





Abstract
The kurtz gene encodes a novel nonvisual arrestin. krz is located at the most-distal end of the chromosome 3R, the third gene in from the telomere. krz is expressed throughout development. During early embryogenesis, krz is expressed ubiquitously and later is localized to the central nervous system, maxillary cirri, and antennal sensory organs. In late third instar larvae, krz message is detected in the fat bodies, the ventral portion of the thoracic-abdominal ganglia, the deuterocerebrum, the eye-antennal imaginal disc, and the wing imaginal disc. The krz1 mutation contains a P-element insertion within the only intron of this gene and results in a severe reduction of function. Mutations in krz have a broad lethal phase extending from late embryogenesis to the third larval instar. The fat bodies of krz1 larva precociously dissociate during the midthird instar. krz1 is a type 1 melanotic tumor gene; the fat body is the primary site of melanotic tumor formation during the third instar. We have functionally rescued these phenotypes with both genomic and cDNA transgenes. Importantly, the expression of a full-length krz cDNA within the CNS rescues the krz1 lethality. These experiments establish the krz nonvisual arrestin as an essential neural gene in Drosophila.
G-protein-coupled receptors (GPCRs) transduce many extracelluar signals including odorants, light, hormones, neurotransmitters, and neuromodulators (Watson and Arkinstall 1994). These signals evoke a conformational change in their cognate receptors, resulting in the catalytic dissociation of heterotrimeric GTP binding proteins. The G protein α and βγ subunits subsequently interact with and regulate many cellular signaling pathways including potassium channels, kinases, and secondary messenger systems. Once bound to an agonist, GPCRs undergo rapid desensitization through the coordinated activities of G-protein-coupled receptor kinases (GRKs) and arrestins (Ferguson et al. 1996; Krupnick and Benovic 1998). This process of downregulating GPCR activity is alternatively referred to as homologous desensitization or agonist-dependent desensitization.
There are four classes of identified vertebrate arrestins that are further subdivided into two functional categories (Craft and Whitmore 1995). The visual arrestins, rod arrestin and cone arrestin, are located almost exclusively in photoreceptor cells. Rod arrestin is solely involved with the desensitization of rhodopsin (Gurevich et al. 1995). In contrast, the nonvisual arrestins, βARR1 and βARR2, are expressed in most tissues and can desensitize a large number of GPCRs (Parruti et al. 1993a,b; Sterne-Marr et al. 1993; Gurevich et al. 1995). In addition to these vertebrate arrestins, there are two visual arrestins identified from Drosophila and one from blow fly (Hyde et al. 1990; Krishnan and Ganguly 1990; Levine et al. 1990; Lieb et al. 1991; Bentrop et al. 1993). Two arrestins have also been identified from the antenna of locust and tobacco budworm (Raming et al. 1993). These latter two arrestins were presumed to be representatives of the insect nonvisual arrestin family (Raming et al. 1993; Craft and Whitmore 1995).
The homologous desensitization of the β2-adrenergic receptor (β2AR) has been well characterized and serves as a useful paradigm for the desensitization of many other nonvisual GPCRs (Zhang et al. 1997; Ferguson and Caron 1998; Ferguson et al. 1998; Krupnick and Benovic 1998). When bound by agonist, β2AR becomes a substrate for GRKs (Benovic et al. 1986, 1988, 1989; Benovic 1991). The GRKs phosphorylate β2AR at several positions in the carboxy-terminal tail. When phosphorylated by GRK, β2AR becomes a high-affinity substrate for binding by the β-arrestin, βARR1, at a 1:1 stoichiometry (Sohlemann et al. 1995). The β-arrestins desensitize β2AR by physically inhibiting further interactions with heterotrimeric G proteins (Lohse et al. 1990, 1992; Attramadal et al. 1992). The βARRs also mediate the sequestration and endocytosis of β2AR through direct interactions with clathrin, β2-adaptin, and phosphoinositides (Goodman et al. 1996, 1997; Krupnick et al. 1997; Gaidarov et al. 1999; Laporte et al. 1999). Once internalized, the endosome becomes acidified, which probably triggers the dephosphorylation of β2AR, βARR is released, and the receptor is recycled back to the plasma membrane or is targeted to the lysosome for degradation (Pitcher et al. 1995; Krueger et al. 1997). Although there appear to be some differences in the process of homologous desensitization and receptor recycling among the many GPCRs, most data indicate that the arrestin-dependent desensitization and recovery from desensitization of β2AR is a general phenomenon (Zhang et al. 1997; Ferguson and Caron 1998; Krupnick and Benovic 1998).
There is growing evidence that a number of agonist-bound GPCRs regulate tyrosine kinase activation of mitogenic signal-transduction cascades (Daaka et al. 1998; Luttrell et al. 1999a,b). In at least one case, this transactivation has been demonstrated to require β2AR homologous desensitization (Luttrell et al. 1999b). Stimulation of β2AR with isoproterenol in HEK-293 cells results in the recruitment of c-Src to the plasma membrane, and this mobilization is mediated by βARR1 (Luttrell et al. 1999b). There are two potential SH3 binding sites within βARR1 that, when mutated, abolish c-Src binding and β2AR-dependent phosphorylation of the ERK1 and ERK2 kinases without affecting receptor-arrestin interaction (Luttrell et al. 1999b). Thus, the process of GPCR homologous desensitization may also act as a focus for the initiation and coordination of mitogenic signaling cascades within a given cell.
Despite the clear importance of nonvisual arrestins in the regulation of G-protein signaling, very little is known of the in vivo requirement for arrestin activity (Conner et al. 1997). A βARR1 mouse knock-out line has been created and appears to be overtly normal (Conner et al. 1997). These animals were viable and fertile, with no apparent defects in development, life expectancy, blood chemistry, or in the number of blood cells or lymphocytes. Nevertheless, the βARR1 −/− mice did show a significant increase in the left ventricular ejection fraction after isoproterenol infusion, consistent with a physiological role for this protein in the desensitization of β2AR (Conner et al. 1997). Since βARR2 is coexpressed with βARR1 in many tissues, additional phenotypes may be concealed by functional redundancy.
In this article, we report the identification and characterization of the kurtz (krz) gene,1 a new nonvisual arrestin in Drosophila melanogaster. During late embryogenesis and the third larval instar, krz is found in a number of neuronal tissues. Mutants disrupted in krz activity have three distinctive phenotypes. These mutants precociously disaggregate their larval fat bodies, form melanotic tumors, and have a broad lethal phase. Melanotic tumor formation is part of the cellular immune response to aberrant tissue or infectious agents (Sparrow 1978). In the krz1 melanotic tumors, hemocytes surround and encapsulate the dissociating fat body cells. We have also shown that the lethality of the krz mutants is specifically due to the loss of arrestin activity within the nervous system.
MATERIALS AND METHODS
Strains: All stocks were raised and maintained on standard yeast-cornmeal-agar media at room temperature. The krz1/TM3, krz2/TM3 l(3)041303/TM3, l(3)024314/TM3, l(3)099801/TM3, and l(3)134408/TM3 lines were obtained from Péter Deák and David Glover (Dundee, UK; Deák et al. 1997). The Df(3R)faf-BP/TM6B line was obtained from the Umeå Stock center. The modP1795/TM3 (ms(3)100EF), c155, P{w+GAL4-Hsp70.PB}89-2-1, Df(3R)04661/TM3, l(3)rH304/TM3, l(3)06886/TM3, and l(3)06497/TM3 lines were obtained from the Bloomington Stock Center. The Df(3R)td106/TM3 and modlethal8/TM3 mutations were the gift of J. Pradel (Marseille, France). The modlethal8/TM6, modlethal3/TM6, and P{w+Sph}; modlethal8 lines were a gift of A. Pereira (Worcester, MA). The CyO, P{w+ KrGFP30} and TM3, P{w+ KrGFP4} green balancers were a gift of D. Casso and T. Kornberg (Berkeley, CA). The TM3-pAct-GFP green balancer was obtained from the Bloomington Stock Center. The following transposons were generated in this study: P{UARRT5}, P{UARRT4}, P{b5.8T12}, and P{b5.8T13}. These P elements and the krz1, krz2, and modlethal8 mutations were all backcrossed six times to a w1118, Canton-S line prior to phenotypic analysis.
Molecular analysis: We isolated the 478/19 flanking genomic DNA through plasmid rescue. The flanking DNA was used to identify cDNA clones from a Canton-S adult head-specific cDNA library (C. Hall and R. Davis) and a Canton-S genomic λ library (M. Eberwine and R. Davis). The DS01476 and DS05238 P1 clones were identified by hybridizing the 478/19 flanking genomic DNA to a gridded P1 library (Genome Systems Inc., St. Louis). The P1 DNA was prepared for restriction analysis and subcloning by standard alkaline lysis protocol, with one modification. The DNA was extracted once with equal volume phenol/chloroform/isoamyl alcohol (25:24:1) after alkaline lysis and prior to isopropanol precipitation. Homology searches with cDNA and genomic sequences were performed with BLASTN and BLASTX (Altschul et al. 1990). DNA and protein sequence analysis was performed with the GCG suite of programs (Devereux et al. 1984).
