





Abstract
Haloperidol is an effective antipsychotic agent, but it causes Parkinsonian-like extrapyramidal symptoms in the majority of treated subjects. To address this treatment-limiting toxicity, we analyzed a murine genetic model of haloperidol-induced toxicity (HIT). Analysis of a panel of consomic strains indicated that a genetic factor on chromosome 10 had a significant effect on susceptibility to HIT. We analyzed a whole-genome SNP database to identify allelic variants that were uniquely present on chromosome 10 in the strain that was previously shown to exhibit the highest level of susceptibility to HIT. This analysis implicated allelic variation within pantetheinase genes (Vnn1 and Vnn3), which we propose impaired the biosynthesis of cysteamine, could affect susceptibility to HIT. We demonstrate that administration of cystamine, which is rapidly metabolized to cysteamine, could completely prevent HIT in the murine model. Many of the haloperidol-induced gene expression changes in the striatum of the susceptible strain were reversed by cystamine coadministration. Since cystamine administration has previously been shown to have other neuroprotective actions, we investigated whether cystamine administration could have a broader neuroprotective effect. Cystamine administration caused a 23% reduction in infarct volume after experimentally induced cerebral ischemia. Characterization of this novel pharmacogenetic factor for HIT has identified a new approach for preventing the treatment-limiting toxicity of an antipsychotic agent, which could also be used to reduce the extent of brain damage after stroke.
HALOPERIDOL is a potent D2 dopamine receptor (DRD2) antagonist that is used to treat psychotic disorders (Beresford and Ward 1987). However, haloperidol-induced alterations in the extrapyramidal motor system depress the ability to initiate voluntary movements. As a consequence, characteristic extrapyramidal symptoms (EPS), which include tremors, Parkinsonian rigidity, and decreased spontaneous movement, develop in 40–76% of human subjects that are chronically treated with haloperidol (Parkes 1982; Soares-Weiser and Fernandez 2007). We recently analyzed a murine genetic model of haloperidol-induced toxicity (HIT) (Zheng et al. 2015) in which the Parkinsonian-like symptoms developing in haloperidol-treated mice resemble those observed in drug-treated human patients (Crowley et al. 2012a). The 27 inbred strains evaluated in this genetic model exhibit substantial variability in susceptibility to HIT (Crowley et al. 2012b). For example, C57BL/6 mice are completely resistant, while A/J mice are highly susceptible to the Parkinsonian hypokinesia associated with HIT. We found that allelic differences in Abcb5, a member of the ABC-transporter gene family, affected susceptibility to this manifestation of HIT by regulating the level of haloperidol in the brain (Zheng et al. 2015). This pharmacogenetic effect is of importance, since haloperidol is oxidatively metabolized to a pyridinium species (HPP+) that is present in brain tissue of haloperidol-treated humans (Eyles et al. 1997) and rodents (Igarashi et al. 1995). HPP+ is a potent inhibitor of mitochondrial complex I activity (Rollema et al. 1994) and is toxic to dopaminergic neurons (Bloomquist et al. 1994; Rollema et al. 1994; Eyles et al. 1997).
Given the extreme differences in the extent of HIT manifested by the inbred strains and the fact that Abcb5 alleles only partially (∼33%) account for their susceptibility, it was likely that other genetic factors also affected this response. Since relatively little is known about the genetic mechanisms affecting susceptibility to drug-induced CNS toxicities, we comprehensively characterized the genetic architecture underlying susceptibility to HIT. We identified a second pharmacogenetic factor that affects susceptibility to HIT by inhibiting cysteamine biosynthesis. This genetic discovery enables a novel therapeutic approach for preventing HIT and possibly for reducing brain damage after ischemic injury.
Materials and Methods
Haloperidol studies
All animal experiments were performed according to protocols approved by the Stanford Institutional Animal Care and Use Committee and the results are reported according to the Animal Research: Reporting of In Vivo Experiments guidelines (Kilkenny et al. 2010). C57BL/6J, A/J, and all consomic mice were obtained from the Jackson Laboratory (Bar Harbor, ME) and were used at 15 weeks of age. Nineteen different consomic strains were evaluated, and each consomic strain is homosomic for a single specified A/J chromosome on an otherwise C57BL/6 genetic background (Nadeau et al. 2000). A haloperidol (Sigma-Aldrich, St. Louis) stock solution of 1.0 mg/ml was made in phosphate buffered saline (PBS) containing 10% ethanol and 0.5% acetic acid, and the pH was adjusted to 7.0, using sodium hydroxide. A cystamine dihydrochloride (Sigma-Aldrich) stock solution of 1 mg/ml was made in PBS. The mice were treated with haloperidol (10 mg⋅kg−1⋅day−1 ip) for 4 days and the extent of HIT was measured as previously described (Zheng et al. 2015). Some mice were treated with cystamine (10 mg⋅kg−1⋅day−1 ip) for 3 days before haloperidol treatment was initiated. The HPP+ levels in brain, liver, and muscle were measured using the mass spectroscopy methods described in Zheng et al. (2015).
Microarray analysis
Whole brain was collected 4 hr after the last drug dose and stored in RNAlater (Life Technologies, San Diego). Coronal blocks of striatum were microdissected using a mouse brain slicer matrix (Zivic Instruments, Pittsburgh). RNA was extracted from the striatal tissue, using an RNAeasy Plus Universal Kit (QIAGEN, San Diego), and stored at −80° prior to analysis. Gene expression profiling of striatal tissue RNA was performed using Illumina (San Diego) Mouse WG-6 v2.0 Expression BeadChips, and the arrays were processed at the Stanford Functional Genomics Facility. Raw microarray data of striatum samples from untreated C57BL/6J mice (n = 3), C57BL/6J mice treated with haloperidol (n = 4), untreated A/J mice (n = 3), A/J mice treated with haloperidol (n = 4), and A/J mice treated with both haloperidol and cysteamine (n = 4) were obtained. The raw expression intensities were processed and normalized among all the included arrays by the quantile-normalization method, using the LIMMA package (Smyth 2004) and R (www.r-project.org).
Our goal was to identify genes that were differentially altered by haloperidol treatment in A/J relative to C57BL/6 mice and whose response to haloperidol treatment was fully or partially reversed by coadministration of cystamine. In other words, we performed a trend analysis to identify those genes whose expression levels displayed a correlation with the toxic effect of haloperidol and the protective effect of cystamine coadministration. A full-reversal pattern can be represented as a 0-1-0 pattern: the gene had a baseline expression level under the control condition, haloperidol treatment induced a change in the expression level, and cystamine coadministration returned its expression back to the baseline level. Similarly, a partial-reversal trend can be represented as a 0-1-0.5 pattern. To identify these genes, two linear regression models with prespecified covariates (i.e., 0/1/0 for baseline/haloperidol only/haloperidol coadministered with cystamine, respectively, for the full-reversal scenario and 0/1/0.5 for the partial-reversal scenario) were fitted to the gene expression data. Since the linear regression models were applied to all the genes (∼25,000) present on the array simultaneously, an empirical Bayes method (Smyth 2004) was applied to obtain more robust estimation of the residual standard deviations, which was accomplished using the LIMMA package in R. P-values were then adjusted for multiple-testing correction, using the Benjamini–Hochberg method (Benjamini and Hochberg 1995). Since expression changes in the brain were usually very small, none of the genes reached statistical significance after multiple-testing correction. Therefore, we elected to filter the gene expression changes measured on the microarray by a two-step process. First, we analyzed the microarray data, using unadjusted P-values. Then, RT-PCR results were used to rigorously confirm whether the genes identified were indeed differentially expressed. This two-step approach was employed because application of a multiple-testing correction for microarray data (25,000 gene probes) would be overly conservative sand would falsely remove genes that were truly differentially expressed. Genes with an absolute fold change >1.5-fold (haloperidol vs. control) and an unadjusted P-value <0.1 were identified as differentially expressed in each of the models. Then, the genes identified by either model were combined into a single list that contained the genes whose expression was altered by haloperidol treatment, which was reversed by cysteamine treatment (Supplemental Material, Table S2). Second, since A/J and C57BL/6 mice had different responses to haloperidol treatment, we sought to identify genes that were differentially induced by haloperidol treatment in A/J, but not in C57BL/6 mice. This was accomplished by fitting the expression data to an ANOVA model, which contained an interaction term (expression ∼ strain + treatment + strain * treatment). When this ANOVA model was fitted to genes whose expression levels were differentially induced by haloperidol in A/J relative to C57BL/6 mice, the interaction effect was expected to be large and significant. We then evaluated the ANOVA model for the ∼25,000 genes on the array simultaneously and applied the empirical Bayes method to obtain a more robust estimation of the residual standard deviations, using the LIMMA package as described above. Next, the subset of these genes with a significant interaction effect (strain * treatment) was identified. Genes with an absolute fold change >1.5 in A/J relative to C57BL/6 striatum and unadjusted P-value <0.1 were identified as the genes whose expression was altered by haloperidol in a strain-specific manner (Table S3). The genes common to both lists (Table S1 and Table S2) were identified (Table 1). These genes, along with their fold change values (haloperidol treated vs. control A/J mice), were then analyzed using the Ingenuity Pathway Analysis software package (QIAGEN) for functional enrichment analysis.
