





Abstract
Animal microRNAs (miRNA) are implicated in the control of nearly all cellular functions. Due to high sequence redundancy within the miRNA gene pool, loss of most of these 21- to 24-bp long RNAs individually does not cause a phenotype. Thus, only very few miRNAs have been associated with clear functional roles. We constructed a transgenic UAS-miRNA library in Drosophila melanogaster that contains 180 fly miRNAs. This library circumvents the redundancy issues by facilitating the controlled misexpression of individual miRNAs and is a useful tool to complement loss-of-function approaches. Demonstrating the effectiveness of our library, 78 miRNAs induced clear phenotypes. Most of these miRNAs were previously unstudied. Furthermore, we present a simple system to create GFP sensors to monitor miRNA expression and test direct functional interactions in vivo. Finally, we focus on the miR-92 family and identify a direct target gene that is responsible for the specific wing phenotype induced by the misexpression of miR-92 family members.
DEVELOPMENT of multicellular organisms requires precisely regulated gene expression. In the past decade, microRNAs (miRNAs) have been found to be essential in fine-tuning gene expression by post-transcriptional regulation (Ambros 2004). Most miRNAs are transcribed by RNA polymerase II. The primary transcript (pri-miRNA) is processed within the nucleus by Pasha and Drosha into the short pre-miRNA hairpin (Krol et al. 2010). A second class of small RNAs called “mirtrons” derive from an alternative pathway by the splicing of short intronic hairpins (Ruby et al. 2007; Okamura et al. 2009). These mirtrons bypass Drosha processing, and their biogenesis merges with the canonical miRNA pathway during nuclear export of the hairpin. Both types of hairpins are exported to the cytoplasm and further processed by Dicer into the mature miRNA. One of the two strands is incorporated into the RNA-induced silencing complex (RISC), while the other strand is degraded. The mature miRNAs are 21–24 bp long and guide the RISC to target messenger RNAs (mRNAs) by binding to partially complementary sequences in the target 3′ UTR (Brennecke et al. 2005). These binding events block the translation of mature protein from the mRNA, and recent studies have indicated that many miRNAs also induce the rapid decay of target mRNAs (Bagga et al. 2005; Lim et al. 2005; Wu and Belasco 2008). Directly or indirectly, individual miRNAs may modulate the translation of hundreds of genes (Selbach et al. 2008). A large number of functions are affected by these changes. In contrast to their important regulatory role in virtually all cellular processes, loss-of-function mutations of only a few miRNAs cause obvious phenotypic consequences (Alvarez-Saavedra and Horvitz 2010). This may be due to redundancy within the miRNA pool or to the existence of alternative regulatory pathways. Loss-of-function phenotypes become more apparent when the entire family is knocked out, demonstrating how critical redundancy is (Alvarez-Saavedra and Horvitz 2010). Membership in a miRNA family is defined by the seed sequence, which consists of nucleotides 2–8 at the 5′ end of the mature miRNA. The seed sequence is thought to be crucial for target recognition (Brennecke et al. 2005). However, in higher organisms it is nearly impossible to knock out larger miRNA families.
The miRNA pool of Drosophila is significantly smaller and less redundant than it is in higher vertebrates (240 fly miRNAs compared to >1500 in humans, according to miRBase version 18) (Griffiths-Jones 2004, 2006; Griffiths-Jones et al. 2007; Kozomara and Griffiths-Jones 2010). However, all vertebrate miRNA families have representatives in Drosophila, and >80 Drosophila miRNA have clear human homologs (Ibáñez-Ventoso et al. 2008). We focused on a set of 180 annotated miRNAs that includes all evolutionarily conserved and highly abundant miRNAs as well as many previously unstudied miRNAs.
Since it circumvents the issue of redundancy, overexpression is an attractive approach to studying miRNA function. One caveat of overexpression studies could be that miRNAs with exaggerated expression might bind to nonphysiological targets. However, it was shown previously that overexpression of miRNAs can reveal physiologically relevant targets. In fact, overexpression might be better suited for RNA profiling since it can induce larger expression changes in target mRNAs than knock-down experiments with the same miRNA (Baek et al. 2008; Selbach et al. 2008). Furthermore, miRNA overexpression is easier to achieve than the creation of loss-of-function mutants for each miRNA, which typically requires very labor-intensive targeted deletions by homologous recombination. Frequently, miRNA loci are transcribed from a polycistronic gene structure, giving rise to RNA precursors with extensive secondary structure. These structures might be disrupted by deletion of individual miRNAs and thereby potentially also affect the expression of other miRNAs (Ambros 2004; He et al. 2005). Deletion of entire clusters was applied to such situations, but this also complicates the functional analysis as miRNAs in some clusters can have opposing effects, e.g., the miR-17-92 cluster (Bonauer and Dimmeler 2009), which targets both positive and negative cell cycle regulators. A further disadvantage of gene deletion is the complete absence of miRNA product. Phenotypes will manifest themselves in the tissue that first requires the specific function of the miRNA, thereby potentially masking additional functions later in development or in other tissues.
Also, miRNAs with highly specific expression patterns in few cells might not cause easily observable loss-of-function phenotypes, while misexpression of these miRNAs might induce more widespread knock-down of potential target genes revealing loss- or reduction-of-function phenotypes associated with these target genes. Thus, miRNAs might induce more interpretable phenotypes by misexpression than by loss of function, thereby facilitating the identification of their biological roles.
