





Abstract
Excision of a Mos1 transposon in the germline of Caenorhabditis elegans generates a double-strand break in the chromosome. We demonstrate that breaks are most prominently repaired by gene conversion from the homolog, but also rarely by nonhomologous end-joining. In some cases, gene conversion events are resolved by crossing over. Surprisingly, expression of the transposase using an intestine-specific promoter can induce repair, raising the possibility that activation of transposase expression in somatic cells can lead to transposition of Mos1 in the germline.
DNA transposons move through a “cut-and-paste” mechanism, which generates a double-strand break in the chromosome. The impact of such a break on genome stability depends on the repair mechanisms at work in the germline. Breaks can be repaired by simply rejoining the broken ends or by homologous recombination (for reviews see Haber 2000; Hefferin and Tomkinson 2005; Chen et al. 2007). End-joining generates blunt ends and ligates the breaks; such events can be recognized by the presence of short footprints. In some cases, a broken end can scan for microhomologies in the other end; such events usually generate small deletions. By contrast, homologous recombination restores broken DNA with high fidelity by using template-dependent DNA synthesis. In the “Szostak model” (Szostak et al. 1983), the broken chromosome and the repair template transiently form a double Holliday junction. Resolution of such structure can potentially produce crossover and noncrossover outcomes but recent works (reviewed in Cromie and Smith 2007) suggest that the resolution of double Holliday junction structure is strongly biased toward crossover formation, which leads to the exchange of chromosome arms. Synthesis-dependent strand annealing (SDSA) uses a template for repair and is also considered conservative, although it can occasionally introduce small deletions or insertions as it has been observed after P-element excision in the germline of Drosophila (Nassif et al. 1994; McVey et al. 2004a). DNA synthesis starts from the ends of the break using homologous sequences as templates. Replication under these circumstances is not fully processive and the strands tend to “fall off” the template. If the newly extended strands overlap, then the DNA can anneal and the gaps can be filled in by further DNA synthesis. When the displaced strand from the template DNA is extensive enough to allow the annealing of the free 5′-ends from the break, collapse of this intermediate leads to perfect repair of the break. In that case, there is nonreciprocal transfer of DNA from the donor to the recipient broken allele, a process called gene conversion. Because 3′ strands can invade at short regions of homology and because synthesis is not processive, SDSA can also introduce small duplications or deletions into the genome.
Double-strand break repair has been extensively characterized in yeast and in somatic cells of metazoans (for reviews see Kanaar et al. 1998; Paques and Haber 1999; Sonoda et al. 2006; Brugmans et al. 2007). However, relatively little is known about double-strand break repair in the germline (Engels et al. 1990; Gloor et al. 1991; McVey et al. 2004b; Clejan et al. 2006). Breaks can be generated at specific sites in chromosomes by transposon excision. The genome of Caenorhabditis elegans contains seven active types of Tc/mariner DNA transposons; however, transposition and thus the resulting DNA breaks are repressed in the germline of standard laboratory strains (Bessereau 2006). In mutator backgrounds, germline transposition is derepressed and breaks occur in the germline of these strains. Analysis of these transposition-induced breaks has indicated that they are repaired either by end-joining or by SDSA (Eide and Anderson 1988; Kiff et al. 1988; Plasterk 1991; Plasterk and Groenen 1992; Zwaal et al. 1993; Fischer et al. 2003). However, these studies were not configured to compare different repair pathways quantitatively or to detect gene conversion and crossing over.
In this study, we activated the heterologous Mos1 transposon (Bessereau et al. 2001) to analyze double-strand break repair in the germline of wild-type C. elegans. Mos1 is a member of the Tc1/mariner family in Drosophila (Jacobson and Hartl 1985; Jacobson et al. 1986). Expression of the Mos transposase in C. elegans is able to catalyze the insertion of Mos1 into the genome or to excise Mos1 from a chromosomal locus. We triggered germline remobilization of specific Mos1 insertions and separately assayed the repair of the chromosomal breaks by end-joining as well as by homologous recombination (Figure 1A). The induced double-strand breaks were mostly repaired by homologous recombination, including by the generation of interhomolog crossovers.
