





Abstract
Silencing provides a critical means of repressing transcription through the assembly and modification of chromatin proteins. The NAD+-dependent deacetylation of histones by the Sir2p family of proteins lends mechanistic insight into how SIR2 contributes to silencing. Here we describe three locus-specific sir2 mutants that have a spectrum of silencing phenotypes in yeast. These mutants are dependent on SIR1 for silencing function at the HM silent mating-type loci, display distinct phenotypes at the rDNA, and have dominant silencing defects at the telomeres. Telomeric silencing is restored if the mutant proteins are directly tethered to subtelomeric regions, via a Gal4p DNA-binding domain (GBD), or are recruited by tethered GBD-Sir1p. These sir2 mutations are found within conserved residues of the SIR2 family and lead to defects in catalytic activity. Since one of the mutations lies outside the previously defined minimal catalytic core, our results show that additional regions of Sir2p can be important for enzymatic activity and that differences in levels of activity may have distinct effects at the silenced loci.
SILENCING is a process by which transcriptional repression occurs in a regional, promoter-nonspecific manner. Chemical modifications to DNA, histones, and other nuclear proteins can lead to specific alterations in chromatin structure that may disrupt or promote transcriptional silencing (reviewed in Wolffe and Guschin 2000; Rice and Allis 2001). In Saccharomyces cerevisiae at least three loci are subject to silencing: telomeres, the silent mating-type loci (HMR and HML, the HM loci), and the ribosomal DNA (rDNA) repeats. Although many cis- and trans-acting factors have been implicated in silencing in yeast (reviewed in Gartenberg 2000), few factors appear required at all three loci; among these is the silent information regulator, Sir2p.
The Sir2 protein is an NAD+-dependent protein deacetylase that is conserved across all kingdoms of life (reviewed in Gottschling 2000; Guarente 2000; Shore 2000; Moazed 2001). The Sir2 family of proteins is the first example of enzymes that couple NADase and deacetylase activities. This unique activity may be what renders cells with aberrant levels of Sir2p defective in a wide range of cellular processes such as silencing, chromosome segregation, DNA repair and recombination, cell cycle checkpoints, and senescence (reviewed in Shore 2000). Sir2p is found in at least two complexes, the regulator of nucleolar silencing and telophase exit (RENT) complex and the Sir2/4 complex, which appear, respectively, to act within the rDNA array or at telomeres (Shou et al. 1999; Straight et al. 1999; Ghidelli et al. 2001; Hoppe et al. 2002). The Sir2, Sir3, and Sir4 proteins also mediate silencing at the HM loci with the aid of the targeting protein Sir1p.
In contrast to the variegated silencing states observed for PolII transcribed genes at the telomeres and within the rDNA array, the HM loci are distinct in that they normally appear completely repressed. This difference may be due in part to the interactions between Sir1p and Orc1p and to their participation in silencing at the HM loci (Pillus and Rine 1989; Triolo and Sternglanz 1996; Fox et al. 1997; Gardner et al. 1999). One puzzle about sir1Δ mutant phenotypes has been that a subpopulation of cells maintains normal silencing. This suggests that other SIR1-independent mechanisms are involved in the establishment of silencing at HML and HMR. To gain insight into these mechanisms, a genetic screen to isolate enhancers of the sir1Δ mating defect was performed, yielding several alleles of genes known to function in silencing, including SIR2, SIR3, and SIR4 (Reifsnyder et al. 1996; Stone et al. 2000). The work presented here describes the characterization of the sir2eso (enhancers of sir-one) mutants.
In previous studies, several sir2 alleles were demonstrated to have a wide range of silencing defects that could be locus specific, dominant, and in some cases correlated with decreases in enzymatic activity (Sherman et al. 1999; Tanny et al. 1999; Cuperus et al. 2000; Imai et al. 2000; Perrod et al. 2001; Armstrong et al. 2002; Hoppe et al. 2002). In this study, we have characterized a unique class of conditional SIR2 alleles that can silence the HM loci only in the presence of Sir1p. Although two of the mutants contain mutations within the conserved catalytic core domain, all are dominantly defective in silencing at the telomeres, but can function if targeted to the locus as Gal4p DNA-binding domain (GBD) fusion proteins or via GBD-Sir1p. We demonstrate that robust enzymatic activity is not necessary for silencing telomeric and rDNA reporter genes and propose that wild-type levels of activity may be required for initiating stable silencing at the telomeres but not within the rDNA array.
MATERIALS AND METHODS
Yeast methods and strains: Yeast strains and plasmids are listed in Table 1 or described below. Strains were grown at 30° and standard manipulations were performed as described (Rose et al. 1989). Yeast extract/peptone/dextrose (YPD), synthetic selective media, and minimal media were prepared as described (Sherman 1991).