The presented library of inducible UAS-miRNA expression lines will allow the experimental examination of individual miRNA in a systematic and comprehensive way. The inducible Gal4-UAS system for transgene induction enables complete spatio-temporal control of gene expression. Our approach complements loss-of-function studies, either by knock-out or sponge-mediated knock-down (Loya et al. 2009). Together, these techniques will facilitate detailed investigation of the entire complement of miRNAs in a complex organism.
Materials and Methods
Cloning of miRNA precursors into pW20
pVALIUM20 [from the Transgenic RNAi Project (TRiP) Harvard Medical School] was digested with HindIII to remove the vermilion marker gene. A white mini-gene was PCR-amplified from a pUASTattB vector (GenBank accession no. EF362409) and cloned into the pV20 backbone to create pW20. To clone the miRNA precursor sequences, pW20 was digested with EcoRI and NheI and gel-purified. Customized forward and reverse oligonucleotides (Microsynth AG, Balgach, Switzerland) were heated to 95° for 5 min and cooled to room temperature. Oligos were designed to create sticky ends for the respective restriction sites on both sides for efficient cloning and spanned the entire predicted stem-loop structures listed in miRBase. Annealed oligos were diluted 1:200 in TE buffer, pooled, and cloned into pW20. Bacterial colonies were genotyped by colony PCR and subsequent sequencing. For miRNAs longer than 119 nucleotides, corresponding oligonucleotides were annealed according to the protocol above. In addition to the restriction sites, these oligos contain extra base pairs at the ends to allow for efficient digest. To fill in the single-stranded overhangs, 10 cycles of PCR were performed using standard PCR conditions on the annealed oligonucleotides. The resulting product was purified on a minicolumn and digested using EcoRI and NheI. Restriction enzymes were heat-inactivated at 65° for 20 min, and the oligos were cloned into pW20. This method was applied for mir-31b, -307b, -956, -989, -998, -2491, -3645, -4951, -4966, -4968, and -4969. mir-997 required an additional step of PCR amplification due to its length. After filling in the overhangs as mentioned above, mir-997 was PCR-amplified using primers mir-997-fw and mir-997-rv and then digested and cloned as mentioned above.
GFP sensor cloning and quantification
A pUASgattB vector was digested using BglII and XhoI. A copy of eGFP was PCR-amplified from pGFPattB using primers BglII-GFP-fw and XhoI-GFP-rv and subloned and inserted into the pUASgattB backbone, creating pGFPattB. 3′ UTRs were PCR-amplified from genomic DNA and cloned into pGFPattB digested with XhoI and HindIII or XhoI only (in the case of 3′ UTRs, which contain a HindIII site).
GFP intensity was quantified with ImageJ. Pictures were taken at identical microscope settings. Pixel intensity was measured in a defined region and normalized to the background. Control samples were set to 100% and relative changes in the experimental samples are shown (average ± SD).
Generation of transgenic fly strains
Plasmids obtained through MiniPrep were pooled, purified on a MidiPrep column, and diluted to 100 ng/μl. Pooled plasmids were injected into embryos of the line ZH-86Fb, carrying an attP site on chromosome arm 3R. Two males were crossed to four y w virgins, and the offspring were screened for white-positive animals. These white-rescued flies were balanced over TM3, Sb to generate the final stocks. Correct insertion of the transgene into the landing site was controlled by PCR using the primers 86Fb-gen-rv and white-F4.
Single-fly DNA preparation
To prepare genomic DNA, single flies were placed in PCR tubes and frozen at −80° for 30 min. The flies were squashed using 50nμl of squishing buffer (10 mM Tris–Cl (pH 8.2), 1 mM EDTA, 25 mM NaCl, 0.2% (w/v) Triton X100, and 300 μg/ml proteinase K). The flies were then incubated at 37° for 1 hr, and subsequently proteinase K was inactivated at 95° for 5 min.
Fly stocks
The following fly stocks were used: actin-5C-Gal4: y w; Sp/CyO; actin-5C-Gal4/TM6B [y+]; MS1096-Gal4: y w MS1096-Gal4; +; +; ey-Gal4: +; ey-Gal4; +; +; TM6B: y w ; +; MKRS/TM6B [y+]; TM3, Sb: y w; +; D gl3/TM3 Sb Ser; UAS-sha: w; P{UAS-sha.GFP}3 (Bloomington 32096) UAS-ck: w; +; UAS-hGFP-ck.
Results
Library construction
To construct our miRNA-overexpression library, we employed the phC31-mediated integration system and used the attp landing site 86Fb on chromosome 3 to create identical integrations for all transgenes (Bischof et al. 2007). Cloning of the miRNA-hairpin precursor sequences was performed by fusion of customized oligos as previously reported (Haley et al. 2008). We used a modified Valium 20 vector (Ni et al. 2011) in which we replaced the vermillion marker gene with a white marker to enable easier screening for positive transformants (Figure 1A). All miRNAs are under the control of the UAS promoter, thereby enabling spatio-temporal control of expression by the established Gal4-UAS system (Duffy 2002). We also included 18 mirtrons that are annotated in miRBase (Supporting Information, Table S1). This miRNA library complements our efforts to also establish a genome-wide UAS-cDNA library (J. Bischof, M. Björklund, E. Furger, C. Schertel, J. Taipale, and K. Basler, unpublished results; C. Schertel, D. Huang, M. Björklund, J. Bischof, D. Yin, R. Li, R. Zeng, J. Wu, J. Taipale, H. Song, and K. Basler, published results). UAS-cDNA lines can be used to try to suppress miRNA overexpression phenotypes when a specific target gene is suspected to cause the phenotype.