Gene expression changes associated with haloperidol-induced toxicity
| A/J hal vs. ctrl | A/J hal + cys vs. ctrl | C57BL/6 hal vs. ctrl | ||||
|---|---|---|---|---|---|---|
| Gene symbol | FC | P | FC | P | FC | P |
| 1110059M19Rik | −1.72 | 0.02 | −1.61 | 0.04 | 1.16 | 0.66 |
| 2810046M22Rik | −1.69 | 0.08 | −1.59 | 0.12 | 1.67 | 0.24 |
| 2900017F05Rik | −1.86 | 0.03 | −1.41 | 0.19 | 1.29 | 0.30 |
| A130082M07Rik | −1.73 | 0.09 | −1.40 | 0.28 | −1.00 | 0.98 |
| A430103B12Rik | −1.69 | 0.03 | −1.36 | 0.17 | 1.07 | 0.69 |
| Aqp1 | −2.14 | 0.01 | −2.21 | 0.01 | 1.47 | 0.41 |
| Arc | 1.65 | 0.16 | 1.04 | 0.90 | −1.66 | 0.04 |
| Calml4 | −1.81 | 0.08 | −1.61 | 0.15 | 1.21 | 0.50 |
| Car14 | −1.59 | 0.01 | −1.41 | 0.04 | 1.05 | 0.69 |
| Cdkn1c | −1.63 | 0.02 | −1.47 | 0.05 | 1.35 | 0.34 |
| Cit | −1.86 | 0.06 | −1.25 | 0.46 | 1.01 | 0.89 |
| Cldn2 | −2.27 | 0.01 | −2.30 | 0.01 | 1.21 | 0.62 |
| Col8a1 | −1.50 | 0.04 | −1.38 | 0.09 | 1.33 | 0.21 |
| Col8a2 | −1.63 | 0.04 | −1.58 | 0.05 | 1.17 | 0.49 |
| D230046H12Rik | −2.32 | 0.03 | −1.94 | 0.07 | 1.22 | 0.53 |
| Enpp2 | −2.25 | 0.01 | −1.81 | 0.03 | 1.28 | 0.48 |
| Fam171a1 | 2.10 | 0.01 | 2.08 | 0.01 | −1.33 | 0.42 |
| Folr1 | −2.69 | 0.01 | −2.60 | 0.02 | 1.29 | 0.60 |
| Fosb | 1.88 | 0.02 | 1.46 | 0.14 | 1.06 | 0.68 |
| Irs2 | −1.71 | 0.04 | −1.15 | 0.57 | 1.10 | 0.58 |
| Ncor1 | 1.83 | 0.01 | 1.84 | 0.01 | −1.10 | 0.74 |
| Nrgn | 2.64 | 0.10 | 1.70 | 0.35 | −1.18 | 0.69 |
| Peg3 | −1.76 | 0.02 | −1.21 | 0.39 | 1.09 | 0.48 |
| Pmch | −9.77 | 0.08 | −2.16 | 0.52 | 1.60 | 0.66 |
| Ptk2b | 1.94 | 0.06 | 1.40 | 0.30 | −1.03 | 0.90 |
| Pwwp2b | 1.56 | 0.07 | 1.35 | 0.20 | −1.07 | 0.65 |
| Scn1a | −1.70 | 0.06 | −1.19 | 0.51 | 1.20 | 0.43 |
| Slc16a8 | −1.56 | 0.01 | −1.48 | 0.02 | 1.30 | 0.43 |
| Slc4a2 | −1.56 | 0.01 | −1.29 | 0.13 | 1.06 | 0.76 |
| Sostdc1 | −3.78 | 0.02 | −4.14 | 0.01 | 1.20 | 0.70 |
| Wfdc2 | −2.59 | 0.01 | −2.38 | 0.01 | 1.28 | 0.67 |
| Wipf3 | 2.09 | 0.08 | 1.27 | 0.54 | −1.14 | 0.58 |
| A/J hal vs. ctrl | A/J hal + cys vs. ctrl | C57BL/6 hal vs. ctrl | ||||
|---|---|---|---|---|---|---|
| Gene symbol | FC | P | FC | P | FC | P |
| 1110059M19Rik | −1.72 | 0.02 | −1.61 | 0.04 | 1.16 | 0.66 |
| 2810046M22Rik | −1.69 | 0.08 | −1.59 | 0.12 | 1.67 | 0.24 |
| 2900017F05Rik | −1.86 | 0.03 | −1.41 | 0.19 | 1.29 | 0.30 |
| A130082M07Rik | −1.73 | 0.09 | −1.40 | 0.28 | −1.00 | 0.98 |
| A430103B12Rik | −1.69 | 0.03 | −1.36 | 0.17 | 1.07 | 0.69 |
| Aqp1 | −2.14 | 0.01 | −2.21 | 0.01 | 1.47 | 0.41 |
| Arc | 1.65 | 0.16 | 1.04 | 0.90 | −1.66 | 0.04 |
| Calml4 | −1.81 | 0.08 | −1.61 | 0.15 | 1.21 | 0.50 |
| Car14 | −1.59 | 0.01 | −1.41 | 0.04 | 1.05 | 0.69 |
| Cdkn1c | −1.63 | 0.02 | −1.47 | 0.05 | 1.35 | 0.34 |
| Cit | −1.86 | 0.06 | −1.25 | 0.46 | 1.01 | 0.89 |
| Cldn2 | −2.27 | 0.01 | −2.30 | 0.01 | 1.21 | 0.62 |
| Col8a1 | −1.50 | 0.04 | −1.38 | 0.09 | 1.33 | 0.21 |
| Col8a2 | −1.63 | 0.04 | −1.58 | 0.05 | 1.17 | 0.49 |
| D230046H12Rik | −2.32 | 0.03 | −1.94 | 0.07 | 1.22 | 0.53 |
| Enpp2 | −2.25 | 0.01 | −1.81 | 0.03 | 1.28 | 0.48 |
| Fam171a1 | 2.10 | 0.01 | 2.08 | 0.01 | −1.33 | 0.42 |
| Folr1 | −2.69 | 0.01 | −2.60 | 0.02 | 1.29 | 0.60 |
| Fosb | 1.88 | 0.02 | 1.46 | 0.14 | 1.06 | 0.68 |
| Irs2 | −1.71 | 0.04 | −1.15 | 0.57 | 1.10 | 0.58 |
| Ncor1 | 1.83 | 0.01 | 1.84 | 0.01 | −1.10 | 0.74 |
| Nrgn | 2.64 | 0.10 | 1.70 | 0.35 | −1.18 | 0.69 |
| Peg3 | −1.76 | 0.02 | −1.21 | 0.39 | 1.09 | 0.48 |
| Pmch | −9.77 | 0.08 | −2.16 | 0.52 | 1.60 | 0.66 |
| Ptk2b | 1.94 | 0.06 | 1.40 | 0.30 | −1.03 | 0.90 |
| Pwwp2b | 1.56 | 0.07 | 1.35 | 0.20 | −1.07 | 0.65 |
| Scn1a | −1.70 | 0.06 | −1.19 | 0.51 | 1.20 | 0.43 |
| Slc16a8 | −1.56 | 0.01 | −1.48 | 0.02 | 1.30 | 0.43 |
| Slc4a2 | −1.56 | 0.01 | −1.29 | 0.13 | 1.06 | 0.76 |
| Sostdc1 | −3.78 | 0.02 | −4.14 | 0.01 | 1.20 | 0.70 |
| Wfdc2 | −2.59 | 0.01 | −2.38 | 0.01 | 1.28 | 0.67 |
| Wipf3 | 2.09 | 0.08 | 1.27 | 0.54 | −1.14 | 0.58 |
Gene expression changes were measured using microarrays in striatal tissue obtained from A/J or C57BL/6 mice after treatment with haloperidol (10 mg⋅kg−1⋅day−1 × 4 days) or from A/J mice treated with cysteamine (10 mg⋅kg−1⋅day−1) for 3 days before and during a 4-day period of haloperidol (10 mg⋅kg−1⋅day−1) treatment. The 32 genes whose level of mRNA expression in striatal tissue was altered by haloperidol treatment in A/J (but not C57BL/6) mice and was reversed by cystamine treatment are shown. The fold change (FC) and unadjusted P-value for the indicated comparisons are shown. A minus (–) indicates that the mRNA level was decreased by haloperidol treatment. hal, haloperidol; ctrl, control; cys, cysteamine.
