-NQO, YUU + MMS, and YUU + bleomycin.
Overexpression of topoisomerase I increases the sensitivity of A. nidulans to DNA-damaging agents in the background of the scaANBS1 mutant: We could not rescue the heat-sensitive defect of the scaA1NBS1 scsA1TOP1 double mutant by overexpressing the topoisomerase I gene. One possible explanation for this is that overexpression of the topoisomerase I gene could cause excessive DNA damage, making it more difficult for the mutant to recover at the restrictive temperature. This effect seems to be dependent on the scaA1 background since the scaA+ scsA- strain is not sensitive to the overexpression of the topoisomerase I gene. Overexpression of yeast topoisomerase I has already been shown to confer hypersensitivity to MMS and other DNA-damaging agents in yeast (Nitiss et al. 2001). To test this hypothesis, the consequences of overexpressing topoisomerase I in the presence of DNA damage were examined in wild type and scaA- strains (Figure 4). In these experiments, we used real-time RT-PCR to quantify the relative transcript levels of A. nidulans scsATOP1. The high-copy plasmid (pRG3-AMA1) carrying the A. nidulans topoisomerase I was transformed into both strains; the control cells for this experiment were the same strains transformed with pRG3-AMA1 vector alone. When topoisomerase I is overexpressed, the wild-type strain grows normally at 28° and 44° while the scaA- grows more slowly at 28° and does not grow at 44° (Figure 4), suggesting that the failure of pRG3-AMA1-top1 to complement the scaA1 scsA1 double mutant could be due to the scaA1 mutation (Figure 4). The overexpression of the topoisomerase I gene did not cause any inhibition of growth of the wild-type strain in the presence of DNA-damaging agents, such as 4-NQO, MMS, and camptothecin (CPT; Figure 4, second to fourth rows, left). In contrast, the scaA- strain was severely inhibited by these DNA-damaging agents (Figure 4, second to fourth rows, right). Taken together, these results indicate that the overexpression of the topoisomerase I gene causes DNA damage that is lethal only in the scaA- background.
The effects of topoisomerase I overexpression could be due to an alteration in the expression levels of genes required for responding to DNA damage. One A. nidulans gene that has been shown to be induced in the presence of DNA damage is uvsCRAD51 (van Heemst et al. 1997). We examined the transcription of the uvsCRAD51 exposed to MMS. scaA+ and scaA- strains, each transformed with pRG3-AMA1-top1 or control pRG3-AMA1 plasmids, were grown in the absence of MMS and then transferred for 3 hr to complete medium containing 0.025% MMS. Mycelia were subsequently collected for RNA extraction, and the RNA was treated with DNAse and subjected to quantitative RT-PCR. The results are expressed as the number of copies of the uvsCRAD51 cDNA divided by the number of copies of the β-tubulin (tubC) cDNA (for calculations, see Semighini et al. 2002). After 3 hr exposure to MMS, there was a 10-fold induction of the uvsCRAD51 gene in the wild-type background (Table 4). Interestingly, the level of uvsCRAD51 gene expression in the scaA- background is 3-fold higher than that in the wild type when both are grown in the absence of MMS, suggesting that the deficiency of scaA causes endogenous DNA damage that is able to induce uvsCRAD51 expression. Overexpression of topoisomerase I causes a 2-fold increase in uvsCRAD51 gene expression in both wild type and scaA-. When the topoisomerase I gene is overexpressed in the presence of MMS, there is a 2.9-fold increase in uvsCRAD51 expression in wild type (or ∼6-fold when compared to the wild type grown without overexpression of the topoisomerase I). However, in the scaA1 mutant, the uvsCRAD51 gene was induced to the same extent (1.9-fold; Table 4), regardless of whether topoisomerase I was overexpressed. Although uvsCRAD51 can be induced by overexpression of topoisomerase I, this mechanism does not appear to explain the sensitivity of scaA1 mutant strains to extra topoisomerase I.