![Construction and evaluation of the UAS-miR library. (A) pW20. Purple bars represent gypsy insulator sequences. Black triangles indicate loxP sites. miRNA precursors are directly cloned into EcoRI and NheI sites. The attB integration site, the white marker, and ampicillin resistance are indicated. (B) pUAST-eGFPattB vector and cloning strategy for 3′ UTR constructs. (C) Correlation of miRNA expression to phenotypic effects caused by miRNA expression. We divided the miRNAs into three classes [top, medium, and lowest 49 miRNAs according to reads in deep-sequencing experiments of imaginal discs (Berezikov et al. 2011)]. (D) Distribution of phenotypes over the miRNA spectrum (black line). miRNAs were grouped into four classes according to their evolutionary conservation and abundance. Class I (bantam to miR-193) contains 44, class II (miR-210 to miR-375) 41, class III (miR-927 to miR-1017) 68, and class IV (miR-2279 to miR-4976) 27 miRNAs. The percentage of lethal phenotypes among all phenotypes in each class is indicated by the red line.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/genetics/192/4/10.1534_genetics.112.145383/7/m_1543fig1.jpeg?Expires=1716580760&Signature=NTYLPZrtSEpw1MZuFg3hCvG5UTjPkje~5jIL-FCOkR3qKCOqC6Rs4VjuJjs-WSNkwWXTlu67FLrVM5WPxi0rfMqAIVq6iHteR~6ACO1-WA5nBEtaaZcIdPEgFOqmlw7S8MGY3Guz1xorAbNxtF6EDTZlIADCcyE9ZszVsJWb~iPIJxQHT32rli0sFHdX5iYUifiIEYffvqBhZfhY~jYBhRNaHFENQ9AUvpl2mV3qJNq9aRqIMvQPk5cXOACYxoqhUtHCrNmzgvBmPC3ewgQua1GY3w13UmUte0Q4CCXgJEdM49a1~DV3ThlChClkNpWO1MQl92qhqp0P8MKysBAgxQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Construction and evaluation of the UAS-miR library. (A) pW20. Purple bars represent gypsy insulator sequences. Black triangles indicate loxP sites. miRNA precursors are directly cloned into EcoRI and NheI sites. The attB integration site, the white marker, and ampicillin resistance are indicated. (B) pUAST-eGFPattB vector and cloning strategy for 3′ UTR constructs. (C) Correlation of miRNA expression to phenotypic effects caused by miRNA expression. We divided the miRNAs into three classes [top, medium, and lowest 49 miRNAs according to reads in deep-sequencing experiments of imaginal discs (Berezikov et al. 2011)]. (D) Distribution of phenotypes over the miRNA spectrum (black line). miRNAs were grouped into four classes according to their evolutionary conservation and abundance. Class I (bantam to miR-193) contains 44, class II (miR-210 to miR-375) 41, class III (miR-927 to miR-1017) 68, and class IV (miR-2279 to miR-4976) 27 miRNAs. The percentage of lethal phenotypes among all phenotypes in each class is indicated by the red line.
During the preparation of this article, additional miRNAs were identified by high-throughput sequencing experiments, raising the number to 240 miRNA in the Drosophila genome (miRBase release 18). However, most of these newly annotated miRNAs were found with only very low abundance of the mature miRNA or the stem loop precursor sequence despite extensive sequencing efforts. Furthermore, none of these additional miRNAs are evolutionarily conserved in higher organisms. Thus, we decided to add only the 5 most abundant of these additional miRNAs (supported by at least 100 read counts) to raise the number of miRNA lines in our transgenic library to 180 (Table S1).