| A/J hal vs. ctrl | A/J hal + cys vs. ctrl | C57BL/6 hal vs. ctrl | ||||
|---|---|---|---|---|---|---|
| Gene symbol | FC | P | FC | P | FC | P |
| 1110059M19Rik | −1.72 | 0.02 | −1.61 | 0.04 | 1.16 | 0.66 |
| 2810046M22Rik | −1.69 | 0.08 | −1.59 | 0.12 | 1.67 | 0.24 |
| 2900017F05Rik | −1.86 | 0.03 | −1.41 | 0.19 | 1.29 | 0.30 |
| A130082M07Rik | −1.73 | 0.09 | −1.40 | 0.28 | −1.00 | 0.98 |
| A430103B12Rik | −1.69 | 0.03 | −1.36 | 0.17 | 1.07 | 0.69 |
| Aqp1 | −2.14 | 0.01 | −2.21 | 0.01 | 1.47 | 0.41 |
| Arc | 1.65 | 0.16 | 1.04 | 0.90 | −1.66 | 0.04 |
| Calml4 | −1.81 | 0.08 | −1.61 | 0.15 | 1.21 | 0.50 |
| Car14 | −1.59 | 0.01 | −1.41 | 0.04 | 1.05 | 0.69 |
| Cdkn1c | −1.63 | 0.02 | −1.47 | 0.05 | 1.35 | 0.34 |
| Cit | −1.86 | 0.06 | −1.25 | 0.46 | 1.01 | 0.89 |
| Cldn2 | −2.27 | 0.01 | −2.30 | 0.01 | 1.21 | 0.62 |
| Col8a1 | −1.50 | 0.04 | −1.38 | 0.09 | 1.33 | 0.21 |
| Col8a2 | −1.63 | 0.04 | −1.58 | 0.05 | 1.17 | 0.49 |
| D230046H12Rik | −2.32 | 0.03 | −1.94 | 0.07 | 1.22 | 0.53 |
| Enpp2 | −2.25 | 0.01 | −1.81 | 0.03 | 1.28 | 0.48 |
| Fam171a1 | 2.10 | 0.01 | 2.08 | 0.01 | −1.33 | 0.42 |
| Folr1 | −2.69 | 0.01 | −2.60 | 0.02 | 1.29 | 0.60 |
| Fosb | 1.88 | 0.02 | 1.46 | 0.14 | 1.06 | 0.68 |
| Irs2 | −1.71 | 0.04 | −1.15 | 0.57 | 1.10 | 0.58 |
| Ncor1 | 1.83 | 0.01 | 1.84 | 0.01 | −1.10 | 0.74 |
| Nrgn | 2.64 | 0.10 | 1.70 | 0.35 | −1.18 | 0.69 |
| Peg3 | −1.76 | 0.02 | −1.21 | 0.39 | 1.09 | 0.48 |
| Pmch | −9.77 | 0.08 | −2.16 | 0.52 | 1.60 | 0.66 |
| Ptk2b | 1.94 | 0.06 | 1.40 | 0.30 | −1.03 | 0.90 |
| Pwwp2b | 1.56 | 0.07 | 1.35 | 0.20 | −1.07 | 0.65 |
| Scn1a | −1.70 | 0.06 | −1.19 | 0.51 | 1.20 | 0.43 |
| Slc16a8 | −1.56 | 0.01 | −1.48 | 0.02 | 1.30 | 0.43 |
| Slc4a2 | −1.56 | 0.01 | −1.29 | 0.13 | 1.06 | 0.76 |
| Sostdc1 | −3.78 | 0.02 | −4.14 | 0.01 | 1.20 | 0.70 |
| Wfdc2 | −2.59 | 0.01 | −2.38 | 0.01 | 1.28 | 0.67 |
| Wipf3 | 2.09 | 0.08 | 1.27 | 0.54 | −1.14 | 0.58 |
| A/J hal vs. ctrl | A/J hal + cys vs. ctrl | C57BL/6 hal vs. ctrl | ||||
|---|---|---|---|---|---|---|
| Gene symbol | FC | P | FC | P | FC | P |
| 1110059M19Rik | −1.72 | 0.02 | −1.61 | 0.04 | 1.16 | 0.66 |
| 2810046M22Rik | −1.69 | 0.08 | −1.59 | 0.12 | 1.67 | 0.24 |
| 2900017F05Rik | −1.86 | 0.03 | −1.41 | 0.19 | 1.29 | 0.30 |
| A130082M07Rik | −1.73 | 0.09 | −1.40 | 0.28 | −1.00 | 0.98 |
| A430103B12Rik | −1.69 | 0.03 | −1.36 | 0.17 | 1.07 | 0.69 |
| Aqp1 | −2.14 | 0.01 | −2.21 | 0.01 | 1.47 | 0.41 |
| Arc | 1.65 | 0.16 | 1.04 | 0.90 | −1.66 | 0.04 |
| Calml4 | −1.81 | 0.08 | −1.61 | 0.15 | 1.21 | 0.50 |
| Car14 | −1.59 | 0.01 | −1.41 | 0.04 | 1.05 | 0.69 |
| Cdkn1c | −1.63 | 0.02 | −1.47 | 0.05 | 1.35 | 0.34 |
| Cit | −1.86 | 0.06 | −1.25 | 0.46 | 1.01 | 0.89 |
| Cldn2 | −2.27 | 0.01 | −2.30 | 0.01 | 1.21 | 0.62 |
| Col8a1 | −1.50 | 0.04 | −1.38 | 0.09 | 1.33 | 0.21 |
| Col8a2 | −1.63 | 0.04 | −1.58 | 0.05 | 1.17 | 0.49 |
| D230046H12Rik | −2.32 | 0.03 | −1.94 | 0.07 | 1.22 | 0.53 |
| Enpp2 | −2.25 | 0.01 | −1.81 | 0.03 | 1.28 | 0.48 |
| Fam171a1 | 2.10 | 0.01 | 2.08 | 0.01 | −1.33 | 0.42 |
| Folr1 | −2.69 | 0.01 | −2.60 | 0.02 | 1.29 | 0.60 |
| Fosb | 1.88 | 0.02 | 1.46 | 0.14 | 1.06 | 0.68 |
| Irs2 | −1.71 | 0.04 | −1.15 | 0.57 | 1.10 | 0.58 |
| Ncor1 | 1.83 | 0.01 | 1.84 | 0.01 | −1.10 | 0.74 |
| Nrgn | 2.64 | 0.10 | 1.70 | 0.35 | −1.18 | 0.69 |
| Peg3 | −1.76 | 0.02 | −1.21 | 0.39 | 1.09 | 0.48 |
| Pmch | −9.77 | 0.08 | −2.16 | 0.52 | 1.60 | 0.66 |
| Ptk2b | 1.94 | 0.06 | 1.40 | 0.30 | −1.03 | 0.90 |
| Pwwp2b | 1.56 | 0.07 | 1.35 | 0.20 | −1.07 | 0.65 |
| Scn1a | −1.70 | 0.06 | −1.19 | 0.51 | 1.20 | 0.43 |
| Slc16a8 | −1.56 | 0.01 | −1.48 | 0.02 | 1.30 | 0.43 |
| Slc4a2 | −1.56 | 0.01 | −1.29 | 0.13 | 1.06 | 0.76 |
| Sostdc1 | −3.78 | 0.02 | −4.14 | 0.01 | 1.20 | 0.70 |
| Wfdc2 | −2.59 | 0.01 | −2.38 | 0.01 | 1.28 | 0.67 |
| Wipf3 | 2.09 | 0.08 | 1.27 | 0.54 | −1.14 | 0.58 |
Gene expression changes were measured using microarrays in striatal tissue obtained from A/J or C57BL/6 mice after treatment with haloperidol (10 mg⋅kg−1⋅day−1 × 4 days) or from A/J mice treated with cysteamine (10 mg⋅kg−1⋅day−1) for 3 days before and during a 4-day period of haloperidol (10 mg⋅kg−1⋅day−1) treatment. The 32 genes whose level of mRNA expression in striatal tissue was altered by haloperidol treatment in A/J (but not C57BL/6) mice and was reversed by cystamine treatment are shown. The fold change (FC) and unadjusted P-value for the indicated comparisons are shown. A minus (–) indicates that the mRNA level was decreased by haloperidol treatment. hal, haloperidol; ctrl, control; cys, cysteamine.