Interaction between scsATOP1 and musNRecQ: Several examples of interactions between helicases and topoisomerases suggest a cooperation between these two classes of enzymes in many aspects of DNA metabolism (for reviews, see Wu and Hickson 2001 and Oakley and Hickson 2002). To determine if the topoisomerase I gene could interact with the A. nidulans musNRecQ gene, encoding a ReqQ helicase, we constructed scsA1 musN227 and scsA1 ΔmusN double mutants. The nature of the musN227 mutation was recently described by Hofmann and Harris (2001). It is a single base deletion that results in premature truncation of the protein just following the helicase domain. As shown in Figure 5, although the scsA1 could suppress camptothecin sensitivity caused by the musN227 mutation, it was unable to suppress the musN deletion. The scsA1 can suppress both the scaA1 mutant and scaA::pyr-4 disruption strains (Figure 5A). Taken together, these results suggest that ScsA could interact physically with the MusN and that the suppression of the camptothecin sensitivity in both sca mutants is probably due to the camptothecin-resistance phenotype conferred by the scsA1 mutation.
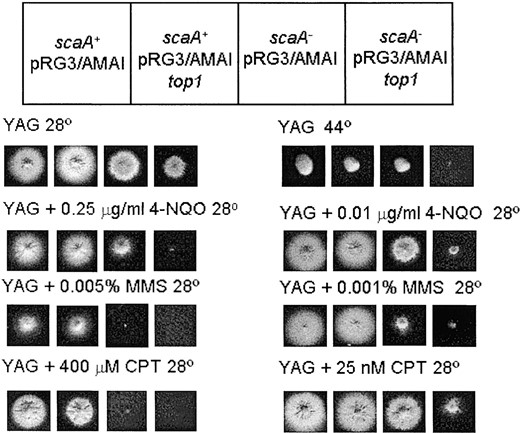
—Overexpression of the scsATOP1 gene blocks the growth of scaA1 mutants exposed to DNA damage. A wild type and scaA1 strains were transformed with the pRG3-AMA1 or the pRG3-AMA1-top1 vectors. The strains were grown at 28° for 72 hr in YAG medium, YAG + 4-NQO, YAG + MMS, YAG + camptothecin, and also in YAG medium at 44°.
The scsA1 musN227 double mutant was much more sensitive to bleomycin than either single mutant was alone, suggesting that ScsA and MusN function in parallel pathways to repair DSBs caused by bleomycin. In contrast, the double mutant did not show increased sensitivity to other DNA-damaging agents.
Quantitation of the uvsCRAD51 expression by real-time RT-PCR
| Strains | -MMS | +MMS | UvsCRAD51 induction ratio |
|---|---|---|---|
| scaA+ AMA1 | 0.07 ± 0.01 0.70 ± 0.02 | ̃10× | |
| scaA+ AMA1-top1 | 0.13 ± 0.01 0.38 ± 0.02 | ̃2.9× | |
| scaA- AMA1 | 0.22 ± 0.01 0.41 ± 0.01 | ̃1.9× | |
| scaA- AMA1-top1 | 0.35 ± 0.01 0.65 ± 0.03 | ̃1.9× |
| Strains | -MMS | +MMS | UvsCRAD51 induction ratio |
|---|---|---|---|
| scaA+ AMA1 | 0.07 ± 0.01 0.70 ± 0.02 | ̃10× | |
| scaA+ AMA1-top1 | 0.13 ± 0.01 0.38 ± 0.02 | ̃2.9× | |
| scaA- AMA1 | 0.22 ± 0.01 0.41 ± 0.01 | ̃1.9× | |
| scaA- AMA1-top1 | 0.35 ± 0.01 0.65 ± 0.03 | ̃1.9× |
The measured quantity of the uvsCRAD51 mRNA in each of the treated samples was normalized using the CT values obtained for the tubC RNA amplifications run in the same plate. The relative quantitation of uvsCRAD51 and tubulin gene expression was determined by a standard curve (i.e., Ct values were plotted against the logarithm of the DNA copy number). The results are the average of four repetitions. The values represent the number of copies of cDNAs of the uvsCRAD51 divided by the number of copies of cDNAs of the tubC.