Library characterization
To validate the miRNA library experimentally, we expressed all miRNAs in the developing embryo by a constitutively active act5C-Gal4 driver. A surprisingly high number of miRNAs (35%, 63/180) caused lethality at different stages of development. An additional five miRNAs induced more specific phenotypes upon constitutive overexpression (Table S1). This is in stark contrast to loss-of-function observations of miRNAs. In Caenorhabditis elegans, only ∼10% of all miRNA deletions had detectable phenotypic consequences (Alvarez-Saavedra and Horvitz 2010). Our high hit rate validates misexpression as an approach to probe miRNA function. Ubiquitous misexpression might lead to unspecific developmental defects due to the simultaneous downregulation of multiple target genes in multiple tissues. To refine our analysis, we further characterized the miRNAs by expressing each in the developing eye (ey-Gal4) and wing (MS1096-Gal4). While these tissues are not required for viability, they allow us to study the defects caused by miRNA expression more precisely and to cover a broader spectrum of endogenously expressed target mRNAs. The number of miRNAs that caused phenotypes in the wing (37%, 67/180) was similar to constitutive expression in the embryo (35%, 63/180). All the miRNAs that caused phenotypes in the eye (16%, 29/178) also induced a phenotype in the wing, indicating that the respective targets of these miRNAs are expressed in both tissues (all primary screening data can be found in Table S1). The larger number of miRNAs causing phenotypes in the wing (cf. the eye) might be due to a stronger induction of the Gal4 driver or to a higher sensitivity of the wing to developmental disturbances. Only 9 of 63 miRNAs induce phenotypes when driven by act5C-Gal4 but had no effect when ectopically expressed in the wing or the eye. Ubiquitous expression of miR-9a, -969, -982, -993, -994, or -1001 causes lethality while miR-4 and miR-6-1 induced bristle defects on the scutellum and miR-960 caused tissue outgrowth on the head. None of these 9 miRNAs induced any observable phenotypes in the wing or the eye. This indicates that the target genes that cause these defects are probably not expressed in the wing or eye or might not exert any function there that causes visible phenotypes. The fact that not all miRNAs induced phenotypes upon misexpression argues that the induced phenotypes are specific. In summary, 43.3% (78/180) of all the miRNAs tested gave rise to a phenotype in at least one of our three screens, indicating that our transgenes are functional (Table S1).
Next we asked if the ability of a miRNA to induce a phenotype is correlated with its endogenous expression level. We used expression data from the modENCODE project (Berezikov et al. 2011) of imaginal-disc-expressed miRNAs and compared the read count to our phenotypic analysis. We find a clear correlation between miRNA read count and induction of a phenotype in our screens (Figure 1C). Over 73% of the 49 miRNAs with the highest read counts induced phenotypes. This number drops to 49% for 49 miRNAs with medium read counts and to 29% for the least sequenced 48 miRNAs. This suggests that ectopic expression does not lead to novel or artificial miRNA–target interactions.
Furthermore, we observed a negative correlation between the miRNA-numbering and phenotypic effects in the wing and eye (Figure 1D). Historically, miRNAs have been discovered by cloning from small RNA libraries (Lee and Ambros 2001). Thus, more abundant miRNAs were discovered earlier than less abundant ones. Consequently, within the system of miRNA nomenclature, the miRNA numbering correlates roughly with abundance and also evolutionary conservation (e.g., miR-1 was discovered earlier than miR-1000 because it is much more abundant and more conserved). Less conserved miRNAs are thought to be still evolving functional relationships to target mRNAs (Chen and Rajewsky 2007; Lu et al. 2008) and are thus less likely to induce phenotypic effects in our screens. For our analysis, we subdivided all miRNAs into four classes according to their numbering (ranging from “highly conserved” and “highly expressed” to “not conserved” and “lowly expressed”). The percentage of induced phenotypes drops from >61.4% in class I to 42.6% in class III. Expressing a class IV miRNA did not have a detectable consequence in our screens (Figure 1D). In contrast, the percentage of embryonic lethality that is caused by miRNA expression stays constant, indicating that these miRNAs target essential genes. When we tested how many conserved miRNAs induce a phenotype in comparison to nonconserved miRNAs, we found a strong bias toward conserved miRNAs. Over 54% of all miRNAs that are conserved between Drosophila melanogaster and Homo sapiens induced phenotypes compared to only 10% of nonconserved miRNAs (conservation based on Ibáñez-Ventoso et al. 2008). Together, these observations confirm that conserved and highly abundant miRNAs are functionally more important than novel and lowly expressed miRNAs.
So far the number of miRNAs with clearly described phenotypes in Drosophila has been rather low. Phenotypes have been reported mainly for a few highly expressed and evolutionarily conserved miRNAs, including bantam, let-7, and miR-1 (Brennecke et al. 2003; Sokol and Ambros 2005; Sokol et al. 2008). In addition to these well-studied examples, only ∼24 miRNA have been associated with loss- or gain-of-function phenotypes in Drosophila (Smibert and Lai 2010). Most of the described phenotypes are rather subtle or restricted to specific tissues. Thus, it is hard to directly compare our phenotypic observations with the published data. We do, however, find that expression of 17 of the 26 miRNAs (65%) with published loss- or gain-of-function phenotypes gave rise to detectable defects, supporting the utility of our approach in uncovering the function of miRNA. In the remaining cases where we did not observe a phenotype in our assays, we might not have investigated the right tissue or developmental stage. Importantly, we observed many new phenotypes for previously unstudied miRNAs (Table S1), raising the overall number of miRNAs that are connected to a phenotypic effect to 78.
Transgenic miRNAs are properly processed into the mature form
The fact that we observe phenotypes indicates that the biogenesis and expression of the transgenic miRNA constructs works properly. Furthermore, the pVALIUM20 vector is well established as a tool to express small RNAs from hairpin precursor structures (Ni et al. 2011). However, it is possible that the overexpression of miRNAs leads to a situation in which the endogenous biogenesis machinery cannot process all precursor miRNAs, leading to an accumulation of these precursors and potentially unspecific effects. To address this question and to directly confirm the expression from our transgenes, we tested the level of miRNA expression by a molecular approach. We chose the members of the miR-92 family and expressed them in the wing imaginal disc by MS1096-Gal4. Total RNA was isolated from discs, and expression was monitored by Northern blot analysis (Figure S1). The results indicate that the mature miRNAs are efficiently produced and expressed in our transgenic lines. We could not detect precursor hairpins, indicating that all expressed miRNA hairpins are efficiently processed. One notable exception is miR-313 for which we could not detect any mature miRNA but the corresponding miR*. Consistent with these results, the annotation of miRBase shows that the star strand is actually much more abundant in deep sequencing experiments, indicating that this hairpin might have experienced an arm-switching event during evolution (Berezikov 2011).