RT-PCR analysis
The level of striatal expression for nine of the genes in Table 1, which were selected because they exhibited the highest level of haloperidol-induced differential expression, was analyzed by RT-PCR. This two-step procedure was performed using the SuperScript III first-strand synthesis system for RT-PCR and the SYBR Select Master Mix (Life Technologies) according to the manufacturer’s instructions. The level of expression of Hprt was used as the internal standard for normalization. The primer sets used for amplification are shown below:
| Gene | Amplicon size (bp) | Forward primer | Reverse primer |
|---|---|---|---|
| Hprt | 142 | TCAGTCAACGGGGGACATAAA | GGGGCTGTACTGCTTAACCAG |
| Pmch | 275 | TCAAACTAAGGATGGCAAAGATG | TTTGGAGCCTGTGTTCTTTG |
| Nrgn | 125 | CCACCCAGCATCGTACAAAC | GCGGGATGTCAAGAATATCG |
| Ptk2b | 147 | TGAGCCGTGTAAAAGTGGG | TGAAGTTCTTCCCTGGGTTG |
| Fosb | 174 | TTTTCCCGGAGACTACGACTC | GTGATTGCGGTGACCGTTG |
| Cit | 125 | AACGGACTGGAACAACATCC | CTGGCACACAGAGGATGAAA |
| Peg3 | 146 | TGTGTGCGTAGAGTGCTGTG | TCAACTGATCTCCCCTTGCT |
| Irs2 | 195 | GCAGCCAGGAGACAAGAAC | GCGCTTCACTCTTTCACGAC |
| Scn1a | 138 | TCAGAGGGAAGCACAGTAGAC | TTCCACGCTGATTTGACAGCA |
| Slc4a2 | 149 | ACCCAGGAGATCTTTGCCTT | GTCATATTGCTGCTGCTGCT |
| Gene | Amplicon size (bp) | Forward primer | Reverse primer |
|---|---|---|---|
| Hprt | 142 | TCAGTCAACGGGGGACATAAA | GGGGCTGTACTGCTTAACCAG |
| Pmch | 275 | TCAAACTAAGGATGGCAAAGATG | TTTGGAGCCTGTGTTCTTTG |
| Nrgn | 125 | CCACCCAGCATCGTACAAAC | GCGGGATGTCAAGAATATCG |
| Ptk2b | 147 | TGAGCCGTGTAAAAGTGGG | TGAAGTTCTTCCCTGGGTTG |
| Fosb | 174 | TTTTCCCGGAGACTACGACTC | GTGATTGCGGTGACCGTTG |
| Cit | 125 | AACGGACTGGAACAACATCC | CTGGCACACAGAGGATGAAA |
| Peg3 | 146 | TGTGTGCGTAGAGTGCTGTG | TCAACTGATCTCCCCTTGCT |
| Irs2 | 195 | GCAGCCAGGAGACAAGAAC | GCGCTTCACTCTTTCACGAC |
| Scn1a | 138 | TCAGAGGGAAGCACAGTAGAC | TTCCACGCTGATTTGACAGCA |
| Slc4a2 | 149 | ACCCAGGAGATCTTTGCCTT | GTCATATTGCTGCTGCTGCT |
| Gene | Amplicon size (bp) | Forward primer | Reverse primer |
|---|---|---|---|
| Hprt | 142 | TCAGTCAACGGGGGACATAAA | GGGGCTGTACTGCTTAACCAG |
| Pmch | 275 | TCAAACTAAGGATGGCAAAGATG | TTTGGAGCCTGTGTTCTTTG |
| Nrgn | 125 | CCACCCAGCATCGTACAAAC | GCGGGATGTCAAGAATATCG |
| Ptk2b | 147 | TGAGCCGTGTAAAAGTGGG | TGAAGTTCTTCCCTGGGTTG |
| Fosb | 174 | TTTTCCCGGAGACTACGACTC | GTGATTGCGGTGACCGTTG |
| Cit | 125 | AACGGACTGGAACAACATCC | CTGGCACACAGAGGATGAAA |
| Peg3 | 146 | TGTGTGCGTAGAGTGCTGTG | TCAACTGATCTCCCCTTGCT |
| Irs2 | 195 | GCAGCCAGGAGACAAGAAC | GCGCTTCACTCTTTCACGAC |
| Scn1a | 138 | TCAGAGGGAAGCACAGTAGAC | TTCCACGCTGATTTGACAGCA |
| Slc4a2 | 149 | ACCCAGGAGATCTTTGCCTT | GTCATATTGCTGCTGCTGCT |
| Gene | Amplicon size (bp) | Forward primer | Reverse primer |
|---|---|---|---|
| Hprt | 142 | TCAGTCAACGGGGGACATAAA | GGGGCTGTACTGCTTAACCAG |
| Pmch | 275 | TCAAACTAAGGATGGCAAAGATG | TTTGGAGCCTGTGTTCTTTG |
| Nrgn | 125 | CCACCCAGCATCGTACAAAC | GCGGGATGTCAAGAATATCG |
| Ptk2b | 147 | TGAGCCGTGTAAAAGTGGG | TGAAGTTCTTCCCTGGGTTG |
| Fosb | 174 | TTTTCCCGGAGACTACGACTC | GTGATTGCGGTGACCGTTG |
| Cit | 125 | AACGGACTGGAACAACATCC | CTGGCACACAGAGGATGAAA |
| Peg3 | 146 | TGTGTGCGTAGAGTGCTGTG | TCAACTGATCTCCCCTTGCT |
| Irs2 | 195 | GCAGCCAGGAGACAAGAAC | GCGCTTCACTCTTTCACGAC |
| Scn1a | 138 | TCAGAGGGAAGCACAGTAGAC | TTCCACGCTGATTTGACAGCA |
| Slc4a2 | 149 | ACCCAGGAGATCTTTGCCTT | GTCATATTGCTGCTGCTGCT |
For analysis of Vnn1 and Vnn3 hepatic mRNA expression, RNA was extracted from the livers of adult male C57/BL6 (n = 2) and A/J (n = 2) mice, using an RNeasy mini kit (QIAGEN). Total cDNA was synthesized as described above, and Vnn1 and Vnn3 mRNAs were amplified using the following primer sets: CCATCTATGTTGTGGCGAAC(forward) and CCATGAAAATGTTTTGCTTATGG (reverse) for Vnn1 and TGGGCAGTGAGTTTTGTCTTC(forward) and AATGATATGTGCACCCTGTCTG (reverse) for Vnn3. Hprt mRNA was used as the internal standard and was amplified using the following primer pairs: TCAGTCAACGGGGGACATAAA (forward) and GGGGCTGTACTGCTTAACCAG (reverse). Real-time PCR quantification was measured on a 7500 Real Time PCR System (ABI). The relative mRNA level was expressed as fold change normalized against the average ΔCt of Vnn1 mRNA relative to Hprt mRNA in C57/BL6 liver.