Quantitation of the uvsCRAD51 expression by real-time RT-PCR
| Strains | -MMS | +MMS | UvsCRAD51 induction ratio |
|---|---|---|---|
| scaA+ AMA1 | 0.07 ± 0.01 0.70 ± 0.02 | ̃10× | |
| scaA+ AMA1-top1 | 0.13 ± 0.01 0.38 ± 0.02 | ̃2.9× | |
| scaA- AMA1 | 0.22 ± 0.01 0.41 ± 0.01 | ̃1.9× | |
| scaA- AMA1-top1 | 0.35 ± 0.01 0.65 ± 0.03 | ̃1.9× |
| Strains | -MMS | +MMS | UvsCRAD51 induction ratio |
|---|---|---|---|
| scaA+ AMA1 | 0.07 ± 0.01 0.70 ± 0.02 | ̃10× | |
| scaA+ AMA1-top1 | 0.13 ± 0.01 0.38 ± 0.02 | ̃2.9× | |
| scaA- AMA1 | 0.22 ± 0.01 0.41 ± 0.01 | ̃1.9× | |
| scaA- AMA1-top1 | 0.35 ± 0.01 0.65 ± 0.03 | ̃1.9× |
The measured quantity of the uvsCRAD51 mRNA in each of the treated samples was normalized using the CT values obtained for the tubC RNA amplifications run in the same plate. The relative quantitation of uvsCRAD51 and tubulin gene expression was determined by a standard curve (i.e., Ct values were plotted against the logarithm of the DNA copy number). The results are the average of four repetitions. The values represent the number of copies of cDNAs of the uvsCRAD51 divided by the number of copies of cDNAs of the tubC.
DISCUSSION
Bruschi et al. (2001) have cloned the A. nidulans analog of the human NBS1, scaA gene by complementing a camptothecin-sensitive mutant. In an effort to further understand how the DSB repair protein ScaANBS1 affects DNA damage response in A. nidulans, we have generated a set of strains containing extragenic suppressors of the camptothecin-sensitive scaA1 mutation. We propose to identify genes encoding proteins that interfere with the pathway or complex in which the ScaA would normally be involved by analyzing mutations that mitigate the scaA1 growth defect in the presence of camptothecin. The conspicuous defect of the scaA1 mutation on colony growth in the presence of camptothecin makes it easy to select revertants and to identify them as intragenic or extragenic mutations. In fact, all 56 revertants to camptothecin resistance were extragenic. Since the scaA1 mutation is a nonsense allele, our failure to identify intragenic suppressors is not a surprise. The scs mutants were sterile when subjected to pairwise crosses. This suggests that the scaANBS1 gene is required for the sexual cycle in A. nidulans. We have further corroborated this hypothesis by showing that the self-cross of the scaA disruption (strain T20) is sterile; however, one copy of the scaA+ is sufficient to restore sexual fertility (Semighini et al. 2003). As a strategy designed to aid in the cloning of these genes, we screened the extragenic suppressors for heat sensitivity. Fifteen of the suppressor mutations exhibited tight heat sensitivity. Most of the conditional lethal mutations are recessive and therefore potentially useful for cloning of the scs genes.
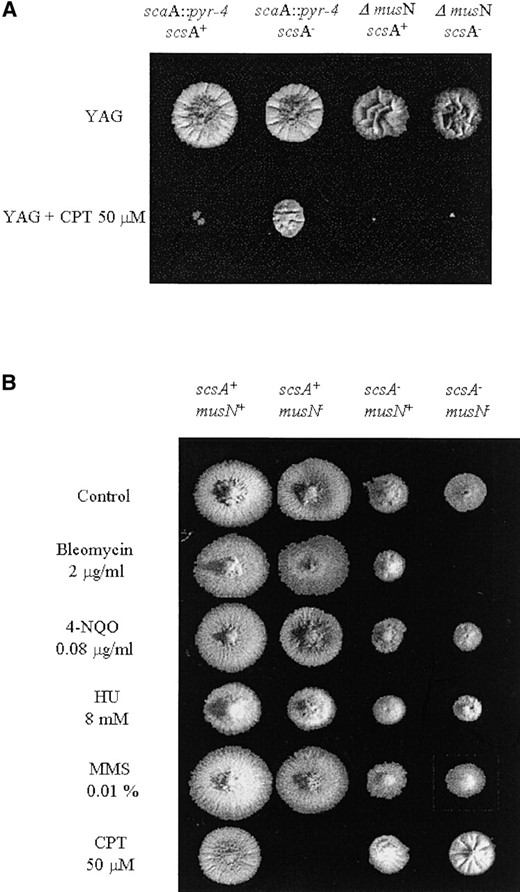
—Growth phenotypes of the scsA musN double mutants. (A) Strains T20 (scaA::pyr4), GG2 (scaA::pyr4 scsA1), AAH16 (ΔmusN), and GG1 (ΔmusN scsA1) were grown for 72 hr at 28° in YUU medium in the presence or absence of camptothecin. (B) Growth phenotypes of the GR5 (wild type), ASH608 (musN227), scs528-180 (scsA1), and ASH610 (scsA1 musN227) strains. Strains were grown for 72 hr at 28° in YAG medium in the presence or absence of bleomycin, 4-NQO, HU, MMS, and camptothecin.