Members of the miR-92 family act like an allelic series to cause wing hair loss
Expression of some members of the miR-92 family with the wing-specific MS1096-Gal4 driver led to a loss of wing hairs in regions adjacent to the longitudinal veins (Figure 2). Due to the presence of the same seed sequence, which defines this family, all six miRNAs should repress an overlapping set of target genes. We observed the strongest effect for miR-92a and miR-92b, while miR-312 caused a qualitatively similar but less pronounced defect (Figure 2, B, C, and F). The other three family members, miR-310, miR-311, and miR-313, do not induce this defect in a hemizygous situation. The wing hair loss is even stronger in homozygous situations when two copies of the miRNA transgenes are induced, indicating a dose effect (data not shown). In this situation, miR-310 and miR-311 also cause a weak loss of wing hairs along the longitudinal vein (Figure 2H and data not shown). We never observed a phenotype for miR-313.
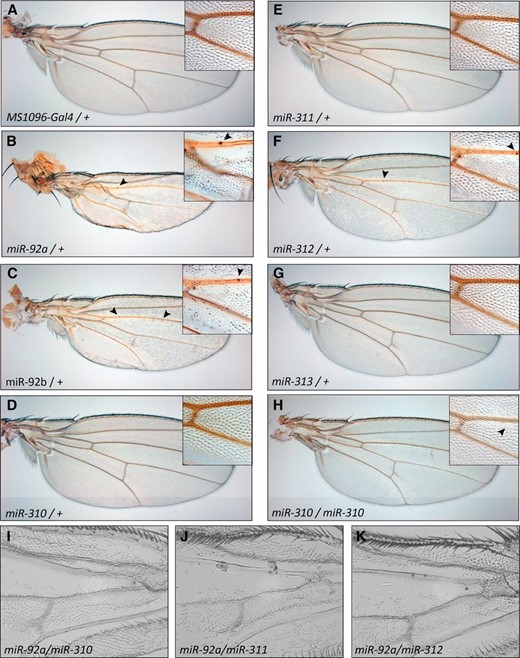
Wing phenotypes caused by expression of the miR-92 family. (A) Control wing. (B) Expression of miR-92a leads to strong wing hair loss, deformation of the wing blade, and formation of ectopic sensillae (arrowheads). Insets show magnifications of the region directly posterior to the anterior cross vein. (C–G) miR-92b and miR-312 expression causes an intermediate phenotype, while miR-310, miR-311, and miR-313 cause no obvious phenotype in a heterozygous state. (H) miR-310 (and miR-311; data not shown) expression leads to mild wing hair loss in homozygous transgenic situations. (I–K) Enhanced phenotypes are induced by co-expression of different miR-92 family members. All transgenes are driven by MS1096-Gal4. All wings were derived from adult females.
To test whether the effect is enhanced by the simultaneous expression of different family members, we tested all pairwise combinations within the miR-92 family by co-expression of the respective transgenes and monitored the induced phenotypes. In most cases, we saw an additive effect of the two transgenes on the phenotype except for the miR-312/miR-313 combination (results are summarized in Table 1).
miR-92 family co-overexpression
| miRNA | Phenotype (one copy) | miR-92a (++) | miR-92b(+) | miR-310 (no) | miR-311 (no) | miR-312 (+) | miR-313 (no) |
|---|---|---|---|---|---|---|---|
| miR-92a | ++ | Lethal | +++ | +++ | +++ | +++ | ++ |
| miR-92b | + | + | ++ | + | + | + | |
| miR-310 | no | + | + | ++ | no | ||
| miR-311 | no | + | + | no | |||
| miR-312 | + | + | no | ||||
| miR-313 | no | no |
| miRNA | Phenotype (one copy) | miR-92a (++) | miR-92b(+) | miR-310 (no) | miR-311 (no) | miR-312 (+) | miR-313 (no) |
|---|---|---|---|---|---|---|---|
| miR-92a | ++ | Lethal | +++ | +++ | +++ | +++ | ++ |
| miR-92b | + | + | ++ | + | + | + | |
| miR-310 | no | + | + | ++ | no | ||
| miR-311 | no | + | + | no | |||
| miR-312 | + | + | no | ||||
| miR-313 | no | no |
The phenotypic classification refers to the severity of the bristle loss (no: no effect, + mild, +++ strong). In some cases, the co-expression phenotype is even stronger than the added effect of each miRNA individually (e.g., miR-92b/miR-310 or miR-310/miR-311). These effects might be caused by overall higher miRNA levels due to induction of two transgenes. Alternatively, the miRNAs could act in parallel on different target sites that synergize in their repressive effect.