Focal cerebral ischemia
These experiments were performed according to protocols approved by the Stanford University Administrative Panel on Laboratory Animal Care. Male A/J mice at 16 weeks of age were used in these experiments. The mice were treated with cystamine (10 mg⋅kg−1⋅day−1 ip) or saline for 4 days, and one additional dose of cystamine (or saline) was administered 3 hr prior to induction of cerebral ischemia, using previously described methods (Zhao et al. 2004). In brief, the mice were anesthetized with isoflurane, the left temporalis muscle was bisected through a scalp incision, and a 2-mm burr hole was made at the junction of the zygomatic arch and squamous bone. The distal middle cerebral artery was exposed and cauterized above the rhinal fissure. The incision was then closed, 100 μl of 0.5% bupivacaine was subcutaneously injected, and the mouse was allowed to recover.
Infarct size measurement
Mice were killed by isoflurane overdose 24 hr after cerebral ischemia was induced. The brains were removed, and coronal sections were prepared at 2-mm intervals to generate four slices, which were stained with a 2% solution of 2,3,4-triphenytetrazolium chloride. A computerized image analysis system (NIH image, version 1.61) was used to measure the infarcted area in the four sections prepared from each mouse. The size of the infarct in the ischemic cortex in each slice was expressed as a percentage after normalization relative to the size of the nonischemic cortex and according to the formula [(area of nonischemic cortex – area of remaining ischemic cortex)/area of nonischemic cortex] × 100 as previously described (Zhao et al. 2005). The numbers shown were the average of the four determinations for each mouse. The statistical significance of the treatment effect on infarct size was assessed using Student’s t-test.
Data availability
The authors state that all data necessary for confirming the conclusions presented in the article are represented fully within the article.
Results
A novel pharmacogenetic factor for HIT
The time required for a haloperidol-treated mouse to move all four paws (latency) after it is placed on an inclined screen is a measure of the Parkinsonian-like rigidity caused by HIT. In a previous study (Crowley et al. 2012b), only 3 of 27 inbred strains manifested a substantial amount of toxicity after haloperidol treatment, and the latency in A/J mice was substantially larger (36% and 59%) than that of the other two strains (NZW and NZO). Therefore, it was possible that A/J mice uniquely had an additional pharmacogenetic factor that increased its susceptibility to HIT. Since A/J and C57BL/6 exhibit extreme differences in susceptibility to HIT, we examined the susceptibility of 19 different C57BL/6J-A/J chromosome substitution strains (CSS) to better characterize the genetic architecture underlying susceptibility to HIT. Since each consomic strain is homosomic for a single specified A/J chromosome on an otherwise C57BL/6 genetic background (Nadeau et al. 2000), this genome scan will identify genetic susceptibility factors on any autosome. The prolonged latency in consomic mice with an A/J chromosome 12 on an otherwise C57BL/6 genetic background (CSS12 mice) after 5 (P = 2 × 10−7) and 10 days (P = 2 × 10−5) of haloperidol treatment is consistent with our previous finding that Abcb5 alleles affect susceptibility to HIT (Zheng et al. 2015) (Figure 1). However, this analysis revealed that a genetic factor on chromosome 10 independently contributes to this interstrain difference; CSS10 mice had a significantly prolonged haloperidol-induced latency after 5 (P = 2 × 10−6) and 10 days (P = 4 × 10−5) of drug treatment. In fact, the latency measured in CSS10 mice was twice as large as that of CSS12 mice, and their combined latencies were equivalent to that of A/J mice (Figure 1). This indicates that the pharmacogenetic factors on chromosomes 10 and 12 quantitatively account for the increased susceptibility of A/J mice to HIT. Two other consomic strains (CSS2 and CSS14) exhibited a small increase in latency on the day 10 measurements only. However, no consomic strains (other than CSS10 and CSS12) exhibited latencies after 5 and 10 days of treatment, which were both significantly prolonged (i.e., P < 0.05) relative to those of control mice.
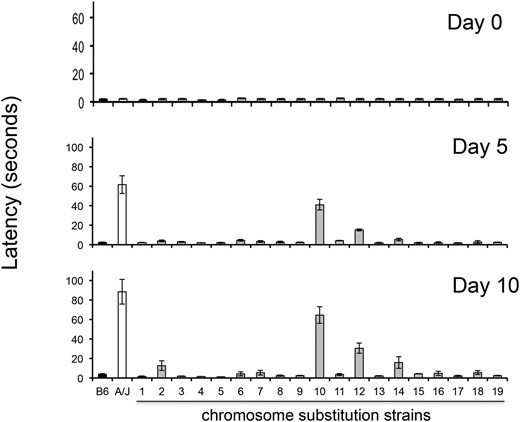
HIT in C57BL/6J, A/J, and 19 chromosome substitution strains. The time (latency) required for a mouse to make a coordinated movement after it is placed on a vertical mesh screen before (day 0) and after treatment with haloperidol (3 mg⋅kg−1⋅day−1 via continuous release from an implanted pellet) for 5 or 10 days is shown. Each bar represents the average ± SEM for three to four mice per strain. Only CSS12 and CSS10 mice (which have A/J alleles for every gene on chromosome 12 or 10, respectively, on an otherwise C57BL/6 background) had significantly increased latencies after both 5 and 10 days of haloperidol treatment. No other CSS mice exhibited a significant increase in latency after treatment day 5; and the small increases in the measured latencies of CSS2 and CSS14 mice after 10 days of haloperidol treatment were not significant (P > 0.05) after correction for the multiple comparisons.
To determine whether differences in the levels of the oxidative metabolite of haloperidol (HPP+) in brain were associated with susceptibility to HIT, we measured HPP+ abundance in brain tissue obtained from C57BL/6, A/J, CSS12, and CSS10 mice after treatment with haloperidol (3 mg⋅kg−1⋅day−1 dose from a continuous release implanted pellet) for 10 days. The concentrations of HPP+ in brain were increased in A/J (P = 3 × 10−5) and CSS12 (P = 0.004) mice relative to C57BL/6 mice. This result is consistent with the mechanism by which Abcb5 alleles on chromosome 12 alter the amount of haloperidol in the brain. However, the amount of HPP+ in brain was not increased in CSS10 mice (P = 0.4) (Figure S1), which indicates that the novel pharmacogenetic factor on chromosome 10 affects HIT by a different mechanism. Although it does not affect the amount of HPP+ in brain, it alters the susceptibility of dopaminergic neurons to HIT.
Identification of a novel pharmacogenetic factor
To identify this factor, we examined our SNP database, which was generated by analysis of whole-genome sequence data obtained from 27 inbred strains (Zheng et al. 2015). We identified 209 genes on chromosome 10 with SNP alleles that were uniquely present in A/J mice, which were not found in the 17 other strains with available genomic sequence and whose susceptibility to HIT was previously analyzed. However, only 6 genes contained a missense (or nonsense) SNP (Table S1), and one was a highly plausible candidate. Vnn3 was the only gene on chromosome 10 where an A/J-specific SNP allele introduced a premature stop codon (Gln494->X), and this produced a 7-amino-acid truncation of the Vnn3 protein. The two pantetheinases (Vnn1 and Vnn3) in the mouse genome are collinear genes on chromosome 10; and both are ectoenzymes that hydrolyze D-pantethein to pantothenic acid (vitamin B5) and cysteamine (Robishaw and Neely 1985). Previously, a complex rearrangement within the Vnn3 promoter region found in A/J mice (and in several other strains) was identified, and it was thought that this promoter rearrangement reduced the level of expression of the adjacent Vnn1 and Vnn3 mRNAs, which produced the complete absence of measured hepatic pantetheinase enzyme activity in A/J mice (Min-Oo et al. 2007). Therefore, we examined hepatic Vnn1 and Vnn3 mRNA levels in A/J and B/6 mice. The normalized level of Vnn1 mRNA in C57BL/6 liver (1.06 ± 0.35 mean ± SE) is 20-fold higher than in A/J liver (0.054 ± 0.009) (Figure S2). Hence, a negligible level of Vnn1 mRNA expression in A/J liver is associated with the promoter rearrangement. In contrast, hepatic Vnn3 mRNA levels were threefold higher than Vnn1 mRNA levels in C57BL/6 mice, and the hepatic Vnn3 mRNA levels (2.99 ± 0.48) in C57BL/6 mice are only fivefold higher (0.65 ± 0.10) than in A/J mice. These results indicate that the combined presence of the promoter rearrangement and of the Vnn3 SNP allele that produces a truncated Vnn3 protein together are responsible for the absence of hepatic pantotheinase activity in A/J mice. None of the other strains, including the two strains with a reduced level of susceptibility to HIT, have the A/J-specific SNP allele that truncates Vnn3. Moreover, the cysteamine produced by these ectoenzymes is required for optimal inflammatory and antioxidative responses (Pitari et al. 2000; Berruyer et al. 2004; Martin et al. 2004). Cysteamine can be exogenously administered as cystamine, which is rapidly metabolized to its reduced form (cysteamine) in vivo (Bousquet et al. 2010). Indeed, it is already known that A/J mice are susceptible to malaria infection because their pantetheinase deficiency reduces their ability to mount an immune response to malarial parasites and that cystamine administration partially reverses their increased susceptibility(Min-Oo et al. 2007).