Plasmids: The pRS313 (pLP60) and pRS315 (pLP62) vectors were used for subcloning (Sikorski and Hieter 1989). Inserting a 2.7-kb BstNI genomic fragment of SIR2 into the SmaI site of pLP60 created the plasmids pLP284 and pLP285. pLP1102, pLP1110, and pLP1112 are described below in Gap repair and sequencing. Inserting an EagI-SalI fragment of pLP285 into pLP62 created the plasmid pLP1237. The plasmids pLP1187, pLP1188, and pLP1189 result from inserting PvuII fragments of the corresponding gap-repaired plasmids pLP1102, pLP1110, and pLP1112, respectively, into pLP62. The pJR1061/pKL5 (GAL4(1-147)-SIR1) vector is also known as pLP114 (Chien et al. 1993). The plasmid pLP118 is vector pYSR102 containing SIR1. The plasmid pLP762 is a 6.6-kb SacII-EcoRI genomic fragment of SIR4 inserted into pRS424 (Sikorski and Hieter 1989). The pGBD-C1 vector (James et al. 1996) is also known as pLP956. The plasmid pLP1073 contains the SIR2 core domain (BclI-NruI sites) cloned into the SmaI site of pLP956. The plasmid pLP1074 contains the ClaI fragment of SIR2 from pLP285 inserted into pGBD-C3 (James et al. 1996), also known as pLP958. The plasmids pLP1369, pLP1370, and pLP1371 were created by inserting a SacI-NcoI fragment from pLP1102, pLP1110, and pLP1112, respectively, into pLP1074. The pGex-4T-1 is also known as pLP1334. The plasmid pDM111a contains wild-type SIR2 inserted into pGex-4T-1 (Straight et al. 1999) and is also known as pLP1275. The plasmids pLP1335, pLP1336, and pLP1337 contain a SacI-NcoI fragment from pLP1102, pLP1110, and pLP1112, respectively, inserted into pLP1275. Mutations in pLP1187, pLP1369, and pLP1335 were confirmed by sequence analysis using primer no. 56 listed below. Mutations in pLP1188, pLP1189, pLP1370, pLP1371, pLP1336, and pLP1337 were confirmed by sequence analysis using primer no. 164 listed below.
The eso screen: Details of the mutagenesis are described elsewhere (Reifsnyder et al. 1996; Stone et al. 2000). Thirty-nine independent mutants were isolated on the basis of their ability to mate in the presence of a SIR1 plasmid but not in its absence. Several of the eso mutants contained mutations in SIR2 as assessed by standard linkage analysis and complementation tests. Five alleles of sir2 were isolated from this screen and were cloned by gap repair as described below. The original sir2eso allele designations prior to gap repair were: LPY655, 6.2k; LPY667, G16a-; LPY727, J9a; LPY733, M5b-; and LPY1418, Gi.
Gap repair and sequencing: To identify the mutations within SIR2 we performed gap repair analysis (Rothstein 1991). The SIR2 wild-type gene on a plasmid was digested in three different ways: Gap1 (G1), pLP284 with ClaI and EcoNI; Gap2 (G2), pLP285 with NcoI and NruI; Gap3 (G3), pLP285 with NruI and BlpI. The mutation within the open reading frame (ORF) of the sir2eso LPY655 was cloned using gaps G1 (pLP1101) and G3 (pLP1102). The mutations in LPY667, LPY733, and LPY1418 were cloned with G2, pLP1112, pLP1111, and pLP1110, respectively. The mutation in LPY727 was cloned with G3 (pLP1137). Plasmids bearing the lesions that resulted in the original eso phenotype were isolated and the entire open reading frames of the gap-repaired plasmids were sequenced. In the course of this sequencing, we identified several polymorphisms between the GenBank sequence data for the S288C background and the W303 background of these studies. Four silent changes were shown with numbering relative to the ORF start: ACA-ACG at nucleotide (nt) 339; CCA-CCG at nt 624; UUG-UUA at nt 774; and CCT-CCC at nt 1404. Four polymorphisms resulted in amino acid substitutions. These included P65T (CCA-ACA, nt 458), K320N (AAC-AAA, nt 1224), M424V (GTG-ATG, nt 1534), and T527A (ACG-GCG, nt 1848). The number of polymorphisms observed from comparative analyses is within the range predicted between these two strain backgrounds (Primig et al. 2000). The oligonucleotides used for sequencing were:
No. 55 5′-ATCGCTTCGGTAGACAC-3′
No. 56 5′-AACGTCTTGGGGATCAT-3′
No. 87 5′-AACGTCTTGGGGATCAT-3′
No. 88 5′-GAAGGAACCAAGCTTACGATTTC-3′
No. 163 5′-TCCTTAACTCATATGGCG-3′
No. 164 5′-TGAAACTATGCAATGGAG-3′.
Mutations were identified by comparison to the wild-type SIR2 sequence reported in the S. cerevisiae Genome Database (http://genome-www.stanford.edu/Saccharomyces/).
Qualitative and quantitative mating assays: For qualitative mating assays, cells were patched onto YPD plates for 12-18 hr and then replica plated to YPD to assay for growth and onto a lawn of cells of the opposite mating type, LPY78 or LPY142, on minimal medium to assay for successful diploid formation. The sir2eso mating efficiencies were determined by performing quantitative assays as described (Stone et al. 2000). Briefly, sir2Δ (LPY1557 and LPY4627), sir1Δ sir2Δ (LPY3439 and LPY3440), wild-type (LPY5 and LPY79), and sir1Δ (LPY6 and LPY80) strains were transformed with vector control, pLP60; wild-type SIR2, pLP285; sir2-R139K, pLP1102; sir2-G270E, pLP1110; or sir2-F296L, pLP1112 (see supplemental data for transformed yeast strain numbers at http://www.genetics.org/supplemental/). Cells were grown to mid-logarithmic phase under selection, serially diluted, and plated onto plates lacking histidine, to assay for growth, or mixed with tester cells of the opposite mating type and plated onto minimal plates, to evaluate mating. Colonies were counted and mating efficiencies were determined by dividing the number of colonies on minimal plates by the number of colonies on selective plates. Mating efficiencies were calculated by averaging three independent experiments done in duplicate. Standard deviations were determined using preset options on Microsoft Excel.