| miRNA | Phenotype (one copy) | miR-92a (++) | miR-92b(+) | miR-310 (no) | miR-311 (no) | miR-312 (+) | miR-313 (no) |
|---|---|---|---|---|---|---|---|
| miR-92a | ++ | Lethal | +++ | +++ | +++ | +++ | ++ |
| miR-92b | + | + | ++ | + | + | + | |
| miR-310 | no | + | + | ++ | no | ||
| miR-311 | no | + | + | no | |||
| miR-312 | + | + | no | ||||
| miR-313 | no | no |
| miRNA | Phenotype (one copy) | miR-92a (++) | miR-92b(+) | miR-310 (no) | miR-311 (no) | miR-312 (+) | miR-313 (no) |
|---|---|---|---|---|---|---|---|
| miR-92a | ++ | Lethal | +++ | +++ | +++ | +++ | ++ |
| miR-92b | + | + | ++ | + | + | + | |
| miR-310 | no | + | + | ++ | no | ||
| miR-311 | no | + | + | no | |||
| miR-312 | + | + | no | ||||
| miR-313 | no | no |
The phenotypic classification refers to the severity of the bristle loss (no: no effect, + mild, +++ strong). In some cases, the co-expression phenotype is even stronger than the added effect of each miRNA individually (e.g., miR-92b/miR-310 or miR-310/miR-311). These effects might be caused by overall higher miRNA levels due to induction of two transgenes. Alternatively, the miRNAs could act in parallel on different target sites that synergize in their repressive effect.
shavenoid is one of the targets that causes wing hair loss
The observed loss of wing hairs resembles the phenotype caused by a loss of function of the gene shavenoid (sha) (Berezikov 2011), which is a predicted target of miR-92, containing five potential binding sites for the miRNA. Furthermore, two other genes that can cause wing hair defects upon inactivation, fritz (frtz) and crinkled (ck), are also among the predicted target genes of miR-92. Thus, we wanted to test if sha is indeed responsible for the loss of wing hairs due to downregulation by the miRNAs. We co-expressed a UAS-sha transgene together with the miRNA in wing discs and monitored the effect on wing hairs in the adult animals. Since the sha transgene does not carry the endogenous 3′ UTR, it should not be subject to miR-92 family regulation itself. Indeed, sha expression could suppress the wing hair loss (Figure 3, D and F), indicating that the phenotype is indeed caused by downregulation of sha. In contrast, a UAS-ck transgene was not able to rescue the wing hair defect observed in miR-92b- or miR-312-expressing flies (Figure S2). We could not test a rescue by frtz co-expression due to the lack of a UAS-frtz transgene. These data suggest that, at least, sha is a direct target of the miRNAs and is also likely to be targeted under physiological conditions.
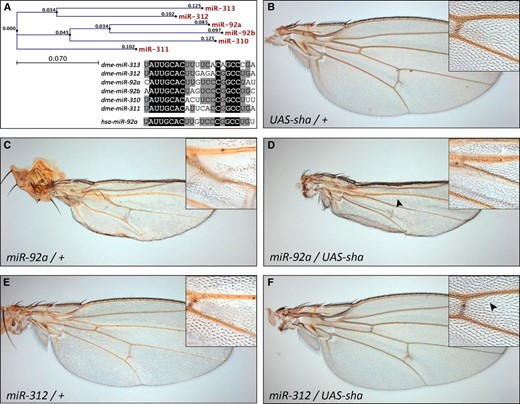
shavenoid co-expression rescues the wing hair loss. (A) Phylogenetic analysis of the miR-92 family and sequence comparison of the six Drosophila (dme) family members and the human (hsa) miR-92a homolog. Identical residues are marked in black and residues shared by at least four family members are shaded in gray. (B–F) Expression of a UAS-sha transgene causes no effect in a wild-type background (B) but rescues the wing hair loss induced by miR-92a (compare C to D) or miR-312 (compare E to F) expression.
Intriguingly, the three miRNAs that cause the strongest phenotypes in the wing contain a G at position 11 of the mature miRNA sequence, while the three that do not cause any effect share a U at this position (Figure 3A). To test whether this single base is particularly important for the miRNA target recognition, we constructed mutant versions of the miRNA expression constructs. We exchanged the G in miR-92a with a U (miR-92aG→U) to test if this would reduce the phenotype. Similarly, we exchanged the U in miR-310 and miR-311 (miR-310U→G and miR-311U→G), both of which showed a weak wing hair loss in a homozygous situation, to test if this change improves the transgene’s capability to induce wing hair loss. It is important to point out that the mutated position does not coincide with the seed region of the respective miRNAs. Upon induction of the transgenes, the mutant versions caused qualitatively and quantitatively similar effects as the wild-type transgenes (data not shown). This indicates that the target recognition capability of the miR-92 family is not significantly impacted by position 11 of the mature miRNA. The experiment illustrates the potential of our system for easy and fast structure–function experiments that connect the primary miRNA sequence with in vivo phenotypic readouts.