Drosophila RNA was isolated for Northern and reverse transcription (RT)-PCR analysis as previously described (Roman et al. 1998). Standard methods were used to isolate poly(A+) RNA (Sambrook et al. 1989). Northern blots were performed using glyoxal-treated poly(A+) RNA (Sambrook et al. 1989). Developmental RT-PCR was performed as previously described with minor modifications (Roman et al. 1998). The krz-specific PCR product was amplified for 10 cycles at 94°, 30 sec; 60°, 30 sec; and 72°, 1 min. After the 10th cycle, rp49 primers were added and the reaction continued for 20 additional cycles. For the RT-PCR reactions of mutant third instar RNAs, 50 μg of poly(A+) RNA was reverse transcribed for each genotype. Reactions containing 1%, 2%, and 4% of the total RT reaction were amplified. For each genotype one RNase control was run using the equivalent of 4% of the total RT reaction. krz and mod products were amplified for 18 cycles, and rp49 was amplified for 10 cycles under the following conditions: 94°, 20 sec; 62°, 20 sec; and 72°, 45 sec. The PCR products were transferred to nylon membranes and probed with radiolabeled gene-specific fragments. Amplification was detected by autoradiography.
Whole mount in situ hybridizations were performed as previously described (Meller et al. 1997). Both sense and antisense riboprobes were generated from the full-length pckrz3 cDNA by in vitro translation using digoxigenin-UTP (Boehringer Mannheim, Indianapolis). A probe concentration of 0.1 ng/λ was used for hybridization. Third instar larval material was dissected and fixed prior to hybridization.
Transgenes: The P{b5.8} construct was generated by cloning in a 5.8-kb XbaI fragment from the genomic λg478.7 clone into a modified P{Casper4} vector (G. Roman, J. He and R. L. Davis, unpublished data). Seven independent transgenes were isolated. The P{b5.8T12} and P{b5.8T13} lines both contained a single X-linked insertion. The P{UARR} construct was generated by directly cloning the pckrz3 cDNA into the KpnI/NotI sites of P{UAST}. Eight independent inserts were isolated. The P{UARRT4} and P{UARRT5} contained single P-element inserts on the X and second chromosome, respectively. We detected significant krz synthesis from both P{UARRT4} and P{UARRT5} transgenes after heat shock when driven by the P{w+GAL4-Hsp70.PB}89-2-1 Gal4 effector (data not shown).
Phenotypic analysis: The krz1, krz2, and modlethal8 homozygous mutants were isolated from a population cage of the respective mutations balanced over the TM3-pAct-GFP balancer (Reichhart and Ferrandon 1998). Mutants were identified as non-green fluorescent protein (GFP)-containing embryos. Embryos older than stage 15 could be unambiguously scored as homozygous mutants. Larvae were staged according to age, mouth hook, and spiracle morphology (Bodenstein 1950). Embryos and larvae were visualized with a Zeiss Axiophot for Nomarski images or Stemi SV6 binocular microscope with GFP modification (Carl Zeiss Inc., Thornwood, NY).
RESULTS
Identification and molecular characterization of the krz locus: The 478/19 line was identified in an enhancer detector screen for genes expressed in a sexually dimorphic manner in the adult head. This line contains a recessive lethal P{lacW} insertion on the third chromosome. In 478/19, LacZ is highly expressed in male, but not female, adult fat bodies (data not shown). On the basis of this unusual pattern of expression, we decided to pursue the identification of the resident gene. We have named this gene kurtz and the 478/19 P-element line as krz1. The genomic sequences flanking the P element in krz1 were cloned by plasmid rescue. The precise cytological position of the krz1 locus was determined by hybridizing the flanking sequences to an arrayed P1 library (Hartl et al. 1994). There are two P1 clones within this library (DS01476 and DS05238) that uniquely hybridize to the 478/19 plasmid rescue; both of these P1 clones have been mapped to position 100F5 at the end of the third chromosome (Hartl et al. 1994). The P1 clones are colinear with genomic DNA at the krz locus, thereby confirming this position (data not shown).
The flanking genomic region was also used to search for genes at the krz locus. We identified two classes of clones from an adult head cDNA library. Genomic clones derived from a Canton-S λ library were also isolated. Sequence and restriction analysis of these clones revealed two genes at this locus: the novel kurtz gene (krz) and the previously identified modulo gene (mod). mod encodes a nucleic acid binding protein that structurally resembles nucleolin and is located in the distal portion of 100F5 (Krejci et al. 1989; Garzino et al. 1992). Two independent krz cDNAs were isolated; the longest was 1986 bp. Both of the krz cDNAs contained a single long open reading frame encoding a predicted protein of 470 amino acids. The krz transcript is divided by a single intron. The P element of krz1 is inserted within this intron (Figure 1B). Interestingly, there are only 130 bp separating the 5′ ends of mod and the longest krz cDNA, and these genes are divergently transcribed (Figure 1B).
The terminus of the right arm of the third chromosome (3R) is variable in length (Levis et al. 1993). Levis et al. (1993) isolated and sequenced an intact telomere from the A4-4 P-element-containing line. The proximity of krz to mod allowed us to align our P1 and genomic maps to the previously mapped end of the third chromosome in this line (Figure 1). krz is the third gene in from this telomere, located ~45 kb from the terminus in the A4-4 line, and is transcribed in a proximal direction. The P1 clones were derived from the iso-1 line; this line lacks the TART telomeric repeats seen in the A4-4 line, indicating these lines have different telomere structures (Levis et al. 1993). From our map of the P1 clones, 3R in iso-1 extends at least 26 kb beyond the terminus of A4-4 (Figure 1A).
The predicted KRZ protein contains significant similarity to the arrestin family of proteins. KRZ was most similar to the human nonvisual arrestin proteins βARR1 and βARR2, with 65% identity and 74% similarity to βARR1 and 62% identity and 72% similarity to βARR2. We have optimally aligned KRZ with βARR1 and βARR2, as well as two arrestins that were isolated from insect antenna cDNA libraries (Figure 2). The regions of identity and similarity are found throughout these proteins. Nevertheless, KRZ has an extended amino terminus that is unique in this family. There are a number of functionally defined structures found within βARR1 or βARR2 that are conserved in KRZ. βARR1 contains two potential SH3 binding domains (PXXP). When both of these domains are mutated, direct interactions with Src are eliminated (Luttrell et al. 1999b). While only the second of these SH3 binding domains is conserved in KRZ, only the first is conserved in βARR2 (Figure 2). The nonvisual arrestins have a clathrin interaction domain that is absent from the visual arrestins (Goodman et al. 1996). This clathrin-binding domain, LIEF/L, is strongly conserved in KRZ with a single glutamine for glutamate substitution (Figure 2). The conservation of the clathrin-binding domain is consistent with KRZ belonging to the nonvisual arrestin family (Craft and Whitmore 1995; Goodman et al. 1996). Additionally, all three of the basic residues identified that are required for βARR1 to bind phosphoinositides are conserved in KRZ. The Locusta and Heliothis proteins have been thought to represent a new class of arthropod nonvisual arrestin in part on the basis of their expression
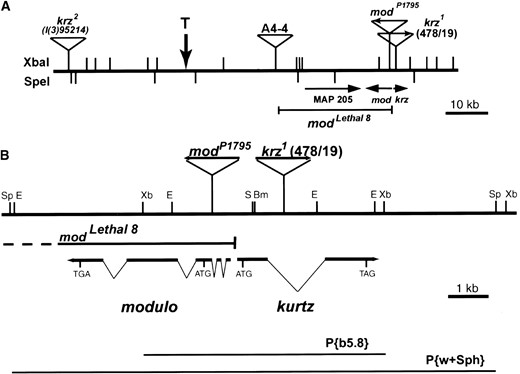
Genomic location and structure of the kurtz locus. (A) Physical map of the DS05238 P1 clone at cytological position 100F5 on 3R. The positions of the A4-4, P1795, and 478/19 P elements are shown. The vertical arrow marked with a T indicates the position of the telomere in the lines carrying modlethal8 and A4-4 (Levis et al. 1993). Beneath the map are the positions and orientations of the distal-most genes on 3R and the modlethal8 deficiency (Pereira et al. 1992; this report). (B) Genomic structure of the kurtz and modulo loci. Genomic and cDNA maps of the mod and krz loci are shown with the positions of mutations. The modlethal8 is a large deletion that terminates 47 bp 5′ of the longest krz cDNA. The 478/19 P{lacW} element in the krz1 mutant is located within the krz intron, whereas the P1795 PZ element is inserted within the second intron of mod. The position of genomic fragments used in the construction of the P{b5.8} and P{w+Sph} transposons is shown below the transcribed regions. Restriction enzymes are as follows: Bm, BamHI; E, EcoRI; S, SacI; Sp, SphI; and Xb, XbaI.
in the antenna (Raming et al. 1993; Craft and Whitmore 1995). Interestingly, the clathrin- and phosphoinositide-binding domains are poorly conserved or absent in these other insect arrestins (Figure 2).