Since cysteamine can cross the blood–brain barrier (Bousquet et al. 2010), the inability of the A/J liver to produce cysteamine reduces the amount of cysteamine available to the brain, which could be needed to protect against the Parkinsonian-like toxicity caused by HPP+. By this proposed genetic mechanism, the cysteamine deficiency caused by the A/J-specific Vnn1/Vnn3 alleles could increase susceptibility to HIT. To test this genetic hypothesis, we investigated whether pharmacological correction for the pantetheinase deficiency would alter the susceptibility of A/J mice to HIT. We examined whether cystamine administration (10 mg⋅kg−1⋅day−1) would decrease the haloperidol-induced latency in A/J mice. A relatively high dose of haloperidol (10 mg⋅kg−1⋅day−1 ip) did not increase the latency measured in resistant C57BL/6 mice, but it significantly increased the latency in A/J mice. To determine whether cystamine could prevent the onset of HIT, cystamine was administered daily for 4 days before haloperidol treatment was initiated (Figure 2A). Cystamine coadministration significantly reduced the latency in A/J mice after 2 (P = 8.0 × 10−3), 3 (P = 9 × 10−5), and 4 (P = 6 × 10−4) days of haloperidol treatment (Figure 2B). Importantly, cystamine coadministration reduced the latency to background levels after 3 or 4 days of haloperidol treatment. To determine whether cystamine treatment affected the susceptibility caused by the pharmacogenetic factor on chromosome 10, the effect of cystamine was examined in CSS10 mice, which have an A/J chromosome 10 on an otherwise C57BL/6 genetic background. Cysteamine coadministration significantly reduced the haloperidol-induced latency in CSS10 mice after 3 (P = 0.02), 4 (P = 3 × 10−4), 5 (P = 0.002), 6 (P = 0.004), and 7 (P = 3 × 10−4) days of haloperidol treatment (Figure 2C). Thus, cystamine coadministration can protect against the genetic susceptibility caused by A/J alleles on chromosome 10.
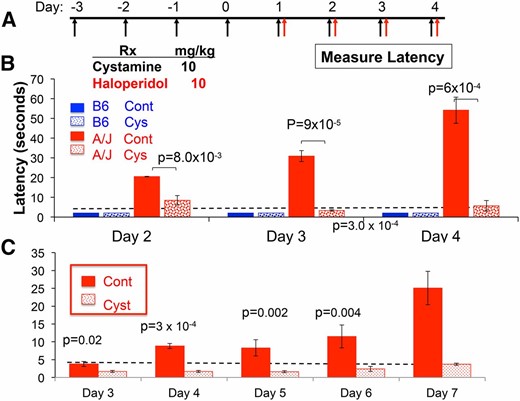
Cystamine coadministration prevents HIT. (A) In the prevention protocol, mice are first treated with cystamine (10 mg⋅kg−1⋅day−1 ip) for 4 days, followed by treatment with haloperidol (10 mg⋅kg−1⋅day−1 ip) and cystamine (10 mg⋅kg−1⋅day−1 ip). A control group of mice received only haloperidol. The haloperidol-induced latency is then measured after ≥2 days of haloperidol treatment in each group of mice. (B and C) Cystamine prevents HIT in A/J (B) and CSS10 (C) mice. The latency measured in C57BL/6 and A/J mice (B), or in CSS10 mice (C), on the indicated day of haloperidol treatment according to the prevention protocol is shown. Each bar represents the average ± SEM for three to four mice per strain, and the dashed horizontal line indicates the average pretreatment latency. Haloperidol increases the latency in A/J and CSS10 mice, but not in C57BL/6, mice. The calculated P-values for comparison of the cystamine-treated A/J (B) or CSS10 mice (C) and control groups are shown for each treatment day.
We next examined whether cystamine administration could reverse the Parkinsonian-like symptoms of HIT after it was established. To do this, A/J mice were treated with haloperidol (10 mg⋅kg−1⋅day−1) alone for 3 days, followed by daily doses of cystamine (10 mg⋅kg−1⋅day−1) and haloperidol (10 mg⋅kg−1⋅day−1) on days 4–13 (reversal protocol). We found that the haloperidol-induced latencies were not altered in mice that were treated with cystamine in this manner (Figure 3). Thus, although cystamine coadministration can prevent the onset of HIT, it cannot reverse the toxicity after it has been established.
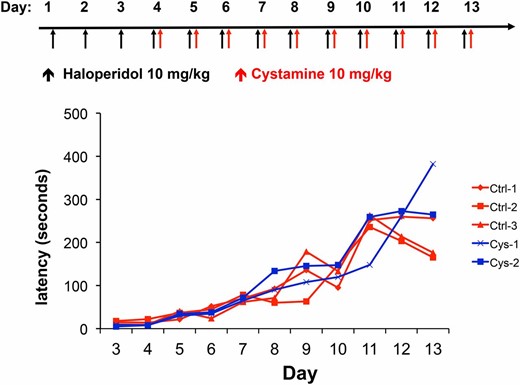
Cystamine cannot reverse HIT after it has been established. In the reversal protocol, A/J mice are first treated with haloperidol (10 mg⋅kg−1⋅day−1 ip) for 3 days and then with haloperidol (10 mg⋅kg−1⋅day−1 ip) ± cystamine (10 mg⋅kg−1⋅day−1 ip) on days 4–13. The haloperidol-induced latency is then measured on the indicated day in each group. A line connects the measurements made on the same mouse on different treatment days.
Investigating the neuroprotective effect of cystamine
Multiple mechanisms have been proposed to explain the neuroprotective effects of cystamine, which have been observed in diverse disease models (Karpuj et al. 2002; Borrell-Pages et al. 2006; Hsu et al. 2008; Pillai et al. 2008; Gibrat et al. 2010). For example, cystamine is a potent antioxidant (Di Leandro et al. 2008), and it is a competitive inhibitor of transglutaminase 2 (Jeitner et al. 2005), which is a multifunctional enzyme whose inappropriate activation is linked with the pathogenesis of multiple disease conditions, including Parkinson’s disease (reviewed in Iismaa et al. 2009; Grosso and Mouradian 2012). However, none of the proposed mechanisms for the neuroprotective effect of cystamine were observed across different models, nor were they sustained across the time points tested in these models. Since HIT has a rapid onset, develops in a strain-specific manner, and is reversed by cystamine coadministration, this model could be used to characterize the neuroprotective effect of cystamine. Because of its reported antioxidant activity (Di Leandro et al. 2008), we first investigated whether cystamine reduces the amount of the oxidative metabolite of haloperidol (HPP+) produced in brain tissue. We found that pretreatment with cystamine, followed by coadministration with each haloperidol dose, did not reduce the abundance of HPP+ in brain tissue obtained from A/J in two separate experiments (P-values = 0.98 and 0.30) (Figure S3, A and B). The liver also generates a significant amount of HPP+, and hepatic HPP+ was also not reduced by cystamine coadministration (P-value = 0.42) (Figure S2B). Thus, we could not find any evidence that the cystamine reduced the production of the toxicity-inducing metabolite in brain or other tissues.