GFP sensors confirm sha as a target of the miR-92 family
To directly monitor the effect of a specific miRNA on its putative target 3′ UTR, we fused the enhanced GFP (eGFP) coding sequence to the endogenous 3′ UTR of the predicted target gene. The 3′ UTR can be PCR-amplified from genomic DNA and is cloned directly into the pUASTattB eGFP vector (Figure 1B). We integrated the transgenes into the same genomic landing site as the miRNA transgenes. Gal4-mediated co-expression of a miRNA with the putative eGFP sensor target determines if there is an interaction in vivo. The interaction between miRNA and the target UTR is reflected in the eGFP intensity. We tested the effect of the miR-92 family on the expression of an eGFP fused with the 3′ UTR of the putative targets sha, frtz, and ck (Figure 4, A–C). The strength of the effects on the sha-eGFP sensor correlates with the severity of the wing hair loss that we observe in the adults. Co-expression of miR-92a or miR-92b caused the strongest downregulation of eGFP (Figure 4A′). miR-312 expression causes clear downregulation of the eGFP signal whereas expressing miR-313 has no effect on the sha-eGFP sensor (Figure 4, A–C). A surprisingly strong suppressive effect is caused by miR-310 and miR-311, which caused only mild wing hair loss even in a homozygous situation. Similar effects were observed with the frtz-eGFP and ck-eGFP sensors: miR-92a, miR-92b, and miR-312 caused strong eGFP downregulation, while miR-310, miR-311, and miR-313 had no effect on eGFP levels as compared to the negative control (Figure 4, B′ and C′).
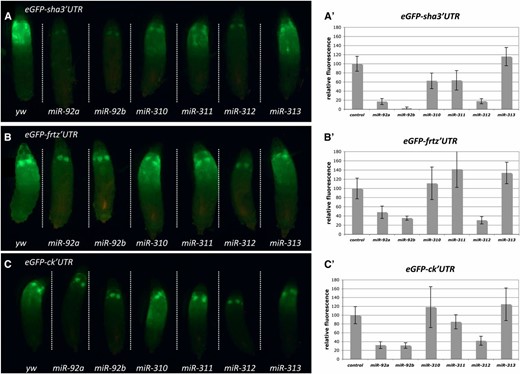
Co-expression of miR-92 family miRNAs with target sensor constructs confirms sha, frtz, and ck as in vivo targets. (A–C) Co-expression of (A) UAS > eGFP-sha 3′UTR, (B) UAS > eGFP-frtz 3′UTR, or (C) UAS > eGFP-ck 3′UTR with miR-92 family members by MS1096-Gal4. All larvae for each 3′ UTR were aligned on a single slide to ensure direct comparison of GFP intensities. (A′–C′) Quantification of GFP intensity using ImageJ software. Between 5 and 11 larvae were measured per data point. Average ± SD is shown, and values are adjusted relative to a control cross (yw), which was set to 100%.
In summary, we present phenotypes for 78 miRNAs, thereby greatly expanding our knowledge about possible roles and functions for this important class of molecules (Table S1). Furthermore, we tested the conserved miR-92 family in more detail and confirmed at least one target gene that is required for the formation of proper wing hair structures in vivo. These results validate our approach to studying miRNA function by misexpression and illustrate how the presented library will serve as a valuable tool for the Drosophila community.
Discussion
Here we present a library of fly lines containing individual transgenes that allow the Gal4-UAS controlled expression of a large fraction of the miRNAs in D. melanogaster. This in vivo library provides an important genetic tool that complements miRNA loss-of-function approaches (e.g., knock-outs, deletions, and miRNA sponges).
While this work was under review, Lai and colleagues described their efforts to generate a similar UAS-miRNA resource (Bejarano et al. 2012). Furthermore, an earlier publication reported a smaller collection of UAS-miRNA lines that was used as a tool to complement genetic deficiency screening (Szuplewski et al. 2012). Our transgenic strategy provides an additional tool that greatly extends the number of experimentally approachable miRNAs. Furthermore, our setup has several advantages. By using site-directed integrations with the identical landing site for all our miRNA transgenes, we avoid issues that arise from the genetic background of the insertion site, or so-called “position effects.” The abundance of the mature miRNA depends only on the processing efficiency of the hairpin precursor by the endogenous biogenesis machinery. Importantly, in contrast to Bejarano et al. (2012), our cloning system focuses only on the pre-miRNA hairpin without adding additional sequences or markers. All our constructs were cloned into identical vectors. The use of customized oligonucleotides is simpler than the PCR-based strategies presented in the other two articles and prevents PCR-based problems. Furthermore, the use of oligonucleotides is an easy way to introduce specific changes and thus to perform structure–function analysis of pre-miRNA hairpins. We examined the biogenesis and induction of our transgenes by Northern blot analysis and found that all (with the exception of the wrongly annotated miR-313) are properly processed into the mature form. Thus, our system allows for the misexpression of individual pre-miRNAs in otherwise identical genetic backgrounds regardless of the genomic situation in which multiple miRNAs might be expressed from a single RNA precursor. However, co-expression of two miRNAs (e.g., from the same or different family) is still possible as we demonstrated by the co-expression of miR-92 family members in the wing. By employing different landing sites, we could even co-express more miRNAs than currently possible with the library presented here. Our library provides the most complete set, comprising 180 miRNAs, thereby significantly raising the number of experimentally amenable miRNAs beyond that of the other two resources.