Genetic analysis of the krz locus: We sought additional mutations for the analysis of krz function by examining extant mutations for complementation of the krz1 lethal phenotype and by excising the P{lacW} element in krz1 (Figure 1; Table 1). Initially, we tested three deficiencies that included 100F5 for genetic complementation of the krz1 lethality (Table 1). The deficiencies Df(3R)faf-BP and Df(3R)04661 both initiate at ~100D2 and extend distally into 100F5 (Fischer-Vize et al. 1992). The Df(3R)td106 is a terminal deletion that includes the mod locus, but whose proximal breakpoint is unknown (Laurenti et al. 1995). All three deficiencies fail to complement krz1, indicating that these large chromosomal deletions also include the krz locus (Table 1).
We also examined eight lethal P-element insertions that were available from the Bloomington Stock Center or through the European chromosome three P-element project (Deák et al. 1997). The l(3)041303, l(3)024314, l(3)099801, l(3)134408, l(3)06886, l(3)rH304, and l(3)06497 mutations all complemented both krz1 and Df(3R)faf-BP (data not shown). These data strongly suggest that the lethal phenotypes of these mutations are located proximal to the 100D breakpoint of Df(3R)faf-BP. The l(3)95214 P-element line, however, failed to complement krz1 and therefore represents a new allele of this gene and has been renamed krz2 (Table 1). We mapped the krz2 P-element insertion by first cloning the flanking DNA sequence by iPCR and then hybridizing this product against the P1 clone DS05238. Surprisingly, the P element is located at the most-distal end of this P1 clone, ~60 kb from the krz locus. This raised the issue of whether the P-element insertion was responsible for the krz lesion or if there was a second site mutation in krz2. We did not detect any polymorphisms by restriction analysis in krz2 within a 12-kb SphI fragment that includes both mod and krz loci. Additionally, we sequenced and failed to detect any changes in ~4.5 kb of the krz gene and promoter region from krz2 mutant animals. This suggests that the l(3)95214 insertion or an undetected second site mutation at the krz locus disrupts the expression of this gene.
From 100 P-element excisions of krz1, we obtained 9 in which the krz1 lethal phenotype is completely reverted. In 7 of these 9 lines, the 478/19 P element had precisely or almost precisely excised from the krz intron. In the other two lines, there was a substantial internal deletion of the P element, leaving a small insertion within the krz intron. Therefore, the 478/19 P-element insertion is responsible for the lethality of krz1. In addition, many lines were identified that had an imprecise excision of the transposon. The lethal krz3 mutation contains a deletion beginning internal to the P element and extending beyond the 3′ end of the gene. We have failed to detect any polymorphism within 15 kb of the

The kurtz gene product is a member of the nonvisual arrestin gene family. An amino acid sequence alignment of several members of the nonvisual arrestin family of proteins is shown. βARR1 (human β-arrestin1; accession no. NP 004032), βARR2 (human β-arrestin2; accession no. NP 004304), KURTZ, (GenBank accession no. AF221066), Locusta (Locusta migratoria arrestin homolog; accession no. P32122), Heliothis (Heliothis virescens arrestin homolog; accession no. B56607) are optimally aligned. Identities between three or more proteins are shown in black with white letters, and conserved substitutions are shadowed in gray. The position of the two sequences identified as possible SH3 binding sites in βARR1 are indicated by the SH3B. The position of the clathrin-binding domain in βARR1 and βARR2 is indicated by CBD. The three basic residues required for binding phosphoinositides are labeled with an asterisk.
Complementation of 100F5 mutants
| krz1 | krz2 | krz3 | krz4 | modL8 | modL3 | modP1795 | Df(3R)04661 | Df(3R)faf-BP | Df(3R)td106 | |
|---|---|---|---|---|---|---|---|---|---|---|
| krz1 | 0 (892) | |||||||||
| krz2 | 0 (215) | 0 (789) | ||||||||
| krz3 | 0 (124) | 0 (86) | 0 (513) | |||||||
| krz4 | 34 (90) | 29 (93) | naa | 31 (93) | ||||||
| modL8 | 0 (246) | 0 (221) | 0 (71) | 31 (101) | 0 (575) | |||||
| modL3 | 0 (410) | 0 (410) | 0 (49) | 18 (51) | 0 (139) | 0 (467) | ||||
| modP1795 | 50 (104) | 44 (102) | 6 (34) | 20 (37) | 57 (125) | 42 (105) | 93 (194) | |||
| Df(3R)04661 | 0 (120) | 0 (127) | na | na | 0 (72) | 0 (170) | 33 (78) | 0 (321) | ||
| Df(3R)faf-BP | 0 (152) | 0 (280) | 0 (88) | 14 (40) | 0 (136) | 0 (54) | 24 (41) | na | 0 (217) | |
| Df(3R)td106 | 0 (180) | 0 (52) | 0 (79) | na | 0 (57) | 0 (64) | 14 (35) | 0 (67) | 0 (48) | 0 (358) |
| krz1 | krz2 | krz3 | krz4 | modL8 | modL3 | modP1795 | Df(3R)04661 | Df(3R)faf-BP | Df(3R)td106 | |
|---|---|---|---|---|---|---|---|---|---|---|
| krz1 | 0 (892) | |||||||||
| krz2 | 0 (215) | 0 (789) | ||||||||
| krz3 | 0 (124) | 0 (86) | 0 (513) | |||||||
| krz4 | 34 (90) | 29 (93) | naa | 31 (93) | ||||||
| modL8 | 0 (246) | 0 (221) | 0 (71) | 31 (101) | 0 (575) | |||||
| modL3 | 0 (410) | 0 (410) | 0 (49) | 18 (51) | 0 (139) | 0 (467) | ||||
| modP1795 | 50 (104) | 44 (102) | 6 (34) | 20 (37) | 57 (125) | 42 (105) | 93 (194) | |||
| Df(3R)04661 | 0 (120) | 0 (127) | na | na | 0 (72) | 0 (170) | 33 (78) | 0 (321) | ||
| Df(3R)faf-BP | 0 (152) | 0 (280) | 0 (88) | 14 (40) | 0 (136) | 0 (54) | 24 (41) | na | 0 (217) | |
| Df(3R)td106 | 0 (180) | 0 (52) | 0 (79) | na | 0 (57) | 0 (64) | 14 (35) | 0 (67) | 0 (48) | 0 (358) |
The top row lists the allele contributed by the first parent and the first column lists the allele contributed by the second parent. The crosses were generally made as follows: allele1/balancer × allele2/balancer. The progeny class of interest is shown as the first number in each cell. In parentheses is the total number of progeny examined for the indicated cross. The frequency of transheterozygotes should be ~33% since homozygous balancers are lethal. The exceptions to this expectation are the crosses involving mod1795; homozygous mod1795 females were used for these crosses, leading to an expected frequency of 50%. The italic entries are statistically significant at the 5% level by χ2 analysis. In all cases the balancers were TM3Sb, Ser, or TM6B.
Data not available.
Complementation of 100F5 mutants
| krz1 | krz2 | krz3 | krz4 | modL8 | modL3 | modP1795 | Df(3R)04661 | Df(3R)faf-BP | Df(3R)td106 | |
|---|---|---|---|---|---|---|---|---|---|---|
| krz1 | 0 (892) | |||||||||
| krz2 | 0 (215) | 0 (789) | ||||||||
| krz3 | 0 (124) | 0 (86) | 0 (513) | |||||||
| krz4 | 34 (90) | 29 (93) | naa | 31 (93) | ||||||
| modL8 | 0 (246) | 0 (221) | 0 (71) | 31 (101) | 0 (575) | |||||
| modL3 | 0 (410) | 0 (410) | 0 (49) | 18 (51) | 0 (139) | 0 (467) | ||||
| modP1795 | 50 (104) | 44 (102) | 6 (34) | 20 (37) | 57 (125) | 42 (105) | 93 (194) | |||
| Df(3R)04661 | 0 (120) | 0 (127) | na | na | 0 (72) | 0 (170) | 33 (78) | 0 (321) | ||
| Df(3R)faf-BP | 0 (152) | 0 (280) | 0 (88) | 14 (40) | 0 (136) | 0 (54) | 24 (41) | na | 0 (217) | |
| Df(3R)td106 | 0 (180) | 0 (52) | 0 (79) | na | 0 (57) | 0 (64) | 14 (35) | 0 (67) | 0 (48) | 0 (358) |
| krz1 | krz2 | krz3 | krz4 | modL8 | modL3 | modP1795 | Df(3R)04661 | Df(3R)faf-BP | Df(3R)td106 | |
|---|---|---|---|---|---|---|---|---|---|---|
| krz1 | 0 (892) | |||||||||
| krz2 | 0 (215) | 0 (789) | ||||||||
| krz3 | 0 (124) | 0 (86) | 0 (513) | |||||||
| krz4 | 34 (90) | 29 (93) | naa | 31 (93) | ||||||
| modL8 | 0 (246) | 0 (221) | 0 (71) | 31 (101) | 0 (575) | |||||
| modL3 | 0 (410) | 0 (410) | 0 (49) | 18 (51) | 0 (139) | 0 (467) | ||||
| modP1795 | 50 (104) | 44 (102) | 6 (34) | 20 (37) | 57 (125) | 42 (105) | 93 (194) | |||
| Df(3R)04661 | 0 (120) | 0 (127) | na | na | 0 (72) | 0 (170) | 33 (78) | 0 (321) | ||
| Df(3R)faf-BP | 0 (152) | 0 (280) | 0 (88) | 14 (40) | 0 (136) | 0 (54) | 24 (41) | na | 0 (217) | |
| Df(3R)td106 | 0 (180) | 0 (52) | 0 (79) | na | 0 (57) | 0 (64) | 14 (35) | 0 (67) | 0 (48) | 0 (358) |
The top row lists the allele contributed by the first parent and the first column lists the allele contributed by the second parent. The crosses were generally made as follows: allele1/balancer × allele2/balancer. The progeny class of interest is shown as the first number in each cell. In parentheses is the total number of progeny examined for the indicated cross. The frequency of transheterozygotes should be ~33% since homozygous balancers are lethal. The exceptions to this expectation are the crosses involving mod1795; homozygous mod1795 females were used for these crosses, leading to an expected frequency of 50%. The italic entries are statistically significant at the 5% level by χ2 analysis. In all cases the balancers were TM3Sb, Ser, or TM6B.