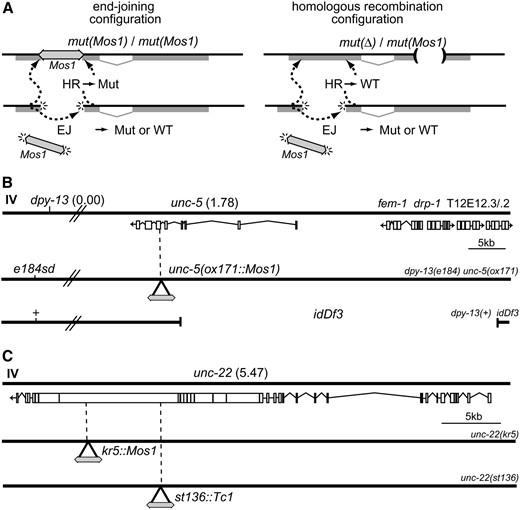
Transposon excision stimulates repair of double-strand breaks in the C. elegans germline. (A) Determining rates of repair. (Left) End-joining (EJ) events can be observed by identifying wild-type revertants in homozygotes for a Mos1 insertion allele. At the DNA level, these repair events generate pseudorevertants; that is, they are phenotypically wild type, but the gene sequence will contain a footprint at the site of excision. Repair by homologous recombination (HR) will regenerate the Mos1 insertion mutation. (Right) Homologous recombination in a heterozygote containing a mutation in a different part of the gene can generate a wild-type allele. In this configuration, end-joining can also generate pseudorevertant alleles but the frequency of such events is much lower. Sequence analysis of the footprints left at the repaired site can identify the repair mechanism for each event (see Table 1). (B) Chromosomes used to analyze Mos1-induced double-strand breaks at the unc-5 locus. unc-5(ox171∷Mos1) is a recessive mutant allele containing a Mos1 element inserted in the 6th exon of unc-5 at position 5,498,642 of chromosome IV (Robert and Bessereau 2007). dpy-13(e184sd) is 1.8 cM left of unc-5. idDf3, previously known as unc-5(ev447), is a deletion. Its left breakpoint is in the middle of unc-5 at position 5,500,882 of chromosome IV. Its right breakpoint was not mapped precisely but idDf3 completely removes sequences of fem-1, drp-1, T12E12.3, and T12E12.2 (A. Spence, personal communication). (C) Chromosomes used to analyze Mos1-induced double-strand breaks at the unc-22 locus. kr5∷Mos1 and st136∷Tc1 (Eide and Anderson 1988) are recessive mutant alleles of unc-22, which contain Mos1 and Tc1 insertions in the 20th and 27th exons of unc-22 at positions 11,979,064 and 11,983,660 of chromosome IV, respectively.
Double-strand breaks can be repaired by end-joining:
Mos1 excision will generate a double-strand break in the chromosome that could be repaired either by end-joining or by homologous recombination. To mobilize the Mos1 transposon, we expressed the Mos transposase under the control of a heat-shock-inducible promoter and analyzed revertants from two loci: unc-5 and unc-22 (Figure 1, B and C). unc-5 encodes the UNC-6/netrin receptor and is required for proper outgrowth and wiring of the nervous system (Hedgecock et al. 1990; Leung-Hagesteijn et al. 1992). The unc-5(ox171) allele contains an insertion of the Mos1 transposon into the coding region and leads to a severely paralyzed animal (Robert and Bessereau 2007). unc-22 encodes twitchin (Benian et al. 1989), a protein required for proper muscle morphology and contraction. unc-22(kr5) contains an insertion of Mos1 in the coding region and these mutants are paralyzed and exhibit a distinctive twitching phenotype.