By misexpression we circumvent the problem of redundancy within miRNA families. Furthermore, we can spatio-temporally control expression using the Gal4-UAS system, while loss-of-function approaches generally work only in the tissue that exhibits endogenous expression of the specific miRNA. Analysis of loss-of-function situations is often restricted to the first phenotype (e.g., lethality), which may mask additional functions later in development of the same or other tissues. Misexpression coupled to transcript profiling can be used to identify miRNA targets that are regulated by mRNA degradation. Since the effect of miRNA misexpression is thought to induce stronger fold changes in target mRNAs than a loss-of-function situation (Selbach et al. 2008), the library will be a useful tool for target identification. In addition to transcriptional profiling, miRNA misexpression is well suited for methods like PAR-CLIP (Photoactivatable-Ribonucleoside-Enhanced Crosslinking and Immunoprecipitation) since it offers easily accessible genetic controls (miRNA expression on or off), which can help to identify Ago-associated mRNAs to a much greater extent in the presence of the misexpressed miRNA (Easow et al. 2007).
On the other hand, one must also keep in mind that there are a number of caveats that may affect our library. First, physiologically relevant miRNA–target mRNA interactions can be identified only among the mRNAs that are present in the tissue where the miRNAs are misexpressed. We demonstrate this point by using different tissues as genetic background for our in vivo screens. Since each tissue (developing embryo, eye, and wing) expresses a different set of genes, we, as expected, observed non-overlapping hit sets. Second, overexpression could cause artificial downregulation of nonphysiological target genes. By increasing the abundance of a specific miRNA over the endogenous level or complete ectopic expression, it could be possible to target 3′ UTRs that contain weak binding sites for the miRNA sequence—UTRs that are not physiological targets. This concern has to be considered in the subsequent validation. To this end, loss-of-function approaches will be required. However, misexpression studies can provide a catalog of potential targets that can be bound by a specific miRNA. This is particularly relevant in pathological conditions where miRNAs are commonly misexpressed, such as tumors. When conserved, the targets will provide insight into the pathology of such tumors and help identify the pathways that are misregulated. Finally, a number of targets are subject to translational repression (Baek et al. 2008; Selbach et al. 2008), which will not be accessible to identification by transcriptional profiling.
The sha-eGFP sensors clearly show that the sha 3′ UTR can be regulated the miR-92 family. Also, the miRNA misexpression phenotype could be rescued by co-expression of the miRNA with its potential target gene. However, sha mRNA abundance was not significantly altered in miR-92a-expressing wing discs (our unpublished results), indicating translational blocking as the main mechanism of repression. Using the tools that we present here this method can be used to test genetic interactions between miRNAs and predicted targets in vivo. Two other genes that cause wing hair loss upon inactivation, frtz and ck, might also contribute to the miR-92 family phenotype since their respective 3′ UTRs are also targeted by at least three family members. While sha is required to determine temporal aspects of wing hair formation, frtz and ck act in parallel by spatial specification through the planar cell polarity pathway (Collier et al. 2005; Ren et al. 2006). Interestingly, misregulation of the closest sha homolog in humans (MAML2) is involved in mucoepidermoid carcinomas by aberrant Notch signaling (Köchert et al. 2011; Von Holstein et al. 2012). Our results suggest the possibility that changes in miRNA abundance, which are also commonly associated with tumor tissues, could be correlated with this regulatory loop.
GFP sensors as we generated for putative target of the miR-92 family can be used in combination with our miRNA library in several ways. One can test if a gene is a potential direct target as we did for sha. One can also take the reverse approach and screen the entire library to identify miRNAs that target the 3′ UTR of a specific gene. This will be a valuable approach in identifying potential miRNA regulators for a gene of interest and for the validation of predicted miRNA–target interactions.
The generation of the library described here provides an important advance in the efforts to identify specific miRNA functions. Our initial analysis assigned phenotypes to 78 miRNAs, most of which have not been studied at this point. The availability of a large number of UAS-cDNA lines (J. Bischof, M. Björklund, E. Furger, C. Schertel, J. Taipale, and K. Basler, unpublished results; C. Schertel, D. Huang, M. Björklund, J. Bischof, D. Yin, R. Li, R. Zeng, J. Wu, J. Taipale, H. Song, and K. Basler, unpublished results) provides a unique opportunity to test functional interactions between miRNA overexpression and the respective target genes in vivo. Combined with miRNA loss-of-function techniques, these tools will facilitate the study of miRNA function in a complex organism in unprecedented detail.
Acknowledgments
We thank Johannes Bischof and Franz Meyer for technical assistance with transgenesis and George Hausman for critical reading and suggestions on the manuscript. We thank the TRiP at Harvard Medical School (National Institutes of Health/National Institute of General Medical Sciences grant R01-GM084947) for providing plasmid vectors used in this study and Daniel Eberl for providing the UAS-ck strain. This work was supported by the Deutsche Forschungsgemeinschaft (SCHE 1641/1-1), the University of Zurich Research Priority Program “Systems Biology,” the National Center of Competence in Research “Frontiers in Genetics,” and the Swiss National Science Foundation and the Canton of Zurich.
Literature Cited
Footnotes
Communicating editor: L. Cooley
Author notes
Supporting information is available online at http://www.genetics.org/lookup/suppl/doi:10.1534/genetics.112.145383/-/DC1/.

{kind=link}
{kind=link}
{kind=link}
{kind=link}