We next investigated whether the protective effect of cysteamine is mediated via an effect on gene transcription. For example, cysteamine is known to be an inhibitor of transglutaminase 2 (Jeitner et al. 2005), whose activity affects the expression of many genes in brain (McConoughey et al. 2010); and cysteamine activates NF-E2 related factor 2 (Nrf2) in brain tissue (Calkins et al. 2010). Through an effect on transglutaminase 2, Nrf2, or other transcription factors, cysteamine could affect the neuronal response to drug or disease-induced insults (reviewed in Grosso and Mouradian 2012). Therefore, whole-transcriptome profiling was used to examine the strain-specific nature of HIT and the neuroprotective effect of cystamine. Gene expression profiling was performed on striatal tissue isolated from five groups of mice (n = 3–4 mice per group): A/J mice treated with (i) haloperidol and cystamine, (ii) haloperidol alone, or (iii) vehicle or C57BL/6J mice treated with (iv) haloperidol or (v) vehicle. The microarray data were fitted to linear regression models, which were designed to identify genes whose expression was altered by haloperidol treatment in a strain-specific manner and haloperidol-induced gene expression changes that were reversed by cystamine treatment. This analysis identified 172 genes whose expression was altered by haloperidol treatment in a strain-specific manner (Table S2) and 110 genes whose expression was altered by cystamine treatment (Table S3). Thus, haloperidol induces a substantial number of gene expression changes in the striatum, which occur in a strain-specific manner.
There are 32 genes commonly present in Table S2 and Table S3, and these represent strain-specific haloperidol-induced gene expression changes that were partially or fully reversed by cystamine treatment (Table 1). These are of particular interest, since they are highly likely to be associated with HIT. Interestingly, the biological functions most highly associated with these 32 genes were body size/feeding behavior (ranks 1, 2, 3, 4, and 6) and neurotransmission or movement disorder (ranks 7 and 8) (Table S4). These categories are consistent with the side effects (movement disorder and weight changes) caused by the DRD2 antagonists used as antipsychotic agents (Parkes 1982). RT-PCR was used to further analyze the expression changes for 9 of the 32 genes in Table 1 that exhibited the highest level of haloperidol-induced differential expression. For 7 of these 9 genes, the RT-PCR-measured expression changes were consistent with those measured by the microarrays (Table S5). The expression changes for two of these genes were of interest, since both were previously associated with Parkinson’s disease (discussed below). The striatal levels of mRNA expression for the precursor of melanin concentrating hormone (Pmch) and the transcription factor FosB were 60-fold decreased (P = 0.04) and 5-fold increased (P = 0.003), respectively, by haloperidol treatment in A/J mice (Figure 4). Moreover, the haloperidol-induced decrease in Pmch mRNA expression occurred only in A/J mice and was reversed by cystamine coadministration (P = 0.57 relative to the vehicle-treated group).
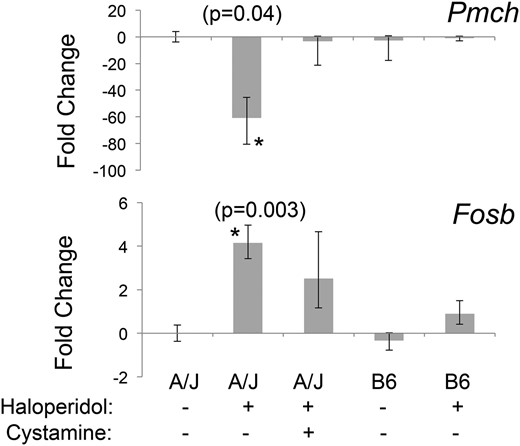
Haloperidol-induced gene expression changes that are strain specific and reversed by cystamine treatment. A/J mice were treated with vehicle (n = 3) or cystamine (10 mg⋅kg−1⋅day−1 ip) for 3 days, followed by treatment with haloperidol (10 mg⋅kg−1⋅day−1 ip) in the presence (n = 4) or absence (n = 4) of cystamine (10 mg⋅kg−1⋅day−1 ip) for 4 days. C57BL/6 mice were treated with haloperidol (10 mg⋅kg−1⋅day−1) (n = 4) or vehicle (n = 3) for 4 days. Four hours after the last treatment, striatal tissue was harvested, and the level of Pmch or Fosb mRNA was assessed by RT-PCR analysis. Haloperidol treatment decreases Pmch and increases Fosb mRNA levels by 60-fold (P = 0.04) and 5-fold (P = 0.003), respectively, in A/J mice. Their expression in C57BL/6 mice was not altered by haloperidol treatment, and the haloperidol-induced expression changes in A/J mice were completely (Pmch) (P = 0.57 relative to vehicle-treated group) or partially (Fosb) (P = 0.07 relative to vehicle-treated group) reversed by cystamine coadministration.
Cystamine protects against ischemic brain injury
Like HIT, the extent of brain injury developing after experimentally induced ischemia is a highly heritable and highly variable (∼30-fold) trait among the inbred strains (Keum and Marchuk 2009). Of note, the extent of ischemic brain damage in A/J mice was reported to be significantly greater than in C57BL/6 mice (Keum and Marchuk 2009). Since cystamine protected dopaminergic neurons in A/J mice against HIT, we used a well-established model of focal cerebral ischemia (Keum and Marchuk 2009) to determine whether cystamine could also protect brain tissue against ischemic damage. To do this, the volume of infarcted cortical tissue was measured 24 hr after occlusion of the distal part of the middle cerebral artery (MCA). To determine whether this response was indeed heritable, we examined the extent of ischemic brain damage in A/J, CSS10, and C57BL/6 mice. Consistent with prior results (Keum and Marchuk 2009), we observed the extent of ischemic brain damage in A/J mice was >2-fold greater than in C57BL/6J mice (P = 2 × 10−5). Moreover, the extent of ischemic damage in CSS10 mice was also increased (1.8-fold, P = 4 × 10−3) relative to that in C57BL/6 mice; and the level of ischemic damage in CSS10 mice was similar to that observed in A/J mice (Figure 5). These results indicate that A/J mice are more susceptible than C57BL/6 mice to ischemic brain injury and that A/J alleles on chromosome 10 could also affect susceptibility to ischemic brain injury. If the Vnn alleles on chromosome 10 affect susceptibility, then cystamine administration should reduce the extent of ischemic brain injury in both A/J and CSS10 mice. MCA occlusion produced a substantial volume of infarction (56.8 ± 2.4%) in control A/J mice, which was dramatically reduced (44.0 ± 3.6%) in mice that were pretreated with cystamine (P = 0.01) (Figure 6). Cystamine administration reduced the infarct volume by 23%, which translates into the preservation of a significant amount of brain tissue after an ischemic insult. The protective effect of cystamine was also demonstrable in CSS10 mice; the infarct size in control CSS10 mice (50.1 ± 4.0%) was reduced to 27.5 ± 3.0% by cystamine treatment (P = 0.02) (Figure 6).
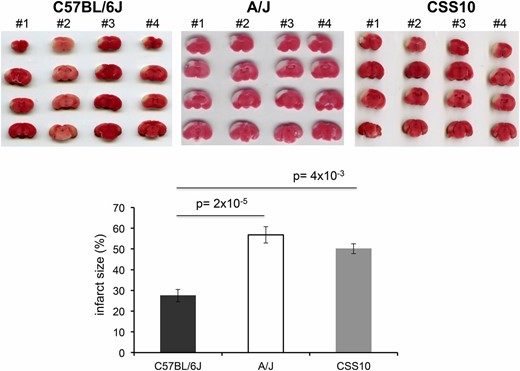
The size of an experimentally induced ischemic brain infarct in A/J and CSS10 mice is greater than in C57BL/6J mice. Infarct size was measured 24 hr postdistal MCA occlusion by 2,3,4-triphenytetrazolium (TTC) staining of whole brain sections prepared from C57BL/6J (n = 4), A/J (n = 4), or CSS10 (n = 4) mice. The extent of ischemic brain damage in A/J mice was twofold greater than in C57BL/6J mice (P = 2 × 10−5). The extent of ischemic damage in CSS10 mice was 1.8-fold increased relative to that in C57BL/6 mice (P = 4 × 10−3) and was similar to that observed in A/J mice. The scanned images of TTC-stained whole brain sections for each animal and the measured infarct size (expressed as the percentage of the cortex) are shown. Each bar represents the average (± SEM) of the infarct volume in the percentage of total cortex volume.