To identify repair by end-joining and other nonconservative mechanisms, we analyzed repair in homozygous animals (Figure 1A, left). In this configuration, repair from the homolog will simply copy the transposon insertion back into the chromosome and no revertant progeny will be observed. However, in some cases end-joining will restore the reading frame of the gene and could potentially generate a functional protein. Parent animals were heat-shocked to induce the excision of Mos1 from the chromosome. Phenotypic revertants were identified in the progeny of the heat-shocked unc-5 and unc-22 parents at a frequency of 1 in 4000 screened animals (Table 1, reversion rate = 2.5 × 10−4 events/F1 offspring). To exclude the possibility that heat-shocking animals causes reversion, we heat-shocked animals that did not contain the hsp∷MosTransposase construct. No revertants were detected in the progeny (Table 1, reversion rate <8.3 × 10−5 events/F1 offspring). Moreover, expressing the Mos transposase under the control of the germline-specific glh-2 promoter induced phenotypic reversion in the absence of heat shock (Table 1, reversion rate = 5 × 10−5 events/F1 offspring), demonstrating that excision can occur without heat shock. Revertant alleles were sequenced and all unc-5 (33/33) and all unc-22 (3/3) reversions were consistent with end-joining of the broken ends (Table 2). Most repaired loci contained short insertions or deletions at the excision site that restored the reading frame of the gene, similar to footprints previously observed (Robert and Bessereau 2007). These results demonstrate that end-joining and other intrachromosomal repair mechanisms are used in the germline to repair Mos1-excision triggered double-strand breaks.
Reversion efficiencies at the unc-5 and unc-22 loci
Transposase | Homozygous background | Wild-type/total footprints | No. of experiments | No.of transgenes | Heterozygous background | Wild-type/total footprints | No. of experiments | No. of transgenes |
|---|---|---|---|---|---|---|---|---|
| unc-5(ox171∷Mos1) | ||||||||
| hsp-16.48∷Tpase (HS+) | 10/40,000 = 2.5 × 10−4 | 2/15 | 16 | 7 | 432/13,000 = 3.3 × 10−2 | 52/52 | 15 | 8 |
| No Tpase (HS+) | 0/12,000 < 8.3 × 10−5 | 7 | 5 | 0/16,500 < 6 × 10−5 | 5 | 5 | ||
| glh-2∷Tpase (HS−) | 3/60,000 = 5 × 10−5 | 6/18 | 3 | 1 | 4/2700 = 1.4 × 10−3 | 4/4 | 1 | 1 |
| vit-2∷Tpase (HS−) | ND | 2 | 2 | 2/8300 = 2.4 × 10−4 | 2/2 | 8 | 3 | |
| unc-22(kr5∷Mos1) | ||||||||
| hsp-16.48∷Tpase (HS+) | 1/4000 = 2.5 × 10−4 | 1/3 | 3 | 1 | 11/700 = 1.6 × 10−2 | 10/10 | 2 | 1 |
| hsp-16.48∷Tpase (HS−) | ND | 0/650 < 1.5 × 10−3 | 1 | 1 |
Transposase | Homozygous background | Wild-type/total footprints | No. of experiments | No.of transgenes | Heterozygous background | Wild-type/total footprints | No. of experiments | No. of transgenes |
|---|---|---|---|---|---|---|---|---|
| unc-5(ox171∷Mos1) | ||||||||
| hsp-16.48∷Tpase (HS+) | 10/40,000 = 2.5 × 10−4 | 2/15 | 16 | 7 | 432/13,000 = 3.3 × 10−2 | 52/52 | 15 | 8 |
| No Tpase (HS+) | 0/12,000 < 8.3 × 10−5 | 7 | 5 | 0/16,500 < 6 × 10−5 | 5 | 5 | ||
| glh-2∷Tpase (HS−) | 3/60,000 = 5 × 10−5 | 6/18 | 3 | 1 | 4/2700 = 1.4 × 10−3 | 4/4 | 1 | 1 |
| vit-2∷Tpase (HS−) | ND | 2 | 2 | 2/8300 = 2.4 × 10−4 | 2/2 | 8 | 3 | |
| unc-22(kr5∷Mos1) | ||||||||
| hsp-16.48∷Tpase (HS+) | 1/4000 = 2.5 × 10−4 | 1/3 | 3 | 1 | 11/700 = 1.6 × 10−2 | 10/10 | 2 | 1 |
| hsp-16.48∷Tpase (HS−) | ND | 0/650 < 1.5 × 10−3 | 1 | 1 |
Reversion experiments were performed in homozygous backgrounds (respectively, ox171∷Mos1/ox171∷Mos1 and kr5∷Mos1/kr5∷Mos1) and in heterozygous backgrounds (respectively, e184sd ox171∷Mos1/idDf3 and kr5∷Mos1/st136∷Tc1). Reversion efficiences were calculated as the ratio between the number of phenotypic revertants and the total number of screened animals. Mos1 excision was triggered by expression of the Mos transposase from different transgenes. hsp-16.48 is a heat-shock-inducible promoter. Reversion assays were performed by heat-shocking parent animals and screening the progeny for revertants as described in Robert and Bessereau (2007). When working with transgenes expressing the Mos transposase under the control of the glh-2 or vit-2 promoters, transgenic L4's were selected and grown at 20° for 5 days before screening their progeny for revertants. “HS+” or “HS−” indicate whether or not parent animals were heat-shocked. “Wild-type/total footprints” indicates the number of wild-type footprints identified among the population analyzed by sequencing. Footprints identified in homozygous backgrounds are listed in Table 2. “No. of experiments” refers to the number of independent experiments. “No. of transgenes” indicates the number of independent trangenes used to express the transposase.