Data not available.
3′ end of krz in heterozygotes, suggesting that the deficiency in this mutation extends beyond this distance (data not shown). krz3 fails to complement krz1 lethality (Table 1). In contrast, the viable krz4 line resulted from an internal P-element deletion and is viable. In this line, ~530 bp of P-element sequences remain at the original site of insertion. The krz4 mutation completely complements the krz1 lethal phenotype (Table 1).
We also examined three mod mutations for genetic interactions with krz1. The modlethal8 and modlethal3 mutations were generated by imprecise excision of the A4-4 P element near the Drosophila telomere (Pereira et al. 1992; Figure 1A). Both of these mod mutations fail to complement krz1 (Table 1). The modlethal3 mutation extends beyond the 5′ region of mod and thus is expected to also disrupt krz (Pereira et al. 1992). The modlethal8 breakpoint was previously mapped to the 5′ region of mod. We isolated this breakpoint by iPCR and the juncture was sequenced to precisely define this mutation. The modlethal8 mutation is a deletion of ~24 kb that has one breakpoint within the rosy sequences of the A4-4 P element and the other breakpoint just 47 bp 5′ to the start of our longest krz cDNA (Figure 1). In addition to modlethal8 and modlethal3, a new mod allele was found in the data of the Drosophila P-element disruption project (Spradling et al. 1999). The modP1795 allele contains a P{PZ} element insertion within the first intron of mod; this allele was originally isolated as ms(3)100EF in a screen for male sterile mutations (Castrillon et al. 1993). Homozygous male modP1795 flies are semisterile, but homozygous females are fertile (Castrillon et al. 1993; Tables 1 and 2). The modP1795 phenotype is more severe when placed in trans with the modlethal8 and modlethal3 null mutations, indicating that modP1795 is a hypomorphic
modulo mutations affect male fertility
| Genotype | Male sterilesa | % male sterility | Minute phenotypeb |
|---|---|---|---|
| modP1795/modP1795 | 49 (59) | 83 | + |
| modL8/modP1795 | 30 (30) | 100 | ++ |
| krz1/modP1795 | 0 (28) | 0 | − |
| krz2/modP1795 | 30 (31) | 97 | + |
| krz3/modP1795 | 1 (11) | 9 | − |
| Df(3R)faf-BP/modP1795 | 41 (41) | 100 | ++ |
| Df(3R)04661/modP1795 | 9 (9) | 100 | ++ |
| Df(3R)td106/modP1795 | 7 (7) | 100 | ++ |
| Genotype | Male sterilesa | % male sterility | Minute phenotypeb |
|---|---|---|---|
| modP1795/modP1795 | 49 (59) | 83 | + |
| modL8/modP1795 | 30 (30) | 100 | ++ |
| krz1/modP1795 | 0 (28) | 0 | − |
| krz2/modP1795 | 30 (31) | 97 | + |
| krz3/modP1795 | 1 (11) | 9 | − |
| Df(3R)faf-BP/modP1795 | 41 (41) | 100 | ++ |
| Df(3R)04661/modP1795 | 9 (9) | 100 | ++ |
| Df(3R)td106/modP1795 | 7 (7) | 100 | ++ |
Sterility was determined by the complete lack of progeny when the male was crossed with three wild-type virgin females.
Flies that were delayed in eclosion and had bristles that were more slender and shorter than wild type were considered to have a Minute phenotype.
modulo mutations affect male fertility
| Genotype | Male sterilesa | % male sterility | Minute phenotypeb |
|---|---|---|---|
| modP1795/modP1795 | 49 (59) | 83 | + |
| modL8/modP1795 | 30 (30) | 100 | ++ |
| krz1/modP1795 | 0 (28) | 0 | − |
| krz2/modP1795 | 30 (31) | 97 | + |
| krz3/modP1795 | 1 (11) | 9 | − |
| Df(3R)faf-BP/modP1795 | 41 (41) | 100 | ++ |
| Df(3R)04661/modP1795 | 9 (9) | 100 | ++ |
| Df(3R)td106/modP1795 | 7 (7) | 100 | ++ |
| Genotype | Male sterilesa | % male sterility | Minute phenotypeb |
|---|---|---|---|
| modP1795/modP1795 | 49 (59) | 83 | + |
| modL8/modP1795 | 30 (30) | 100 | ++ |
| krz1/modP1795 | 0 (28) | 0 | − |
| krz2/modP1795 | 30 (31) | 97 | + |
| krz3/modP1795 | 1 (11) | 9 | − |
| Df(3R)faf-BP/modP1795 | 41 (41) | 100 | ++ |
| Df(3R)04661/modP1795 | 9 (9) | 100 | ++ |
| Df(3R)td106/modP1795 | 7 (7) | 100 | ++ |
Sterility was determined by the complete lack of progeny when the male was crossed with three wild-type virgin females.
Flies that were delayed in eclosion and had bristles that were more slender and shorter than wild type were considered to have a Minute phenotype.
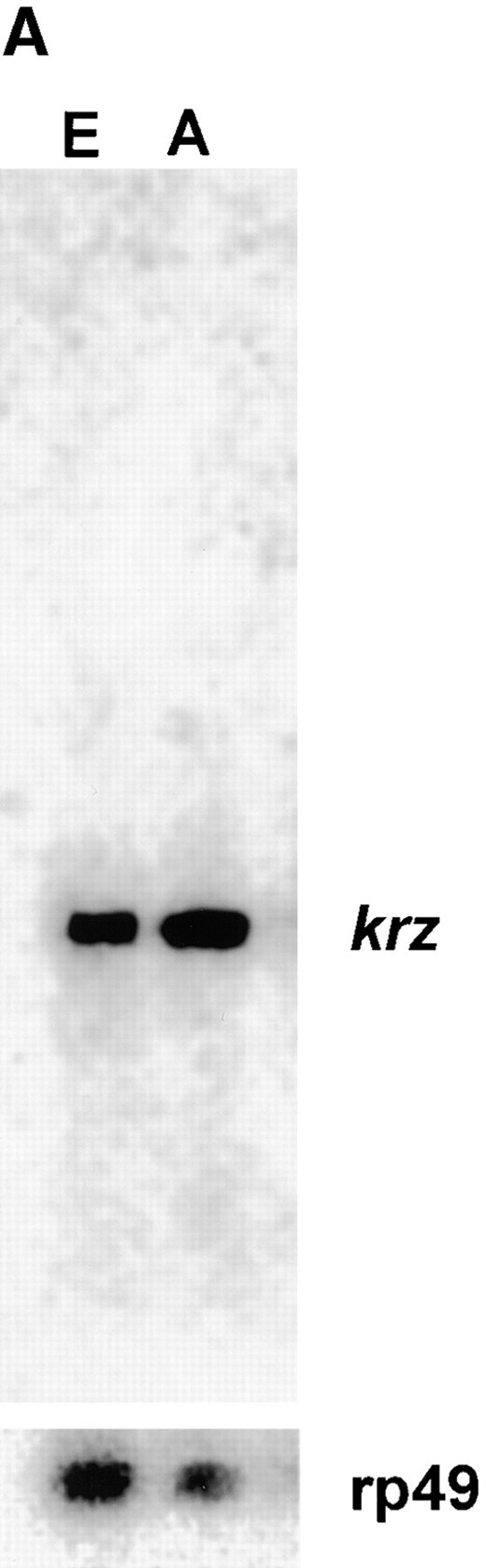
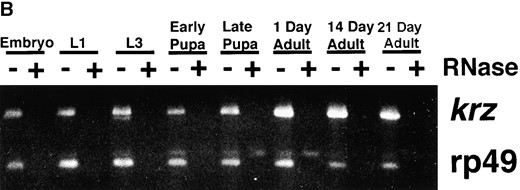
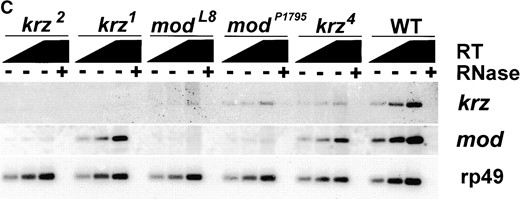
kurtz is expressed throughout development. (A) A Northern blot containing 5 μg poly(A)+ RNA from 0- to 18-hr embryos (E) and 0- to 7-day-old adult flies (A) was probed with a full-length kurtz cDNA. A single 2-kb message was detected. rp49 is shown as a loading control. (B) kurtz message is detected by RT-PCR in RNA from the selected stages of development. rp49 was coamplified as an internal control. The indicated lanes contained PCR reactions from reverse transcription reactions that were pretreated with RNAse. The primers used to amplify krz and rp49 cDNAs were located on either side of the respective introns, thus giving a transcript-specific product size. (C) krz mutants show decrements in transcript levels. RT-PCR analysis of poly(A)+ RNA from third instar larvae. PCR reactions were transferred to nylon and hybridized with radiolabled gene-specific probes. The graded bar indicates an increasing amount of input cDNA into the PCR reaction. In the indicated lanes, RNase was added prior to RT reaction as a control.