Telomeric and rDNA silencing assays: Telomeric and rDNA silencing assays were performed as described previously (Gottschling et al. 1990; Smith and Boeke 1997). Silencing of a URA3 reporter gene placed proximal to telomere VII was assayed as described (Gottschling et al. 1990), with the modification that the strains tested were grown in liquid medium for 4 days at 30° instead of on solid medium prior to testing. For telomeric silencing assays, LPY1953 and LPY1954 were transformed with pLP62, pLP1237, pLP1187, pLP1188, and pLP1189. For telomeric SIR1-tethering silencing assays, LPY4624 and LPY1030 were cotransformed with pLP114 and the same set of plasmids listed above (pLP62-pLP1189). For rDNA silencing assays, LPY2446 and LPY2447 were transformed with the same set of plasmids listed above (pLP62-pLP1189). All transformed strains were grown for 4 days at 30°, serially diluted fivefold, and plated onto selective plates to assay growth. Diluted cells were also plated onto the following silencing test plates: plates lacking leucine supplemented with 0.1% 5-fluoroorotic (5-FOA), plates lacking leucine and histidine containing 0.1% 5-FOA for telomeric silencing assays, and plates lacking uracil for assaying rDNA silencing.
Yeast strains used in this study
| Straina | Genotype | Source |
|---|---|---|
| LPY5 | W303-1a MATa ade2-1 can1-100 his3-11,15 leu2,3,112 trp1-1 ura3-1 | R. Rothstein |
| LPY6 | AMR27 W303-1a sir1Δ::LEU2 | Stone et al. (1991) |
| LPY11 | W303-1a sir2::HIS3 | |
| LPY12 | AMR50 W303-1a sir1::URA3 | R. Sternglanz |
| LPY78 | MATα his4 | P. Schatz |
| LPY79 | W303-1b MATα ade2-1 can1-100 his3-11 leu2,3,112 trp1-1 ura3-1 | R. Rothstein |
| LPY80 | W303-1b sir1Δ::LEU2 | |
| LPY142 | MATa his4 | P. Schatz |
| LPY655 | 6.2k W303-1b sir1Δ::LEU2 sir2eso-R139K | |
| LPY667 | G16a- W303-1a sir1Δ::LEU2 sir2esoF296L | |
| LPY733 | M5b- W303-1a sir1Δ::LEU2 sir2esoG270E | |
| LPY1029 | YDS631-W303-1b adh4::URA3 (C1-3)n | Chien et al. (1993) |
| LPY1030 | YDS634-W303-1b adh4::URA3-1XUAS (C1-3)n | Chien et al. (1993) |
| LPY1398 | W303-1b sir2::HIS3 + pLP37 | |
| LPY1418 | JRY3003-3i W303-1a sir1Δ::LEU2 sir2esoG270E | |
| LPY1557 | W303-1a sir2Δ::TRP1 | |
| LPY1953 | YCB652 MATa his3Δ200 leu2Δ1 lys2Δ202 trp1Δ63 ura3-52 sir2Δ2::TRP1 ADH4::URA3-TEL | Brachmann et al. (1995) |
| LPY1954 | YCB647 MATa his3Δ200 leu2Δ1 lys2Δ202 trp1Δ63 ura3-52 ADH4::URA3-TEL | Brachmann et al. (1995) |
| LPY2446 | JS128 (S6) MATα his3Δ200 leu2Δ1 ura3-52 RDN:: Ty1 -mURA3 | Smith and Boeke (1997) |
| LPY2447 | JS163 (S6) sir2Δ2::HIS3 RDN:: Ty1 -mURA3 | Smith and Boeke (1997) |
| LPY3439 | W303-1a sir1Δ::LEU2 sir2Δ::TRP1 | LPY1557 × LPY80 |
| LPY3440 | W303-1b sir1Δ::LEU2 sir2Δ::TRP1 | LPY1557 × LPY80 |
| LPY3712 | W303-1a sir2esoG270E | LPY1418 × LPY79 |
| LPY4595 | W303-1a sir2Δ::TRP1 (LPY1557) + pLP285 | |
| LPY4621 | WY53-W303-1b net1::Myc9-Net1/LEU2 | Straight et al. (1999) |