Reversion efficiencies at the unc-5 and unc-22 loci
Transposase | Homozygous background | Wild-type/total footprints | No. of experiments | No.of transgenes | Heterozygous background | Wild-type/total footprints | No. of experiments | No. of transgenes |
|---|---|---|---|---|---|---|---|---|
| unc-5(ox171∷Mos1) | ||||||||
| hsp-16.48∷Tpase (HS+) | 10/40,000 = 2.5 × 10−4 | 2/15 | 16 | 7 | 432/13,000 = 3.3 × 10−2 | 52/52 | 15 | 8 |
| No Tpase (HS+) | 0/12,000 < 8.3 × 10−5 | 7 | 5 | 0/16,500 < 6 × 10−5 | 5 | 5 | ||
| glh-2∷Tpase (HS−) | 3/60,000 = 5 × 10−5 | 6/18 | 3 | 1 | 4/2700 = 1.4 × 10−3 | 4/4 | 1 | 1 |
| vit-2∷Tpase (HS−) | ND | 2 | 2 | 2/8300 = 2.4 × 10−4 | 2/2 | 8 | 3 | |
| unc-22(kr5∷Mos1) | ||||||||
| hsp-16.48∷Tpase (HS+) | 1/4000 = 2.5 × 10−4 | 1/3 | 3 | 1 | 11/700 = 1.6 × 10−2 | 10/10 | 2 | 1 |
| hsp-16.48∷Tpase (HS−) | ND | 0/650 < 1.5 × 10−3 | 1 | 1 |
Transposase | Homozygous background | Wild-type/total footprints | No. of experiments | No.of transgenes | Heterozygous background | Wild-type/total footprints | No. of experiments | No. of transgenes |
|---|---|---|---|---|---|---|---|---|
| unc-5(ox171∷Mos1) | ||||||||
| hsp-16.48∷Tpase (HS+) | 10/40,000 = 2.5 × 10−4 | 2/15 | 16 | 7 | 432/13,000 = 3.3 × 10−2 | 52/52 | 15 | 8 |
| No Tpase (HS+) | 0/12,000 < 8.3 × 10−5 | 7 | 5 | 0/16,500 < 6 × 10−5 | 5 | 5 | ||
| glh-2∷Tpase (HS−) | 3/60,000 = 5 × 10−5 | 6/18 | 3 | 1 | 4/2700 = 1.4 × 10−3 | 4/4 | 1 | 1 |
| vit-2∷Tpase (HS−) | ND | 2 | 2 | 2/8300 = 2.4 × 10−4 | 2/2 | 8 | 3 | |
| unc-22(kr5∷Mos1) | ||||||||
| hsp-16.48∷Tpase (HS+) | 1/4000 = 2.5 × 10−4 | 1/3 | 3 | 1 | 11/700 = 1.6 × 10−2 | 10/10 | 2 | 1 |
| hsp-16.48∷Tpase (HS−) | ND | 0/650 < 1.5 × 10−3 | 1 | 1 |
Reversion experiments were performed in homozygous backgrounds (respectively, ox171∷Mos1/ox171∷Mos1 and kr5∷Mos1/kr5∷Mos1) and in heterozygous backgrounds (respectively, e184sd ox171∷Mos1/idDf3 and kr5∷Mos1/st136∷Tc1). Reversion efficiences were calculated as the ratio between the number of phenotypic revertants and the total number of screened animals. Mos1 excision was triggered by expression of the Mos transposase from different transgenes. hsp-16.48 is a heat-shock-inducible promoter. Reversion assays were performed by heat-shocking parent animals and screening the progeny for revertants as described in Robert and Bessereau (2007). When working with transgenes expressing the Mos transposase under the control of the glh-2 or vit-2 promoters, transgenic L4's were selected and grown at 20° for 5 days before screening their progeny for revertants. “HS+” or “HS−” indicate whether or not parent animals were heat-shocked. “Wild-type/total footprints” indicates the number of wild-type footprints identified among the population analyzed by sequencing. Footprints identified in homozygous backgrounds are listed in Table 2. “No. of experiments” refers to the number of independent experiments. “No. of transgenes” indicates the number of independent trangenes used to express the transposase.