allele (Table 2). The krz1 mutation fully complements the male sterility of modP1795. Therefore, the krz1 mutation does not appreciably affect mod function, and the modlethal8 and modlethal3 mutations significantly reduce krz gene activity. Interestingly, krz2 fails to complement the semisterile phenotype of modP1795, suggesting that the krz2 mutation affects the function of both mod and krz loci.
Analysis of krz transcripts: A single 2.0-kb message was detected in both embryos and adults by Northern blots hybridized with a full-length krz cDNA probe (Figure 3A). These data suggest that we have identified a full-length cDNA clone. The krz transcript is detected by RT-PCR at each developmental stage and these messages are also found in aged adults (Figure 3B). Additionally, we utilized semiquantitative PCR to examine the relative levels of transcripts of krz and mod present in third instar larvae homozygous for the different mutations (Figure 3C). krz1, krz2, and modlethal8 are deficient in krz expression, and steady state mod transcript levels are reduced or completely missing in krz2, modlethal8, and modP1795. The relatively high level of mod transcripts we detected in modP1795 was initially unexpected since this mutation contains a 17-kb P element within the first intron of mod. However, it has been suggested that mod may have more than one transcription start site (Alexandre et al. 1996). If this is true, it may be that modP1795 disrupts only one of the mod transcriptional units, resulting in the hypomorphic phenotype. krz1 contains approximately wild-type levels of mod transcript at this stage. The krz4 mutation has only a minor reduction in krz expression. These data support our genetic finding that the krz1 mutation is a severe reduction of function that disrupts krz gene activity, while not significantly affecting mod expression. The krz2 and modlethal8 mutations are less specific, reducing or eliminating the expression of both krz and mod genes.
kurtz is expressed throughout development: The krz spatial expression pattern in embryos and late third instar larvae was examined by in situ hybridization (Figure 4 and 5). krz message is expressed in nurse cells and deposited in the developing oocytes (data not shown). Consistent with this result, we find abundant krz message in preblastoderm embryos (Figure 4A). krz remains ubiquitously expressed throughout gastrulation (Figure 4). In later stage embryos, krz becomes increasingly more localized. In stage 16 embryos, expression is primarily detected throughout the central nervous system. In stage 17 embryos, krz expression is also seen in the trachea and the dorsal pouch (Figure 4). The region of the dorsal pouch detected by krz antisense riboprobe does not include the eye-antennal disc anlagen (Younossi-Hartenstein et al. 1993). krz expression is also first detected during stage 17 in the antennal sense organ and the maxillary cirri (Figure 4). We also find significant staining to the cephalopharyngeal skeleton, including the mouth hooks and the cephalopharyngeal plates (Figure 4). We have not seen krz hybridization to fat bodies in late embryos.
In late third instar larvae, krz expression within the thoracic-abdominal ganglia is detected only in the ventral half (Figure 5). Within the brain of third instar larvae, krz expression is limited to the deuterocerebrum, which lies behind the eye-antennal imaginal disc (Figure 5).
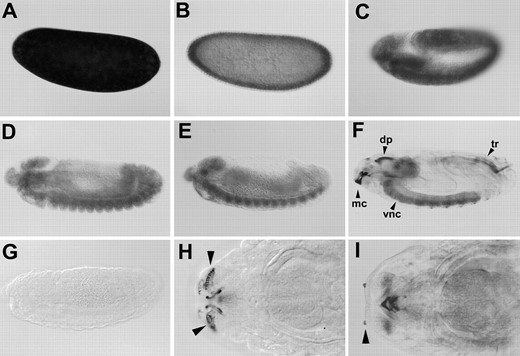
Spatial localization of krz transcripts within the developing embryo. In situ hybridizations with krz antisense riboprobe to embryos of different developmental stages are shown. The embryos are oriented such that the dorsal side is on the top and posterior is on the right. (A) An early preblastoderm embryo. (B) A stage 3 embryo. krz message is enriched in the cellular cortex layer. (C) An approximately stage 10 embryo. (D) A stage 13 embryo. (E) A stage 14 embryo. (F) A stage 17 embryo. Staining of trachea is seen only in the posterior of this animal. Typically, all trachea at this stage would stain. krz message is also detected throughout the CNS, the maxillary cirri, and the dorsal pouch. Staining is also detected within the pharynx at this time. (G) A stage 13 embryo hybridized with a krz sense riboprobe. (H) A ventral view of the maxillary cirri and mouth hooks of a stage 17 embryo. Arrows point to the maxillary cirri. (I) The antennal sensory organ of a stage 17 embryo. Magnification for (A) through (G) is ×20. Magnification for (H) and (I) is ×40. mc, maxillary cirri; dp, dorsal pouch; tr, trachea; vnc, ventral nerve chord.
krz is also expressed highly in the fat bodies (Figure 5). Expression is also seen in the eye-antennal and wing imaginal disc. krz is found throughout the eye-antennal disc, but the expression within the wing imaginal disc is more defined; krz is expressed uniformly in the notum region of the wing disc, but is also expressed in a thin stripe that winds through the wing primordium (Figure 5). krz message was not found to be sexually dimorphic in any larval tissues.
kurtz mutants have a broad lethal phase and form melanotic tumors: The lethal period of homozygous krz mutant animals was determined by directly observing homozygotes during development. The krz1, krz2, and modlethal8 mutations were placed over the TM3-pAct-GFP balancer (Reichhart and Ferrandon 1998). Homozygous mutants were selected as late-stage embryos lacking GFP. We analyzed 286, 187, 237, and 600 embryos of krz1, krz2, modlethal8, and wild type, respectively. krz1 and modlethal8 homozygous embryos were detected at ~25% of the total embryos screened. In contrast, the number of krz2 homozygous embryos was significantly lower than the expected 25% (χ2 = 8.5, P < 0.01). The krz2 mutation may therefore have a slight defect in transmission through gametes or lethality during early embryogenesis, but the effect appears small. A significant fraction (18%) of the control wild-type animals failed to hatch, but after hatching there were no further fatalities among these wild-type animals (Figure 6). In contrast, both krz1 and krz2 animals died throughout development (Figure 6). The majority of these mutants failed to hatch, and none of the surviving larvae managed to live to puparium formation. The modlethal8 homozygotes were slightly more robust than the krz1 and krz2 animals; a greater percentage hatched and 16% survived the third instar and died as early pupae (Figure 6). modlethal8 larvae also demonstrated significant delays in development. In fact, surviving modlethal8 homozygotes pupated up to 7 days after their heterozygous siblings.
Prior to death, krz1, krz2, and modlethal8 mutant larvae became immobile and flaccid. Mutant third instar larvae of all three genotypes formed melanotic tumors (Figure 7A). In krz1, krz2, and modlethal8 mutants, these tumors would form throughout the body cavity, but would most frequently collect in the posterior of the larvae as seen in Figure 7A. These tumors are frequently found floating freely in the haemocoel. In krz1 third instar larvae, using Nomarski optics on living specimens, large numbers of lamellocytes were seen circulating through the haemoceol. These flattened disc-shaped cells typically form in wild-type pupae (data not shown; Rizki 1957a).