| LPY4624 | W303-1a sir2::TRP1 adh4::URA3-1XUASGBD (C1-3A)n | LPY3439 × LPY1030 |
| LPY4627 | W303-1b sir2Δ::TRP1 | LPY3439 × LPY1030 |
| LPY4724 | W303-1a sir2::HIS3 net1::Myc9-Net1/LEU2 | LPY4621 × LPY1398 |
| LPY5378 | GCY62-W303-1b sir2::KanMX4 RDN1::4XUASg-mURA3-HIS3 | Cuperus et al. (2000) |
| LPY5611 | W303-1a sir2::HIS3 adh4::URA3-1xUASGBD(C1-3A)n | LPY11 × LPY1030 |
| LPY5615 | W303-1b sir2::TRP1 net1::Myc9-Net1/LEU2 | LPY4595 × LPY4724 |
| LPY6400 | W303-1b sir1::URA3 sir2::TRP1 net1::Myc9-Net1/LEU2 | LPY5615 × LPY12 |
| Straina | Genotype | Source |
|---|---|---|
| LPY5 | W303-1a MATa ade2-1 can1-100 his3-11,15 leu2,3,112 trp1-1 ura3-1 | R. Rothstein |
| LPY6 | AMR27 W303-1a sir1Δ::LEU2 | Stone et al. (1991) |
| LPY11 | W303-1a sir2::HIS3 | |
| LPY12 | AMR50 W303-1a sir1::URA3 | R. Sternglanz |
| LPY78 | MATα his4 | P. Schatz |
| LPY79 | W303-1b MATα ade2-1 can1-100 his3-11 leu2,3,112 trp1-1 ura3-1 | R. Rothstein |
| LPY80 | W303-1b sir1Δ::LEU2 | |
| LPY142 | MATa his4 | P. Schatz |
| LPY655 | 6.2k W303-1b sir1Δ::LEU2 sir2eso-R139K | |
| LPY667 | G16a- W303-1a sir1Δ::LEU2 sir2esoF296L | |
| LPY733 | M5b- W303-1a sir1Δ::LEU2 sir2esoG270E | |
| LPY1029 | YDS631-W303-1b adh4::URA3 (C1-3)n | Chien et al. (1993) |
| LPY1030 | YDS634-W303-1b adh4::URA3-1XUAS (C1-3)n | Chien et al. (1993) |
| LPY1398 | W303-1b sir2::HIS3 + pLP37 | |
| LPY1418 | JRY3003-3i W303-1a sir1Δ::LEU2 sir2esoG270E | |
| LPY1557 | W303-1a sir2Δ::TRP1 | |
| LPY1953 | YCB652 MATa his3Δ200 leu2Δ1 lys2Δ202 trp1Δ63 ura3-52 sir2Δ2::TRP1 ADH4::URA3-TEL | Brachmann et al. (1995) |
| LPY1954 | YCB647 MATa his3Δ200 leu2Δ1 lys2Δ202 trp1Δ63 ura3-52 ADH4::URA3-TEL | Brachmann et al. (1995) |
| LPY2446 | JS128 (S6) MATα his3Δ200 leu2Δ1 ura3-52 RDN:: Ty1 -mURA3 | Smith and Boeke (1997) |
| LPY2447 | JS163 (S6) sir2Δ2::HIS3 RDN:: Ty1 -mURA3 | Smith and Boeke (1997) |
| LPY3439 | W303-1a sir1Δ::LEU2 sir2Δ::TRP1 | LPY1557 × LPY80 |
| LPY3440 | W303-1b sir1Δ::LEU2 sir2Δ::TRP1 | LPY1557 × LPY80 |
| LPY3712 | W303-1a sir2esoG270E | LPY1418 × LPY79 |
| LPY4595 | W303-1a sir2Δ::TRP1 (LPY1557) + pLP285 | |
| LPY4621 | WY53-W303-1b net1::Myc9-Net1/LEU2 | Straight et al. (1999) |
| LPY4624 | W303-1a sir2::TRP1 adh4::URA3-1XUASGBD (C1-3A)n | LPY3439 × LPY1030 |
| LPY4627 | W303-1b sir2Δ::TRP1 | LPY3439 × LPY1030 |
| LPY4724 | W303-1a sir2::HIS3 net1::Myc9-Net1/LEU2 | LPY4621 × LPY1398 |
| LPY5378 | GCY62-W303-1b sir2::KanMX4 RDN1::4XUASg-mURA3-HIS3 | Cuperus et al. (2000) |
| LPY5611 | W303-1a sir2::HIS3 adh4::URA3-1xUASGBD(C1-3A)n | LPY11 × LPY1030 |
| LPY5615 | W303-1b sir2::TRP1 net1::Myc9-Net1/LEU2 | LPY4595 × LPY4724 |
| LPY6400 | W303-1b sir1::URA3 sir2::TRP1 net1::Myc9-Net1/LEU2 | LPY5615 × LPY12 |
Except where indicated, strains used were constructed in the course of these experiments or are part of the lab collection. The LPY numbers for transformed strains used in this study can be found in the supplemental data at http://www.genetics.org/supplemental/.