Footprints identified in homozygous unc-5(ox171) and unc-22(kr5) backgrounds
 |
|
Footprints are represented and classified according to the rules defined by Robert and Bessereau (2007). Briefly, class I events are typical end-joining footprints. In subclass Ia footprints, it is possible to recognize the TA dinucleotide duplicated during Mos1 insertion and some Mos1 sequences left during excision. In subclass Ib footprints, at least one copy of the duplicated TA dinucleotide is missing, as expected if processing of the ends of the break occurs before ligation. Class III footprints contain duplication of the genomic sequences adjacent to the broken site which might result from intrachromosomal recombination (see Figure S1 in Robert and Bessereau 2007).
Footprints identified in homozygous unc-5(ox171) and unc-22(kr5) backgrounds
|
|
Footprints are represented and classified according to the rules defined by Robert and Bessereau (2007). Briefly, class I events are typical end-joining footprints. In subclass Ia footprints, it is possible to recognize the TA dinucleotide duplicated during Mos1 insertion and some Mos1 sequences left during excision. In subclass Ib footprints, at least one copy of the duplicated TA dinucleotide is missing, as expected if processing of the ends of the break occurs before ligation. Class III footprints contain duplication of the genomic sequences adjacent to the broken site which might result from intrachromosomal recombination (see Figure S1 in Robert and Bessereau 2007).
Many germline breaks are repaired by homologous recombination:
To measure repair by mechanisms that operate using the homolog as template DNA, we assayed repair after Mos1 excision in heterozygotes. These strains carry the Mos1 insertion allele in trans to a different allele of the same gene (Figure 1A, right). These genes are large and the second allele was at least 2 kb away from the Mos1 insertion site. Repair by homologous recombination would copy wild-type sequences into the broken chromosome and generate an intact copy of the gene. Such events will lead to wild-type animals among the mutant progeny. For excision from unc-5(ox171∷Mos1), we made heterozygotes with idDf3 [previously known as unc-5(ev447)], which deletes the 5′ region of the unc-5 locus and the neighboring locus fem-1 (Figure 1B). For excision from unc-22(kr5∷Mos1), we made heterozygotes with unc-22(st136∷Tc1), a recessive allele of unc-22 containing a Tc1 insertion 4596 nt upstream of the Mos1 insertion site (Figure 1C). These strains also carried a transgene in which the Mos transposase is under the control of the heat-shock promoter. These strains were heat-shocked to activate Mos1 excision in the germline. Wild-type animals were found at high frequency among the uncoordinated progeny of the heat-shocked animals: reversion was observed in 1 of 30 animals at the unc-5 locus [Table 1; 432/13,000 revertants from unc-5(ox171∷Mos)/idDf3 parents, reversion rate = 3.3 × 10−2 events/F1 offspring]. Reversion was observed in 1 of 63 animals from heterozygotes at the unc-22 locus [11/700 revertants from unc-22(kr5∷Mos)/unc-22(st136∷ Tc1), reversion rate = 1.6 × 10−2 events/F1 offspring]. Reversion was dependent on the activation of the Mos transposase since no revertants were observed if the strains were not heat-shocked (Table 1; reversion rate <1.5 × 10−3 events/F1 offspring). In addition, reversion was not caused simply by heat shock since heat-shocked animals that lacked the transposase array did not generate revertants (Table 1; reversion rate <6 × 10−5 events/F1 offspring).