The presence of melanotic tumors in krz mutants, at least in part, can be ascribed to the precocious disaggregation of fat bodies in the third instar larvae (Figure 7C). The larval fat body is a monolayer of cells that grow in multilobed sheets, extending the length of the larvae (Butterworth et al. 1965). This adipose tissue performs functions analogous to the vertebrate liver. Shortly after the second molt, the fat bodies of krz1 larvae have a rough appearance when compared to wild type. At ~18–20 hr after the second molt the fat bodies within surviving krz1 larvae begin to dissociate into individual cells (Figure 7, C and F). The wild-type larval fat body
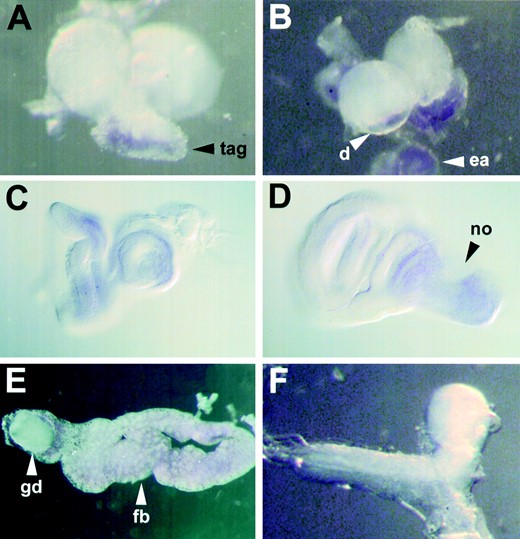
Spatial localization of krz transcripts within third instar larvae. In situ hybridizations with krz antisense riboprobe to dissected third instar larvae are shown. (A) A lateral/posterior view of a third instar larval CNS. krz transcripts are present in the ventral portion of the ventral nerve chord, but not in the posterior lobes of the brain. (B) A dorsal view of the brain with the eye-antennal imaginal disc removed. krz is detected in the antennal lobe region beneath the eye-antennal disc. (C) Eye-antennal imaginal disc visualized with Nomarski optics. (D) Wing imaginal disc visualized with Nomarski optics. (E) Dissected fat body with a gonadal disc. krz is expressed in all fat body cells of late third instar larvae. (F) Ventral view of a krz1 mutant CNS. No hybridization was detected in krz1 homozygotes. tag, thoracic-abdominal ganglia; d, deuterocerebrum; ea, eye-antennal disc; no, notum anlagen; gd, gonadal disc; fb, fat body.
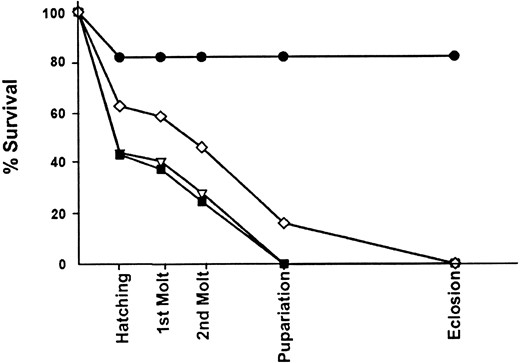
A death curve of krz mutants. Animals were selected as late-stage embryos and allowed to develop. The percentage of homozygous mutant animals that survived the indicated developmental events are shown. The genotypes are as follows: wild type, solid circles; krz1, open triangles; krz2, solid squares; modlethal8, open diamonds.
normally does not dissociate until after pupation (Butterworth et al. 1965). Melanotic tumors begin to form on the surface of the mutant fat bodies shortly after the second molt (Figure 7C). Early in their formation, these growths are irregularly shaped brown masses (Figure 7B). In the majority of krz1 third instar larvae examined these tumors continue to grow larger and darker until the death of the animal (Figure 7A). The formation of melanotic tumors and fat body disaggregation in krz2 is identical to krz1; however, modlethal8 larvae tend to form fewer melanotic tumors, and the fat body disaggregation is less severe.
Despite the precocious disaggregation of the fat bodies in the krz1 mutant larvae, this tissue maintains its identity during mid third instar. The Krüppel gene is transcriptionally activated in the fat bodies of mid third instar larvae (Hoshizaki 1994). We followed this activation with the CyO, P{w+ KrGFP30} balancer (Casso et al. 1999). This chromosome carries two P elements; in the first transposon Gal4 is expressed from a Krüppel promoter fragment and the second transposon carries an upstream activation sequence (UAS)-GFP reporter. Using this balancer, GFP was initially detected within the
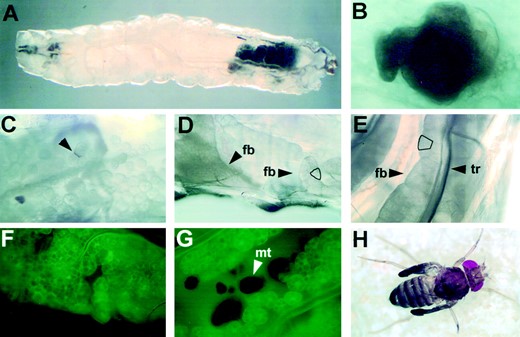
krz mutants have defects in fat body integrity and CNS function that can be functionally rescued by krz-specific transgenes. All images were taken of living animals. (A) A typical krz1 third instar larva is shown at ×5 magnification. The melanotic tumors in this animal have accumulated in the posterior end. (B) A Nomarski image of a recently formed melanotic tumor. This tumor was located within the fat body of a krz1 third instar larva. Melanotic tumors are frequently uneven in shape and formed by layers of lamellocytes. Magnification: ×100. (C) A Nomarski image of a krz1 homozygous late third instar larva. The fat bodies in this mutant animal have lost their integrity and are beginning to disaggregate. The arrow shows a melanotic tumor forming between two dissociated fat body cells. Magnification: ×20. (D) A Nomarski image of wild-type late third instar larval fat bodies. The left arrow shows the position of a fat body sheet. The right arrow indicates a fat body lobe near the cuticle wall. A representative fat body cell has been outlined. Magnification: ×20. (E) A Nomarski image of a P{b5.8T13}; krz1 late third instar larva. The 5.8-kb genomic XbaI fragment completely rescues the precocious dissociation of fat bodies in krz1 mutant animals. The trachea (tr) is seen on top of the fat body in this image. A representative fat body cell has been outlined. Magnification: ×20. (F) A CyO, P{w+,KrGFP30}/+; krz1 homozygous third instar larva ~20 hr after the second molt. In this line, GFP is expressed specifically in the fat bodies at this stage of development. Magnification: ×40. (G) A late third instar PUARRT5/CyO, P{w+,KrGFP30}; krz1 larva. In this animal the krüpple promoter/Gal4 construct drives the expression of both GFP and the krz cDNA in the fat body. The fat bodies have started to dissociate and melanotic tumors have formed. Magnification: ×40. (H) Expression of a krz cDNA in the nervous system rescues krz1 mutants. A 7-day-old c155/+; P{UARRT5}/+; krz1 adult is shown. Approximately one-third of rescued flies with this genotype are unable to unfurl their wings after eclosion. fb, fat body; tr, trachea; mt, melanotic tumor.
fat bodies of both wild-type and krz1 larvae at ~18–20 hr after the second molt (Figure 7, F and G). The fat bodies of krz1 third instar larvae are therefore still responding to the signals responsible for activation of the Krüppel promoter. Perhaps significantly, the start of fat body disaggregation roughly coincided with the activation of this promoter in krz1 mutant larvae.
Transgenic rescue of krz mutant phenotypes: To verify that the lethal and melanotic tumor phenotypes result from loss of krz activity, we functionally rescued the krz1 mutant with krz-specific transgenes. The lethal phenotype of modlethal8 was previously rescued by a 12-kb SphI genomic fragment that includes both the mod and krz genomic region (Pereira et al. 1992; Figure 1). We found that this same transgene, P{w+Sph}, also rescues krz1 and krz2 lethality. To further define the genomic region responsible for this lethality, we generated several transgenes that are missing the 3′ end of mod. Specifically, transgenes were generated containing a 5.8-kb XbaI fragment that begins within the mod coding region and terminates just 3′ to the krz locus (Figure 1). Two independent X-linked transgenes, P{b5.8T12} and P{b5.8T13}, completely rescued the krz1 lethal phenotype. Additionally, both the disaggregation of fat bodies and the formation of melanotic tumors in krz1 homozygous larvae are also complemented by these genomic constructs (Figure 7E). The P{b5.8T12}; modlethal8 animals do not form melanotic tumors; however, they fail to eclose and die as pharate adults. These animals also pupate up to 7 days after their P{b5.8T12}; modlethal8/TM6B siblings and have a distinct minute phenotype. Thus the fat body and larval lethal phenotypes of these mutations are due specifically to the reduction of krz gene activity. The inability of P{b5.8T12} to completely rescue modlethal8 lethality indicates that mod activity is also necessary for full viability and the loss of mod activity is responsible for the minute phenotype of this mutation. The lethality of krz3 was not rescued by either P{b5.8T12} or P{b5.8T13}, indicating that this mutation is affecting additional loci proximal to the krz locus.