Yeast strains used in this study
| Straina | Genotype | Source |
|---|---|---|
| LPY5 | W303-1a MATa ade2-1 can1-100 his3-11,15 leu2,3,112 trp1-1 ura3-1 | R. Rothstein |
| LPY6 | AMR27 W303-1a sir1Δ::LEU2 | Stone et al. (1991) |
| LPY11 | W303-1a sir2::HIS3 | |
| LPY12 | AMR50 W303-1a sir1::URA3 | R. Sternglanz |
| LPY78 | MATα his4 | P. Schatz |
| LPY79 | W303-1b MATα ade2-1 can1-100 his3-11 leu2,3,112 trp1-1 ura3-1 | R. Rothstein |
| LPY80 | W303-1b sir1Δ::LEU2 | |
| LPY142 | MATa his4 | P. Schatz |
| LPY655 | 6.2k W303-1b sir1Δ::LEU2 sir2eso-R139K | |
| LPY667 | G16a- W303-1a sir1Δ::LEU2 sir2esoF296L | |
| LPY733 | M5b- W303-1a sir1Δ::LEU2 sir2esoG270E | |
| LPY1029 | YDS631-W303-1b adh4::URA3 (C1-3)n | Chien et al. (1993) |
| LPY1030 | YDS634-W303-1b adh4::URA3-1XUAS (C1-3)n | Chien et al. (1993) |
| LPY1398 | W303-1b sir2::HIS3 + pLP37 | |
| LPY1418 | JRY3003-3i W303-1a sir1Δ::LEU2 sir2esoG270E | |
| LPY1557 | W303-1a sir2Δ::TRP1 | |
| LPY1953 | YCB652 MATa his3Δ200 leu2Δ1 lys2Δ202 trp1Δ63 ura3-52 sir2Δ2::TRP1 ADH4::URA3-TEL | Brachmann et al. (1995) |
| LPY1954 | YCB647 MATa his3Δ200 leu2Δ1 lys2Δ202 trp1Δ63 ura3-52 ADH4::URA3-TEL | Brachmann et al. (1995) |
| LPY2446 | JS128 (S6) MATα his3Δ200 leu2Δ1 ura3-52 RDN:: Ty1 -mURA3 | Smith and Boeke (1997) |
| LPY2447 | JS163 (S6) sir2Δ2::HIS3 RDN:: Ty1 -mURA3 | Smith and Boeke (1997) |
| LPY3439 | W303-1a sir1Δ::LEU2 sir2Δ::TRP1 | LPY1557 × LPY80 |
| LPY3440 | W303-1b sir1Δ::LEU2 sir2Δ::TRP1 | LPY1557 × LPY80 |
| LPY3712 | W303-1a sir2esoG270E | LPY1418 × LPY79 |
| LPY4595 | W303-1a sir2Δ::TRP1 (LPY1557) + pLP285 | |
| LPY4621 | WY53-W303-1b net1::Myc9-Net1/LEU2 | Straight et al. (1999) |
| LPY4624 | W303-1a sir2::TRP1 adh4::URA3-1XUASGBD (C1-3A)n | LPY3439 × LPY1030 |
| LPY4627 | W303-1b sir2Δ::TRP1 | LPY3439 × LPY1030 |
| LPY4724 | W303-1a sir2::HIS3 net1::Myc9-Net1/LEU2 | LPY4621 × LPY1398 |
| LPY5378 | GCY62-W303-1b sir2::KanMX4 RDN1::4XUASg-mURA3-HIS3 | Cuperus et al. (2000) |
| LPY5611 | W303-1a sir2::HIS3 adh4::URA3-1xUASGBD(C1-3A)n | LPY11 × LPY1030 |
| LPY5615 | W303-1b sir2::TRP1 net1::Myc9-Net1/LEU2 | LPY4595 × LPY4724 |
| LPY6400 | W303-1b sir1::URA3 sir2::TRP1 net1::Myc9-Net1/LEU2 | LPY5615 × LPY12 |
| Straina | Genotype | Source |
|---|---|---|
| LPY5 | W303-1a MATa ade2-1 can1-100 his3-11,15 leu2,3,112 trp1-1 ura3-1 | R. Rothstein |
| LPY6 | AMR27 W303-1a sir1Δ::LEU2 | Stone et al. (1991) |
| LPY11 | W303-1a sir2::HIS3 | |
| LPY12 | AMR50 W303-1a sir1::URA3 | R. Sternglanz |
| LPY78 | MATα his4 | P. Schatz |
| LPY79 | W303-1b MATα ade2-1 can1-100 his3-11 leu2,3,112 trp1-1 ura3-1 | R. Rothstein |
| LPY80 | W303-1b sir1Δ::LEU2 | |
| LPY142 | MATa his4 | P. Schatz |
| LPY655 | 6.2k W303-1b sir1Δ::LEU2 sir2eso-R139K | |
| LPY667 | G16a- W303-1a sir1Δ::LEU2 sir2esoF296L | |
| LPY733 | M5b- W303-1a sir1Δ::LEU2 sir2esoG270E | |
| LPY1029 | YDS631-W303-1b adh4::URA3 (C1-3)n | Chien et al. (1993) |
| LPY1030 | YDS634-W303-1b adh4::URA3-1XUAS (C1-3)n | Chien et al. (1993) |
| LPY1398 | W303-1b sir2::HIS3 + pLP37 | |
| LPY1418 | JRY3003-3i W303-1a sir1Δ::LEU2 sir2esoG270E | |
| LPY1557 | W303-1a sir2Δ::TRP1 | |
| LPY1953 | YCB652 MATa his3Δ200 leu2Δ1 lys2Δ202 trp1Δ63 ura3-52 sir2Δ2::TRP1 ADH4::URA3-TEL | Brachmann et al. (1995) |
| LPY1954 | YCB647 MATa his3Δ200 leu2Δ1 lys2Δ202 trp1Δ63 ura3-52 ADH4::URA3-TEL | Brachmann et al. (1995) |