To determine if the reversions were the result of bona fide gene conversion events, the loci from the wild-type progeny were amplified by PCR and sequenced. All unc-5 revertant alleles (52/52, Table 1) and all unc-22 revertant alleles (10/10, Table 1) were repaired to the wild type. Since heat shock was performed in adult hermaphrodites after spermatogenesis was completed, excision and repair events most likely occur in female germ cells only. Because only half of the chromosomes contained a Mos1 insertion, the reversion rate observed at the unc-5 locus suggests that at least 1 chromosome in 15 is broken by Mos1 excision and subsequently repaired by homologous recombination. This rate of reversion is substantially higher than the observed rate of end-joining. However, the number of chromosomes repaired by end-joining was underestimated since we could detect only events that regenerated an in-frame coding sequence. Even after correcting for such “invisible repair events,” gene conversion occurs 100 times more frequently than end-joining. Thus, these data suggest that homologous recombination is the most prominent pathway used in the germline to repair double-strand breaks.
Crossing over caused by double-strand break repair in the germline:
During meiosis, breaks in chromosomes are used as the substrates for crossovers. Is transposon excision able to trigger crossing over? To monitor the formation of crossovers, we performed reversion experiments using a marked template chromosome. The unc-5(ox171∷Mos1)-containing chromosome was marked with the mutation dpy-13(e184sd) and Mos1 excision was induced in heterozygous animals dpy-13(e184sd) unc-5(ox171∷Mos1)/idDf3. The dpy-13 marker was no longer linked in cis to the repaired unc-5 gene in 8.5% (14/164) of the revertant progeny. This represents an increase over the expected frequencies on the basis of both the published dpy-13 unc-5 map distance (1.8 cM) and our own measurements of crossover frequency in this interval (2.6%; data not shown). These recombination events were identified as individual revertants in the progeny of independent P0 animals, suggesting that recombination occurred in meiotic germ cells rather than in mitotically dividing germ cells. We verified that heat shock had no significant effect on the recombination frequency between these two loci (data not shown). We conclude that double-strand breaks triggered by transposon excision serve as substrates for meiotic recombination machinery.
Recombinational repair can potentially be deleterious if recombination is initiated at ectopic homologous sites and causes chromosomal rearrangements. We were previously able to analyze recombination at ectopic sites when establishing MosTIC, a technique developed to engineer the C. elegans genome by homologous recombination (Robert and Bessereau 2007). During MosTIC experiments, ectopic repair templates were carried by extrachromosomal arrays. MosTIC-engineered alleles were obtained with frequencies varying from 10−4 to 7 × 10−4 after excision of Mos1 from the unc-5 locus. These recombination events were ∼100 times less frequent than the ones that we observed in the e184sd ox171/idDf3 background using the homologous chromosome as a repair template (see above). To compare recombination at sites on either the homolog or the extrachromosomal array in the same experiment, we performed MosTIC experiments in an e184sd ox171/idDf3 background. Sixty-three revertant animals among 1114 F1's (reversion rate = 5.6 × 10−2) were identified and none had copied the polymorphisms contained in the repair template (data not shown). These results confirm that most of the recombination events initiated to repair a Mos1 excision-induced double-strand break use the homologous chromosome as a repair template.