scsATOP1 encodes DNA topoisomerase I: The scsA1 mutation was complemented by DNA-mediated transformation and sequence analysis revealed that the scsA gene encodes DNA topoisomerase I. Topoisomerases are enzymes that modify and regulate the topological state of DNA (Wang 1996). Since they are required for replication, transcription, recombination, and chromosome segregation, they have an important role in the maintenance of genome integrity (Liu 1989; Berger 1998). During the cleavage reaction, topoisomerases covalently attach to newly generated DNA 5′ (members of the type IA subfamily, including E. coli topoisomerases I and III, eukaryotic topoisomerase III, and archaeal reverse gyrase) or 3′ (members of the type IB subfamily, including eukaryotic topoisomerase I, archaeal topoisomerase V, and the poxvirus type I topoisomerases) phosphotyrosyl bonds (Liu 1989, 1994; Froelich-Ammon and Osheroff 1995; Gupta et al. 1995; Wang 1996). In normal circumstances, these covalent enzyme-DNA cleavage complexes are fleeting catalytic intermediates present in low steady-state concentrations that are tolerated by the cell. However, conditions that significantly increase the physiological concentration or life time of these breaks, such as exposition to camptothecin, cause several deleterious side effects, including mutations, insertions, deletions, and chromosomal aberrations.
In numerous cell lines, mutations that decrease the sensitivity of topoisomerase I to drug-induced DNA cleavage, or simply reduce the cellular content of the enzyme, are known to cause camptothecin resistance (for a review, see Larsen and Skladanowski 1998). In the yeast Saccharomyces cerevisiae, deletion of the TOP1 gene has little effect on cell viability, yet renders the cells completely resistant to camptothecin (Nitiss and Wang 1988). These experiments clearly identified Top1p as the cellular target of camptothecin and suggested that the drug causes cell death by converting it into a cellular poison rather than by simply inhibiting enzyme activity (Reid et al. 1998). Specific amino acid substitutions in yeast and human DNA topoisomerase I that promote camptothecin resistance in yeast have been defined (Knab et al. 1993, 1995; Levin et al. 1993; Megonigal et al. 1997). We sequenced the scsA1TOP1 allele from the scs528 mutant and found that the mutation changed a triplet codon that encodes an amino acid located in the DNA-binding domain of topoisomerase I. This region of the enzyme is highly conserved among eukaryotic species, and it has already been shown that mutations in this region of human topoisomerase I can confer camptothecin resistance (Andoh et al. 1987; Benedetti et al. 1993; Rubin et al. 1994). This could block the formation of the ternary complex, camptothecin-topoisomerase I-DNA, making the extragenic suppressor as resistant to camptothecin as the wild type. This is further corroborated by the fact that the scsA1 can suppress both the scaA1- mutant and scaA::pyr-4 disruption strains, suggesting that the suppression of the camptothecin sensitivity in both double mutants is probably due to the phenotype conferred by the scsA1 mutation.