| LPY2446 | JS128 (S6) MATα his3Δ200 leu2Δ1 ura3-52 RDN:: Ty1 -mURA3 | Smith and Boeke (1997) |
| LPY2447 | JS163 (S6) sir2Δ2::HIS3 RDN:: Ty1 -mURA3 | Smith and Boeke (1997) |
| LPY3439 | W303-1a sir1Δ::LEU2 sir2Δ::TRP1 | LPY1557 × LPY80 |
| LPY3440 | W303-1b sir1Δ::LEU2 sir2Δ::TRP1 | LPY1557 × LPY80 |
| LPY3712 | W303-1a sir2esoG270E | LPY1418 × LPY79 |
| LPY4595 | W303-1a sir2Δ::TRP1 (LPY1557) + pLP285 | |
| LPY4621 | WY53-W303-1b net1::Myc9-Net1/LEU2 | Straight et al. (1999) |
| LPY4624 | W303-1a sir2::TRP1 adh4::URA3-1XUASGBD (C1-3A)n | LPY3439 × LPY1030 |
| LPY4627 | W303-1b sir2Δ::TRP1 | LPY3439 × LPY1030 |
| LPY4724 | W303-1a sir2::HIS3 net1::Myc9-Net1/LEU2 | LPY4621 × LPY1398 |
| LPY5378 | GCY62-W303-1b sir2::KanMX4 RDN1::4XUASg-mURA3-HIS3 | Cuperus et al. (2000) |
| LPY5611 | W303-1a sir2::HIS3 adh4::URA3-1xUASGBD(C1-3A)n | LPY11 × LPY1030 |
| LPY5615 | W303-1b sir2::TRP1 net1::Myc9-Net1/LEU2 | LPY4595 × LPY4724 |
| LPY6400 | W303-1b sir1::URA3 sir2::TRP1 net1::Myc9-Net1/LEU2 | LPY5615 × LPY12 |
Except where indicated, strains used were constructed in the course of these experiments or are part of the lab collection. The LPY numbers for transformed strains used in this study can be found in the supplemental data at http://www.genetics.org/supplemental/.
Targeting assays: Yeast strains LPY1030, LPY5611, and LPY5378 were transformed with pLP956, pLP1073, pLP1074, pLP1369, pLP1370, and pLP1371 and assayed for telomeric and rDNA silencing as described above. All transformed strains were grown for 4 days at 30° and serially diluted fivefold on plates lacking tryptophan to assay growth and onto test plates lacking tryptophan and containing 0.1% 5-FOA. To be certain that the mutants expressed at high-copy (2μ) levels retained their eso phenotype, the strains were tested for mating ability in a sir1Δ sir2Δ and SIR1 sir2Δ background. As expected, they were able to mate only in the presence of Sir1p (data not shown).
Immunoblot and immunofluorescence analyses: Levels of Sir2p and mutant sir2eso proteins were detected by immunoblot analysis as described (Stone and Pillus 1996). Cell extracts from ∼2.0 × 107 cells were loaded per well. Sir2p was detected using a 1:1000 dilution of the polyclonal antiserum (2916/8) raised to a C-terminal peptide of Sir2p (Smith et al. 1998). Goat anti-rabbit secondary antibody was used at 1:10,000 and Western blots were developed using a standard alkaline phosphatase detection system (Promega, Madison, WI).
Centromeric plasmids bearing the sir2eso mutations and wild-type SIR2 were transformed into sir2Δ (LPY1557) and sir1Δ sir2Δ (LPY3439) strains. Immunofluorescence using affinity-purified antibody raised to the C terminus of Sir2p (2916/8) was performed as described (Stone and Pillus 1996; Gotta et al. 1997; Ersfeld 1999). The other antibody used was anti-Nop1p (D77; Aris and Blobel 1988). Fluorescein-conjugated anti-rabbit and Texas-red-conjugated anti-mouse secondary antibodies (Jackson Immunoresearch, West Grove, PA) were preadsorbed against spheroplasted mutant and wild-type yeast cells. Microscopy was performed on an Applied Precision optical sectioning microscope to collect images spaced at 0.2-μm increments. The images were deconvolved using the Delta Vision deconvolution software as previously described (Pogliano et al. 1999).