We have generated additional transgenic lines that contain the full-length krz cDNA behind the synthetic Gal4/UAS promoter. The P{UARRT4} transgene is X linked, and the P{UARRT5} element is on the second chromosome. Temporal control of krz expression was achieved with the P{w+GAL4-Hsp70.PB}89-2-1 Gal4 effector (Brand and Perrimon 1993). This Gal4 effector, located on the third chromosome, was recombined onto
Rescue of kurtz1 lethality with induced expression of a krz cDNA
| Genotype | Heat shocka | Stage treatedb | Adult survivors (%) | Total no. of animals |
|---|---|---|---|---|
| + | 1st instar | 6 (14.6) | 41 | |
| − | 0 (0) | 57 | ||
| + | 1st instar | 0 (0) | 25 | |
| − | 0 (0) | 40 | ||
| + | 2nd instar | 5 (14.7) | 34 | |
| − | 0 (0) | 119 | ||
| + | 2nd instar | 0 (0) | 25 | |
| − | 0 (0) | 62 | ||
| + | Early 3rd instar | 27 (27.8) | 97 | |
| − | 0 (0) | 150 | ||
| + | Early 3rd instar | 11 (14.6) | 75 | |
| − | 0 (0) | 100 | ||
| + | Late 3rd instar | 72 (43.6) | 165 | |
| − | 0 (0) | 150 | ||
| + | Late 3rd instar | 27 (54) | 50 | |
| − | 0 (0) | 100 | ||
| + | Late 3rd instar | 0 (0) | 100 | |
| + | Late 3rd instar | 0 (0) | 100 |
| Genotype | Heat shocka | Stage treatedb | Adult survivors (%) | Total no. of animals |
|---|---|---|---|---|
| + | 1st instar | 6 (14.6) | 41 | |
| − | 0 (0) | 57 | ||
| + | 1st instar | 0 (0) | 25 | |
| − | 0 (0) | 40 | ||
| + | 2nd instar | 5 (14.7) | 34 | |
| − | 0 (0) | 119 | ||
| + | 2nd instar | 0 (0) | 25 | |
| − | 0 (0) | 62 | ||
| + | Early 3rd instar | 27 (27.8) | 97 | |
| − | 0 (0) | 150 | ||
| + | Early 3rd instar | 11 (14.6) | 75 | |
| − | 0 (0) | 100 | ||
| + | Late 3rd instar | 72 (43.6) | 165 | |
| − | 0 (0) | 150 | ||
| + | Late 3rd instar | 27 (54) | 50 | |
| − | 0 (0) | 100 | ||
| + | Late 3rd instar | 0 (0) | 100 | |
| + | Late 3rd instar | 0 (0) | 100 |
Larvae were heat shocked at 37° for 1 hr and then returned immediately to 23° (+) or left at 23° (−).
The larvae were selected at the indicated stages for heat-shock or control treatments. Early third instar represents 0–12 hr after the second molt. Late third instar larvae were collected and treated 24–48 hr after the second molt.
Rescue of kurtz1 lethality with induced expression of a krz cDNA
| Genotype | Heat shocka | Stage treatedb | Adult survivors (%) | Total no. of animals |
|---|---|---|---|---|
| + | 1st instar | 6 (14.6) | 41 | |
| − | 0 (0) | 57 | ||
| + | 1st instar | 0 (0) | 25 | |
| − | 0 (0) | 40 | ||
| + | 2nd instar | 5 (14.7) | 34 | |
| − | 0 (0) | 119 | ||
| + | 2nd instar | 0 (0) | 25 | |
| − | 0 (0) | 62 | ||
| + | Early 3rd instar | 27 (27.8) | 97 | |
| − | 0 (0) | 150 | ||
| + | Early 3rd instar | 11 (14.6) | 75 | |
| − | 0 (0) | 100 | ||
| + | Late 3rd instar | 72 (43.6) | 165 | |
| − | 0 (0) | 150 | ||
| + | Late 3rd instar | 27 (54) | 50 | |
| − | 0 (0) | 100 | ||
| + | Late 3rd instar | 0 (0) | 100 | |
| + | Late 3rd instar | 0 (0) | 100 |
| Genotype | Heat shocka | Stage treatedb | Adult survivors (%) | Total no. of animals |
|---|---|---|---|---|
| + | 1st instar | 6 (14.6) | 41 | |
| − | 0 (0) | 57 | ||
| + | 1st instar | 0 (0) | 25 | |
| − | 0 (0) | 40 | ||
| + | 2nd instar | 5 (14.7) | 34 | |
| − | 0 (0) | 119 | ||
| + | 2nd instar | 0 (0) | 25 | |
| − | 0 (0) | 62 | ||
| + | Early 3rd instar | 27 (27.8) | 97 | |
| − | 0 (0) | 150 | ||
| + | Early 3rd instar | 11 (14.6) | 75 | |
| − | 0 (0) | 100 | ||
| + | Late 3rd instar | 72 (43.6) | 165 | |
| − | 0 (0) | 150 | ||
| + | Late 3rd instar | 27 (54) | 50 | |
| − | 0 (0) | 100 | ||
| + | Late 3rd instar | 0 (0) | 100 | |
| + | Late 3rd instar | 0 (0) | 100 |
Larvae were heat shocked at 37° for 1 hr and then returned immediately to 23° (+) or left at 23° (−).
The larvae were selected at the indicated stages for heat-shock or control treatments. Early third instar represents 0–12 hr after the second molt. Late third instar larvae were collected and treated 24–48 hr after the second molt.
the same chromosome as the krz1 mutation and balanced by TM6B. We have found that heat-induced krz activity is sufficient to functionally rescue some of the krz1 mutants at each larval instar (Table 3). Furthermore, the functional rescue of lethality became significantly more efficient at older stages. In the majority of late third instar larvae, melanotic tumors were visible throughout the body prior to heat-shock induction of krz. Despite the presence of large numbers of tumors, substantial rescue was achieved.
Since krz expression during mid to late third instar was primarily restricted to the fat bodies and a subset of the central nervous system, we examined whether expression in these tissues could rescue viability and the precocious disaggregation of the larval fat bodies. In the CyO, P{w+ KrGFP30} balancer, Gal4 is expressed behind the Krüppel promoter (Casso et al. 1999). Gal4 expression in this line begins within the fat bodies at ~18–20 hr after the second molt. Neither Kr-Gal4-P{UARRT4} nor Kr-Gal4-P{UARRT5} expression rescued krz1 lethality. In fact, these animals continued to develop melanotic tumors and their fat bodies continued to dissociate in a manner indistinguishable from krz1 (Figure 7E). The expression of krz in these animals was therefore too little or too late to affect the fat body disaggregation.
To see if krz expression within the nervous system is sufficient for viability, we used the neural-specific c155 Gal4 effector to drive krz expression. Gal4 is constitutively expressed pan-neurally during development in c155 (Lin and Goodman 1994). Additionally, Gal4 activity is not detected in larval fat bodies within c155 (G. Roman and R. L. Davis, unpublished observation). From the cross c155/Y; krz1/TM6B males by P{UARRT4}; krz1/TM6B females, we recovered 16 c155/P{UARRT4}; krz1 adult females out of 47 total females. When this cross was repeated with P{UARRT5}; krz1/TM6B females, there were only 8 c155/+; P{UARRT5}/+; krz1 females out of 46 total females. We did not detect any male krz1 homozygotes in these two crosses; the male progeny from these crosses will not carry the c155 Gal4 effector and therefore should not be rescued. The rescued krz1 adults did not display any gross developmental abnormalities and lived for several weeks after eclosion (Figure 7F). Approximately one-third of these rescued females failed to expand their wings after eclosion (Figure 7E). Additionally, melanotic tumors were visible in the abdomens of most of the rescued females (data not shown). Thus, the expression of krz within neurons is essential for viability. This gene is also required for the integrity of larval fat bodies during the third instar and functions in wing expansion.
DISCUSSION
In this article, we present the molecular and genetic characterization of the kurtz gene from D. melanogaster. krz encodes a novel member of the nonvisual arrestin family of adaptor proteins. This gene is expressed ubiquitously during early embryogenesis and later becomes more localized to several tissues including the larval fat bodies and the central nervous system in both embryos and third instar larvae. We have characterized several mutations that disrupt krz. krz1 is specific severe reduction-of-function mutation caused by the insertion of a P{lacW} within this gene's intron. Mutations that disrupt krz expression have a broad lethal phase. The targeted expression of krz within the nervous system functionally rescues the lethality of the krz1 mutation. Additionally, the third instar fat bodies of krz1 mutants precociously disaggregate into single cells. These fat bodies are the site of significant melanotic tumor formation.
The krz gene was serendipitously identified in an enhancer detector screen by virtue of reporter expression in male, but not female, fat bodies. krz messages were detected in adult and larval fat body of both sexes by in situ hybridization (data not shown). We also failed to detect any sexual dimorphism in krz expression in other larval somatic tissues. We decided to pursue this gene because of the expected importance of nonvisual arrestins in regulating nervous system function. The arrestins are subdivided into two categories: the visual and nonvisual arrestins (Craft and Whitmore 1995). The visual arrestins are more specialized, desensitizing photoreceptors in the vertebrate retina, whereas the nonvisual arrestins are ubiquitously expressed and interact with GPCRs from several families (Gurevich et al. 1995). The previously identified antennal arrestins from the insects Locusta and Heliothis were thought to form a third group of arthropod arrestins, perhaps specific for olfactory receptors (Raming et al. 1993). The krz arrestin is more similar to the vertebrate βARR1 and βARR2 than to these insect arrestins. We have classified krz as a novel nonvisual arrestin on the basis of two criteria. First, krz is expressed widely in nonvisual tissue, including the central nervous system. Second, krz shares structural features found in the βARR1 and βARR2 nonvisual arrestins, but not in rod or cone arrestins. These features include the clathrin-binding domain and the phosphoinositide interaction domain (Goodman et al. 1996; Gaidarov et al. 1999). We suspect that, similar to βARR1 and βARR2, krz will interact with a broad array of GPCRs. Additionally, these conserved domains also suggest that krz will interact with clathrin and phosphoinositides to sequester the GPCRs after desensitization. One of the two potential SH3 binding sites present in βARR1 is conserved within krz. Thus it is possible that krz can transactivate mitogenic signaling pathways analogous to βARR1.