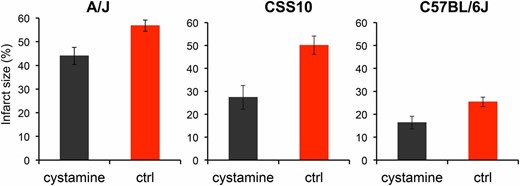
Cystamine administration reduces the infarct volume after ischemic injury in A/J, CSS10, and C57BL/6J mice. Male A/J, CSS10, and C57BL/6J mice were treated with cystamine (10 mg⋅kg−1⋅day−1 ip) (n = 5, 4, and 8 mice per group, respectively) or saline (n = 8, 4, and 7 mice per group, respectively) for 5 days. Then, 24 hr after MCA occlusion, whole brains were obtained to assess the infarct size. Each bar represents the average (± SEM) of the infarct size. The infarct size in the control group was significantly reduced by cystamine administration in A/J mice (56.8% vs. 44.0%, P = 0.01), CSS10 mice (50.1% vs. 27.5%, P = 0.02) and C57BL/6J mice (25.4% vs. 16.4%, P = 0.028).
C57BL/6J mice have Vnn alleles that confer normal pantetheinase enzyme activity and had lower levels of ischemic brain injury in the MCA occlusion model. Therefore, we investigated whether cystamine supplementation, which produces supraphysiologic levels of cysteamine in the recipients, would reduce the extent of ischemic brain injury after MCA occlusion in C57BL/6 mice. Cystamine administration had a protective effect against ischemic brain damage in C57BL/6 mice; the infarct size after MCA occlusion was reduced from 25.4 ± 2.1% in the control group to 16.4 ± 2.8% in the cystamine-treated group (P = 0.028) (Figure 6). Of note, the level of ischemic damage in untreated C57BL/6 mice was ∼50% of that in A/J or CSS10 mice, and cystamine treatment conferred a lesser degree of protection in C57BL/6 mice relative to that in A/J or CSS10 mice. Nevertheless, these results indicate that pharmacological levels of cysteamine—which are attained through cystamine supplementation—can protect against ischemic brain injury, even when physiological amounts of cysteamine are being produced.
Discussion
The results in our previous article (Zheng et al. 2015), along with those described here, provide the first complete characterization of pharmacogenetic factors affecting susceptibility to a drug-induced CNS toxicity. The differential susceptibility of inbred strains to HIT is determined by at least two pharmacogenetic factors: (i) Abcb5 alleles, which regulate brain haloperidol levels after a dose of haloperidol (Zheng et al. 2015), and (ii) a locus on chromosome 10, which may include the candidate Vnn1/3 alleles that we propose eliminate cysteamine biosynthesis. Since both of these loci contribute to susceptibility, if a strain does not have A/J Abcb5 alleles, it may not be susceptible to HIT even though it has the A/J Vnn1/3 alleles. While proving that the A/J Vnn1/3 alleles are indeed causal would require further genetic dissection, we demonstrate that cystamine coadministration can completely prevent the onset of a haloperidol-induced movement disorder in mice, which resembles that of human Parkinson’s disease. Since other commonly used drugs [e.g., prochlorperazine (Drotts and Vinson 1999) and metoclopramide (Moos and Hansen 2008)] cause EPS (Orti-Pareja et al. 1999), this raises the possibility that cystamine coadministration could prevent the EPS caused by these drugs. Of note, there are three human orthologs of the murine pantetheinase genes (VNN1, VNN2, and VNN3) (Martin et al. 2001). Human VNN1 alleles have been associated with differences in human VNN1 mRNA expression (Goring et al. 2007; Kaskow et al. 2013) and in high-density lipoprotein cholesterol levels (Goring et al. 2007; Jacobo-Albavera et al. 2012). Thus, it is possible that genetic variation in human pantetheinase genes could also affect susceptibility EPS caused by antipsychotic agents and other drugs.
The strain-specific and cystamine-reversible gene expression changes caused by haloperidol provide novel insight into the mechanisms underlying HIT and the neuroprotective effect of cystamine. First, haloperidol induced a substantial number of strain-specific gene expression changes in the striatum, and many (19%) of these were reversed by cystamine treatment. Although these results do not implicate a specific molecular target, they suggest that the neuroprotective effect of cystamine could be mediated by an effect on transcription. Second, it is striking that haloperidol selectively reduced striatal Pmch mRNA expression in the susceptible strain. A selective loss of MCH neurons has been noted in Parkinson’s disease (Thannickal et al. 2007), and the degree of MCH neuronal loss correlates with the clinical stage of Parkinson’s disease (Thannickal et al. 2008). Third, the haloperidol-induced increase in striatal Fosb mRNA expression is also of interest. Increased striatal expression of a splicing isoform of Fosb (ΔFosb) was associated with progression of Parkinson’s disease (Tekumalla et al. 2001). Moreover, striatal overexpression of ΔFosb in rats (Cao et al. 2010) or of a dominant negative ΔFosb inhibitor in primates (Berton et al. 2009) increased or decreased, respectively, the development of involuntary movement disorders in animal models of Parkinson’s disease. Given these similarities to Parkinson’s disease, our results support the possibility that cystamine administration could prevent or delay the onset of neurodegenerative diseases. Cystamine treatment was previously shown to be of benefit in murine models of Huntington’s disease (Dedeoglu et al. 2002; Karpuj et al. 2002; Wang et al. 2005) and Parkinson’s disease (Sun et al. 2010). Of note, cysteamine (as a bitartrate salt) has received Food and Drug Administration approval for treatment of a genetic disease (cystinosis) (Dohil et al. 2010) and has recently been evaluated as a potential treatment for Huntington’s disease (Borrell-Pages et al. 2006) and for Parkinson’s disease (Gibrat and Cicchetti 2011).
Consistent with the results of multiple prior studies, we also demonstrate that cystamine has a broader neuroprotective effect. In this case, it had a substantial neuroprotective effect in an experimental mouse model of ischemic cerebral stroke. Stroke is a devastating neurological disorder that causes impairment, disability, and death (Tu 2010). A recent study demonstrated that administration of cystamine after ischemic brain injury occurred could improve functional recovery from stroke in mice. However, in that study, cystamine administration had no effect on infarct size, and its effect on recovery was ascribed to an increase in the production of brain-derived neurotrophic factor (BDNF) (Li et al. 2014). Other investigators have also implicated the cystamine-induced increase in BDNF production (and increased axonal growth) as the reason for its beneficial effects (Pillai et al. 2008; Gibrat et al. 2010). However, in our study, the neuroprotective effect of cystamine pretreatment occurred within 24 hr after stroke induction, which is a time period that is far too rapid for its neuroprotective effect to be due to an increase in neural regeneration. Thus, the neuroprotective effects of cystamine—whether for ischemic injury or HIT—occur via another mechanism that has yet to be identified. Additional analysis of the genes with haloperidol-induced changes in their level of expression, especially those that were strain specific and cysteamine reversible, may provide a useful starting point in the search for this uncharacterized mechanism. There is also a need for additional characterization of the relationship between Vnn1/3 and susceptibility to HIT. Given the pleotropic effects of cystamine on neurologic responses, further studies involving gene knockout mice could be used to provide more direct experimental evidence of this relationship.
In summary, characterization of pharmacogenetic factors affecting HIT in a murine model has identified a new approach for preventing the dose-limiting toxicity of an antipsychotic agent. Coadministration of cystamine could prevent the movement disorder caused by haloperidol, which could improve outcomes for individuals with psychotic diseases. Moreover, these results indicate that subsequent clinical studies should be performed to investigate whether cystamine administration could reduce the extent of brain damage in individuals with ischemic stroke.
Acknowledgments
We thank Angela Rogers and Bob Lewis for reviewing this manuscript and for their help in interpreting our data.
Footnotes
Communicating editor: A. A. Palmer
Supplemental material is available online at www.genetics.org/lookup/suppl/doi:10.1534/genetics.115.184648/-/DC1.
Literature Cited
Dohil, R., M. Fidler, J. A. Gangoiti, F. Kaskel, J. A. Schneider et al., 2010 Twice-daily cysteamine bitartrate therapy for children with cystinosis. J. Pediatr. 156: 71–75.
Kaskow, B. J., L. A. Diepeveen, J. Michael Proffitt, A. J. Rea, D. Ulgiati et al., 2013 Molecular prioritization strategies to identify functional genetic variants in the cardiovascular disease-associated expression QTL Vanin-1. Eur. J. Hum. Genet. 22: 688–695.
Smyth, G. K., 2004 Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 3: Article3.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}