NAD+ hydrolysis assays: Glutathione S-transferase (GST; pLP1334), GST-Sir2p (pLP1275), GST-sir2-R139Kp (pLP1335), GST-sir2-G270Ep (pLP1336), and GST-sir2-F296Lp (pLP1337) fusion proteins were expressed in Escherichia coli BL21 (DE3) during a 4- to 5-hr induction with 0.5 mm isopropyl β-d-thiogalactoside at room temperature. Proteins were purified on glutathione Sepharose beads as directed by the manufacturer (Pharmacia, Piscataway, NJ). Purified proteins were dialyzed against 50 mm sodium phosphate (pH 7.2) and stored at 4° in 50 mm sodium phosphate (pH 7.2), 0.5 mm dithiothreitol (DTT), and 10% glycerol (Landry et al. 2000b). Protein concentration was deduced from extinction coefficient measurements as described (Gill and von Hippel 1989) and by SDS-PAGE, comparing Coomassie brilliant blue staining of purified GST-protein samples and various concentrations of the BSA protein standard. NAD+-hydrolysis assays to measure histone deacetylation were performed as described (Landry et al. 2000a). Reactions were carried out in 1 ml with 50 mm glycine (pH 9.0), 0.5 mm DTT, 5 mm tetrasodium pyrophosphate (NaP2O7), 0.1 mg/ml BSA, 1 mg calf thymus histones (Sigma, St. Louis), 2 μCi [4-3H]NAD+ (Amersham TRA298; 4.3 Ci/ mmol, 1 mCi/ml), and 3.7 μg of purified proteins. The reactions were performed in triplicate and incubated at 30°. After 10 min and 3, 7, 24, and 33 hr, 185 μl of the total reaction was transferred to tubes containing 135 μl 0.5 m boric acid (pH 8.0) to quench the reaction. A total of 1 ml of ethyl acetate was added and vortexed for 5 min and 700 μl of the ethyl acetate phase was transferred to 3 ml Ecoscint fluid (National Diagnostics, Atlanta) and analyzed by scintillation counting. Radioactivity released from Sir2p wild-type control reactions lacking histones was subtracted. The slope, or rate of change, for activities through five time points was calculated and shown as percentage of wild-type Sir2p activity with standard deviations indicated.
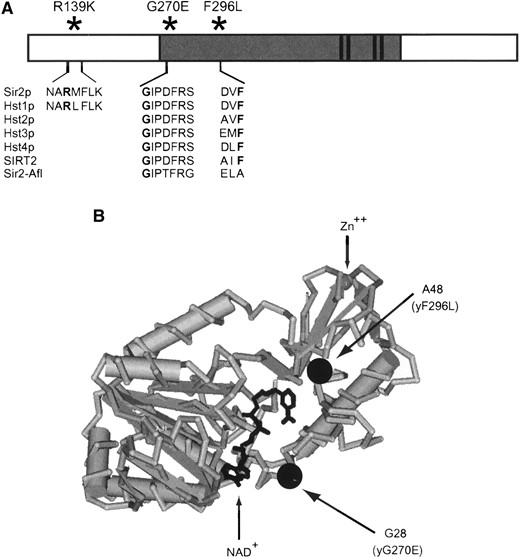
—The sir2eso mutations result in substitutions of highly conserved amino acids, including a subset within the catalytic core domain of Sir2p. (A) The sir2eso alleles were cloned onto plasmids using gap repair (pLP1102-R139K, pLP1110-G270E, and pLP1112-F296L). Sequence analysis of each sir2eso ORF revealed mutations altering amino acids that lie within conserved residues of the Sir2p family. Two mutations (G270E and F296L) changed amino acids within the highly conserved core of SIR2 (dark shading), a domain essential for silencing function. (B) The crystal structure of A. fulgidus-Sir2 with sir2eso amino acid changes mapped using Cn3D, the National Center for Biotechnology Information Entrez structure retrieval and analysis program (http://www.ncbi.nlm.nih.gov/Structure/CN3D/cn3d.shtml). The R139K substitution is not shown since the Sir2-Afl enzyme lacks the N-terminal domain of ySir2p.
Immunoprecipitation reactions: A total of 25 ml of LPY5615 and LPY6400 transformed with pLP60, pLP285, pLP1102, pLP1110, and pLP1112 was grown in medium lacking histidine until it reached an A600 of 0.7-0.8. The cells were harvested and lysed as described (Straight et al. 1999). Three microliters of α-Sir2p polyclonal antiserum (above) or 4 μl of α-Sir4p polyclonal antiserum (7795, raised against a β-gal-Sir4 fusion protein) was used and incubated at 4° for 3-4 hr. One hundred microliters of 10% (w/v) protein-A Sepharose (Pharmacia) in lysis buffer was added and mixed at 4° for 1 hr. Immune complexes were harvested and washed as described (Straight et al. 1999) and resuspended in 50 μl of 2.5× SDS sample buffer. Ten microliters of sample was loaded on 12 cm 9% SDS polyacrylamide. Immunoblots were probed with a 1:20 dilution of α-myc (9E10) hybridoma supernatant, a 1:1000 dilution of α-Sir2p polyclonal antiserum (2916/8), or a 1:1000 dilution of α-Sir4p polyclonal antiserum raised against a Sir4p C-terminal peptide (2913/8; Palladino et al. 1993). Secondary antibodies, horseradish peroxidase-coupled anti-rabbit (for Sir2p and Sir4p) and anti-mouse (for 9E10; Promega) were used at 1:10,000 and detected using the enhanced chemiluminescence reagents (Pharmacia).
RESULTS
Identification of the sir2eso mutations and characterization at the HM loci: Five sir2 mutants were isolated from the eso mutant screen described previously (Reifsnyder et al. 1996; Stone et al. 2000). The sir2eso mutations were cloned onto centromeric (CEN) plasmids using standard gap repair (Rothstein 1991). The repaired plasmids were tested for their ability to confer mating in a SIR1 sir2Δ strain but not in a sir1Δ sir2Δ strain to confirm their eso phenotype. The mutations contained in the five sir2eso alleles were identified by sequence analysis and three are shown in Figure 1A. Two of the strains (LPY655 and LPY667) had mutations affecting amino acids that are highly conserved among Sir2p family members at positions R139K and F296L, respectively. Although the eso mutants were isolated from independently mutagenized cultures, two additional strains (LPY1418 and LPY733) contained an identical mutation changing a highly conserved glycine to a glutamic acid, G270E. Characterization of one strain (LPY1418) was extended as representative of both of these mutants.