The specificity of krz mutations: We have examined the phenotypes of a number of mutations that disrupt krz function. The krz1 mutation appears to be the most specific. In this mutation, a P{lacW} element is inserted within the only intron of this gene, resulting in a severe reduction of function. There is an absence of detectable transcript in late embryos and third instar larvae in krz1 as analyzed by in situ hybridization. In addition, very little transcript can be found by RT-PCR. When krz1 is placed over the deficencies, Df(3R)faf-BP or Df(3R)td106, the third instar phenotype appears unchanged (unpublished data). Despite the close proximity of the krz and mod genes, krz1 appears not to disrupt mod. There are near wild-type levels of mod in krz1 homozygous third instar larvae. Also, krz1 fully complements the male sterility phenotype of modP1795. The krz1 mutant phenotypes have been unambiguously mapped back to the krz gene. Precise excision of the P element in krz1 reverts the lethality and melanotic tumor phenotypes. The functional rescue of krz1 with transgenes containing the 5.8-kb genomic XbaI fragment results in animals with no obvious phenotype. Additionally, we functionally rescued krz1 animals with cDNA transgenes. Taken together, these data demonstrate that the krz1 phenotypes specifically result from the loss of krz activity.
The krz2 and modlethal8 mutations are also strong reduction-of-function alleles of krz but these mutations disrupt mod as well. modlethal8 is a deletion of ~21 kb that includes MAP205K, modulo, and the immediate promoter region of krz. The mod gene product is a nucleic acid binding protein that shares significant similarity to nucleolin (Perrin et al. 1998). This similarity and the subnuclear localization of mod to the nucleolus suggest a role in the synthesis of ribosomal proteins (Perrin et al. 1998, 1999). Mutations in ribosomal proteins frequently give rise to the minute phenotype, characterized by a dominant reduction in bristle length and width and a significant retardation in development (Lambertsson 1998). Using mosaic analysis Perrin et al. (1998) demonstrated that cuticle clones homozygous for modlethal8 have short slender bristles similar to that of the minute phenotype. We have found a delay in pupation in modlethal8 homozygotes and in P{b5.8T12}; modlethal8 animals of up to 7 days. Additionally, P{b5.8T12}; modlethal8 pharate adults have short, slender bristles. These two phenotypes are consistent with mod acting within the nucleolus to regulate synthesis of ribosomal proteins. The modlethal8 mutation is also a dominant suppressor of position-effect variegation (Garzino et al. 1992). This phenotype is most likely to be due to the effect of this mutation of the mod gene. On the other hand, the broad lethal phase and melanotic tumor formation of modlethal8 are due to the effects of this mutation on krz. The modP1795 mutation does not significantly affect krz activity and suggests that mod is required for male fertility. The homozygous modP1795 flies eclose without a significant delay and the bristle phenotype is very subtle. In this mutation, mod levels are detectable but reduced in third instar larvae, indicating a weak reduction of function.
The phenotypic analysis of krz mutations: The larval fat bodies of Drosophila are multilobed monolayer sheets of cells born during embryogenesis. This organ performs larval functions analogous to the vertebrate liver. In wild-type animals, the larval fat bodies remain intact until shortly after pupation, when they begin to disaggregate into single cells (Butterworth et al. 1965). The larval fat bodies begin to die during metamorphosis, but a substantial number survive until shortly after eclosion (Butterworth et al. 1965; Butterworth 1972). In krz1 homozygous animals, this disaggregation begins ~18 hr after the second larval molt. The krz gene is expressed in fat body at mid third instar and probably before this time. The Krüppel gene is activated within third instar larval fat bodies in response to the first pulse of ecdysone at approximately the same time that fat body disaggregation begins in krz1 mutants. The expression of a wild-type krz cDNA within krz1 third instar larval fat bodies at this time cannot appreciably affect the progression of the disaggregation. The failure to affect rescue may be due to inadequate levels of arrestin or to expression after a critical point. In support of the latter of these two possibilities, fat body morphology in krz1 mutants appears to be altered shortly after the second molt, suggesting a role for krz in fat body integrity early in the third instar.
During this early third instar period, melanotic tumors may begin to form on the fat bodies of krz1 larvae. The formation of melanotic tumors is an integral feature of the cellular immune response of Drosophila (Sparrow 1978). These masses form when hemocytes attach to aberrant or foreign tissue. Subsequently, these hemocytes transform to flattened lamellocytes and encapsulate the foreign tissue (Rizki 1957b, 1960). Mutations that result in ectopic melanotic tumor formation have been characterized as class 1, which result from a normal immune response to abnormal tissue, and class 2, in which tumor formation is the result of defects in the hematopoietic organ or the hemocytes (Watson et al. 1991). The melanotic tumor phenotype of krz1 is very similar to the phenotype of the tuw class 1 mutation (Rizki 1957b; Rizki and Rizki 1974). This mutation provides a precedent for understanding the formation of melanotic tumors within krz mutant third instar larvae. tuw is located on the second chromosome, but has not been cloned (Rizki and Rizki 1981). In this mutant strain, the larval fat body basement membrane begins breaking down early in the third instar (Rizki and Rizki 1974). This results in a precocious dissociation of the fat body cells in mid to late third instar. The transformation of plasmatocytes into lamellocytes, which normally occurs after pupariation, occurs shortly after the second molt in tuw homozygous larvae (Rizki 1957b). These lamellocytes begin to encapsulate fat body tissue during the mid third instar (Rizki 1957b, 1960; Rizki and Rizki 1974). The precocious differentiation of lamellocytes also occurs in krz1 early third instar larvae. We did not detect any krz expression in the Drosophila hematopoietic organ, the lymph glands. Since these glands are full of hemocytes, krz is unlikely to be expressed in these cells as well. The dissociation of fat body cells within krz1 would require the breakdown of the basement membrane as seen in the tuw mutation. The precocious differentiation of the lamellocytes in krz1 larvae is most likely induced by the fat bodies that are no longer sheltered by this basement membrane and therefore are substrates for hemocyte attachment. Thus, krz is required for fat body integrity and without arrestin activity the fat bodies dissociate and become substrates for lamellocyte aggregation and melanotic tumor formation.
There is not a single developmental point at which krz1 mutants die, but rather their deaths arrive stochastically. During the three larval instars, animals simply turn flaccid and remain motionless, sometimes for more than an hour, before dying. The dorsal vessel continues to pump hemolymph during this motionless period. The fat body abnormality is not a significant cause of lethality in krz1. This point is demonstrated by the functional rescue of mutant larvae by inducing krz expression in late third instar larvae and through the rescue of mutant larvae with krz expression delimited to the central nervous system (CNS). In both cases, the fat bodies dissociate and melanotic tumors form; these tumors persist through adulthood. The functional rescue of krz1 with the c155 Gal4 effector driving the krz cDNA indicates that lethality is due to a primary nervous system defect. Within the CNS, krz is expressed ubiquitously during late embryogenesis, but by the late third instar is more localized to the abdominal-thoracic ganglia and the dueterocerebrum. The CNS is a major site of GPCR activity. This class of receptors transduces signals from most if not all neurotransmitters and neuromodulators (Watson and Arkinstall 1994). A failure to desensitize GPCRs within the krz1 nervous system may lead to a long-term downregulation of receptors due to heterologous mechanisms such as post-translational modification or may simply lead to increased signaling and hypersensitivity within affected neurons. In either of these scenarios, neuronal excitability would be grossly altered, eventually leading to a lethal failure of nervous system function.
Acknowlegement
We thank D. Kimbrell and V. Hartenstein for helpful discussions. We also thank K. Posey for the developmentally staged RNAs and K. Endo for technical assistance. This research was supported by Grant DR-1344 from the Damon Runyon-Walter Winchell Cancer Research Foundation (G.R.) and by National Institute of Mental Health Grant 1-RO1-H/NS-55230 and the R. P. Doherty-Welch Chair in Science (R.L.D.).
Footnotes
This gene is named kurtz after the character in Joseph Conrad's The Heart of Darkness in which Conrad brilliantly and symbolically uses the contrast of light and dark. The character Kurtz is defined by his darkness and is an excellent metaphor for our arrestin mutants.
Communicating editor: K. Anderson
LITERATURE CITED

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}