Interaction between scaANBS1 and scsATOP1: The scaA1 scsA1 double mutant is more sensitive than scaA1 alone to several DNA-damaging agents, suggesting that these two genes function in parallel DNA repair pathways. Presumably, the accumulation of unrepaired DSBs in the scaA1 mutant is further exacerbated by the loss of topoisomerase I activity, thereby causing increased sensitivity to genotoxins. To our knowledge, there is no previous observation in the literature reporting any kind of interaction between these two genes. Since NBS1 and TOP1 have both been implicated in DSB repair (Wang 1996; Duguet 1997; D’Amours and Jackson 2002), we propose that scaANBS1 and scsATOP1 interact during homologous recombination (HR). HR restores a broken DNA molecule by using an intact homologous sister chromatid as a template. Migration of the branchpoint of the crossed DNA strands, known as Holliday junctions (HJs), allows the generation of heteroduplex DNA, which consists of one strand from each of the parental DNA molecules. The recombined molecules are separated into intact duplex DNAs by resolution of HJs (for a review, see Pâques and Haber 1999). There are three possible pathways for resolving HJs: (i) through resolution, where the best-characterized HJ resolvases are E. coli RuvC and eukaryotic Mus81/Eme1, which cleave HJs (Bennett et al. 1993; Boddy et al. 2001); (ii) through topoisomerase I, as was demonstrated by the vaccinia virus topoisomerase, a eukaryotic type I enzyme that catalyzes resolution of synthetic HJs in vitro (Sekiguchi et al. 1996); and (iii) through reverse branch migration through the concerted action of RecQ helicases and topoisomerases (Chakraverty and Hickson 1999; Oakley and Hickson 2002).
We propose that the scaANBS1 monitors and/or repairs abnormal DNA structures formed during HR. The accumulation of aberrant DNA structures, such as hairpins, could be due to a decrease in mre11 endonuclease activity caused by the scaA1 mutation. In humans, Nbs1 stimulates endonuclease, but not exonuclease, activity (Paull and Gellert 1999). An important source of DNA damage during replication is the formation of hairpin structures that block the progression of replication fork during S phase (Samadashwily et al. 1997). These structures are substrates for the endonucleolytic activity of Mre11 in vitro (Paull and Gellert 1999; Trujillo and Sung 2001). The presence of repeated sequences such as trinucleotide and palindromic repeats increases the formation of such hairpin structures. It has already been shown that trinucleotides and human Alu repeats are unstable in yeast cells that are defective in the Mre11 complex (D’Amours and Jackson 2002). Therefore, we propose that aberrant DNA structures such as hairpins may be present during HR and that the synergistic interaction between the scaA1NBS1 scsA1TOP1 mutations is caused by failure to resolve these structures.
Since the defects observed in the scaA1 scsA1 double mutant could not be complemented when the scsATOP1 gene was provided on a high-copy AMA1 vector, we suspected that overexpression of topoisomerase I might be causing additional DNA damage. Nitiss et al. (2001) have already shown that overexpression of yeast topo-isomerase I in wild-type strains conferred hypersensitivity to MMS and to other DNA-damaging agents. These authors suggested that topoisomerase I can cause DNA damage by forming covalent DNA-protein complexes. In A. nidulans, we have shown that overexpression of scsATOP1 inhibits growth in the presence of DNA-damaging agents, specifically in the scaA1 background, but not in the wild-type strain. This suggests that scaANBS1 could monitor and/or promote the repair of DNA damage caused by topoisomerase I overexpression.
Interaction between musNRecQ and scsATOP1: RecQ helicases, which are conserved from prokaryotes to humans, control HR by regulating the extent of HJ formation (for reviews, see Chakraverty and Hickson 1999; Frei and Gasser 2000; Oakley and Hickson 2002). Genetic and biochemical interactions have been identified between the RecQ family helicases and topoisomerases in both human and budding yeast cells (Chakraverty and Hickson 1999). In A. nidulans, musN encodes a member of the RecQ family (Hofmann and Harris 2001). To test for genetic interactions between scsATOP1 and musNRecQ, we constructed scsA1 musN227 double mutants. The scsA1 mutation could suppress the camptothecin sensitivity caused by the musN227 mutation, but not that caused by the musN deletion, suggesting that the ScsATOP1 could physically interact with the MusNRecQ. In addition, the scsA1 musN227 double mutant was more sensitive to bleomycin than was either single mutant alone, suggesting that ScsATOP1 and MusNRecQ function in parallel to repair DSBs caused by bleomycin. For example, topoisomerase I and RecQ may control alternative pathways for the processing of HJs (Sekiguchi et al. 1996; Oakley and Hickson 2002).
Acknowledgement
We thank Gregory May for providing the AMA1 genomic library used for complementation experiments and the two anonymous referees for useful comments. We thank the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Brazil (G.H.G.), and The American Cancer Society (S.D.H.) for financial support.
Footnotes
Sequence data from this article have been deposited with the National Center for Biotechnology Information under accession nos. AF451327 and AY178192.
Communicating editor: J. J. Loros
LITERATURE CITED

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}