Somatic expression of the Mos transposase:
The reversion rates of ∼1 in 50 progeny suggest that Mos1 excision occurs frequently in the germline. These data suggest that transposase expression under the control of the heat-shock promoter must be appreciable in the germline. However, the hsp-16.48 promoter is known to be mainly active in somatic cells (Stringham et al. 1992). Moreover, expression from repetitive extrachromosomal arrays is strongly repressed in the germline (Kelly et al. 1997; Dernburg et al. 2000; Ketting and Plasterk 2000; Robert et al. 2005). Surprisingly, every transgene containing the hsp-16.48∷MosTransposase construct was able to induce Mos1 transposition in the germline (this study and Robert and Bessereau 2007). It is possible that this construct induces transposon excision, but the heat-shock promoter is not being expressed in the germline. The intestine can actively transport molecules to the germ cells, including RNA, such as the double-stranded RNA that triggers RNA interference (for review see Whangbo and Hunter 2008), and proteins, such as yolk components (Kimble and Sharrock 1983; Hall et al. 1999). Strong expression of the transposase in the intestine may provide a source for transposase mRNA or protein in the germline. To test this hypothesis, we expressed the Mos transposase under the control of the vit-2 promoter. vit-2 encodes vitellogenin, a major yolk component specifically synthesized in the intestine of adult hermaphrodites (Grant and Hirsh 1999). To our surprise, expressing the transposase in the intestine was able to stimulate repair of the unc-5 locus: revertants were found at a rate of 1 animal in 4000 progeny (2/8300; reversion rate = 2.4 × 10−4 events/F1 offspring, Table 1). Considering the complex nature of extrachromosomal transgenes in C. elegans, it is not possible to rule out that some transposase might be expressed directly in the germline in spite of the use of an intestine-specific promoter. However, this hypothesis is not likely since repetitive transgenes are known to be strongly silenced in the germline. Hence, we propose that transposase message or protein expressed in the intestine is able to enter the germline to activate Mos1 transposition.
In conclusion, excision of a Mos1 element from a chromosome generates a double-strand break in the germline, which must be repaired to recover a functional gamete. We do not know what fraction of chromosomes experiences an excision; however, we have seen reversion rates as high as 3.3 × 10−2 events/F1 offspring, suggesting that excision may be fairly efficient. The resulting double-strand breaks can be repaired by a variety of repair mechanisms. End-joining generates only a functional product with a frequency of 2.5 × 10−4 events/F1 offspring. The major mechanism for repair appears to be by template-dependent repair from the homolog. However, we cannot observe repair from the sister strand since that regenerates only the mutant gene product. Importantly, the break can also be repaired from a template found on an extrachromosomal array (Robert and Bessereau 2007). Repair from a transgenic array allows targeted gene changes at genes containing Mos1 insertions.
Interestingly, ∼9% of double-strand breaks are resolved as interhomolog crossovers. These data suggest that breaks may be repaired by the machinery for meiotic recombination. A recent study suggests that C. elegans germ cells use RAD-50 and the crossover machinery to repair breaks induced by ionizing radiation during early pachytene stage of meiosis. Then, at the mid-to-late pachytene transition, the cells undergo a developmentally programmed switch to a less constrained repair mode (Hayashi et al. 2007). In addition, end-joining contributes to double-strand break repair during meiosis when homologous chromosomes are unavailable (Smolikov et al. 2007). It is likely that the interhomolog crossover events that we observed might have been generated during meiosis before the mid-to-late pachytene transition zone. The observed end-joining events might be the result of late repair processes, which occur to clean up double-strand breaks from the genome before the end of meiosis.
Footnotes
Communicating editor: A. Villeneuve
Acknowledgement
We thank Andrew Spence for unc-5(idDf3) and Thomas Boulin for unc-22(kr5∷Mos1). E.M.J. received a Poste Orange fellowship from the Institut National de la Santé et de la Recherche Médicale. This work was supported by a European Union Grant (6th Framework program, PL-503334, code NemageneTAG) and an AVENIR grant from the Institut National de la Santé et de la Recherche Médicale.
References
Bessereau, J. L.,
Chen, J. M., D. N. Cooper, N. Chuzhanova, C. Ferec and G. P. Patrinos,
Eide, D., and P. Anderson,
Hedgecock, E. M., J. G. Culotti and D. H. Hall,
Hefferin, M. L., and A. E. Tomkinson,
Jacobson, J. W., M. M. Medhora and D. L. Hartl,
Nassif, N., J. Penney, S. Pal, W. R. Engels and G. B. Gloor,
Sonoda, E., H. Hochegger, A. Saberi, Y. Taniguchi and S. Takeda,
Zwaal, R. R., A. Broeks, J. van Meurs, J. T. Groenen and R. H. Plasterk,

{kind=link}