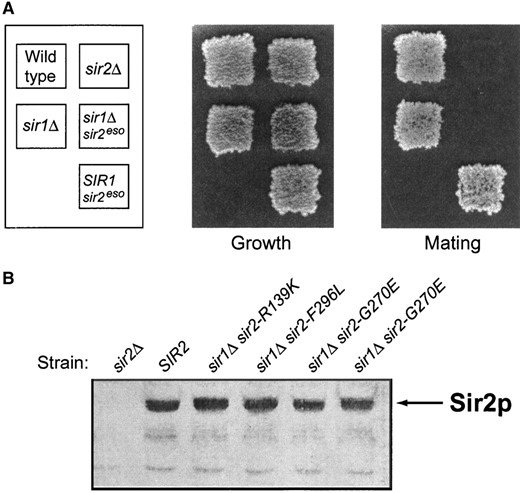
—The sir2eso mutants enhance the sir1Δ defect and the mutant proteins are expressed at wild-type levels. (A) The sir2eso mutants are completely mating defective only in sir1Δ cells. MATa strains were patched and replica plated onto YPD plates (growth control) and onto minimal plates top spread with cells of the opposite mating type. The sir2-G270E mutant, in the absence of Sir1p and in the presence of Sir1p, is shown. The strains used are wild-type, LPY5; sir1Δ, LPY6; sir2Δ, LPY11; sir1Δ sir2-G270E, LPY1418; and sir2-G270E, LPY3712. (B) Immunoblot analysis of sir2eso mutant whole-cell extracts using anti-Sir2p antisera (2916/8). The sir2eso mutant proteins are expressed from their chromosomal locus in a sir1Δ background. The three sir2eso alleles characterized in this article are expressed at levels comparable to wild type in both the SIR1 wild-type and sir1Δ backgrounds (the sir1Δ background is shown). The strains from left to right are sir2Δ (LPY11), wild type (LPY5), sir1Δ sir2-R139K (LPY655), sir1Δ sir2-F296L (LPY667), sir1Δ sir2-G270E (LPY1418), and sir1Δ sir2-G270E (LPY733).
Another mutant isolated from the screen, LPY727, contained a nonsense mutation at amino acid 15. The mutant protein was presumed to be translated using a downstream methionine since a truncated version of the protein was detected by immunoblot analysis (data not shown). This mutant protein also appeared to be expressed at lower levels, perhaps due to three additional mutations found in the promoter region. Previous evidence indicates that silencing is sensitive to Sir2p dosage (Holmes et al. 1997; Smith et al. 1998). Therefore, it is possible that this mutant’s eso phenotype is due to some combination of decreases in levels of expression and altered N-terminal sequence. This mutant has not been pursued further. Immunoblot analysis showed that levels of the other sir2 mutant proteins were equivalent to wild type in the presence and absence of Sir1p (sir1Δ background shown in Figure 2B). Since the sir2 mutant proteins appear to be expressed comparably to wild type, the phenotypes observed cannot be due to decreased expression or to instability leading to grossly lowered steady-state levels. Thus, it appears that the mutant defects are due to more specific influences on silencing.
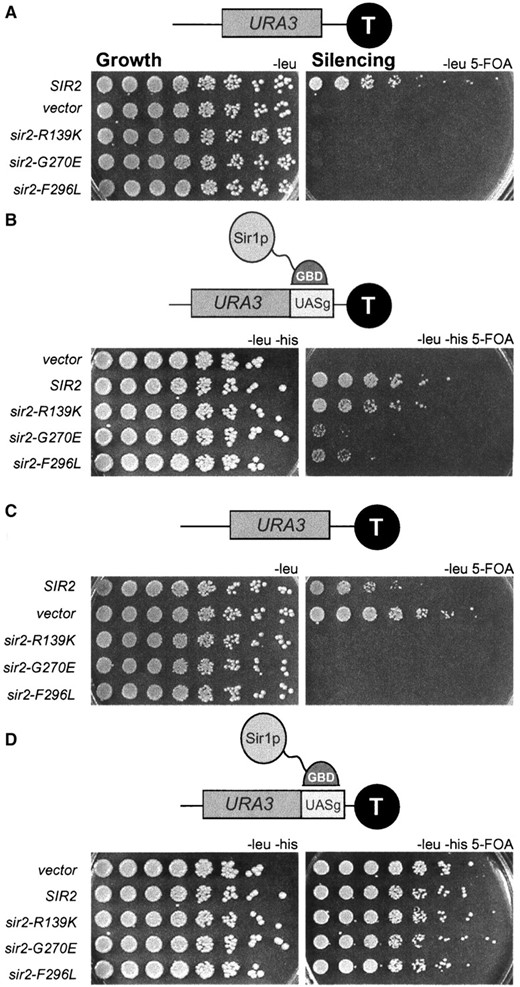
—The sir2eso mutants show defects in TPE. (A) A sir2Δ strain (LPY1953) containing a URA3 marker proximal to telomere VII was transformed with a LEU2 CEN vector (LPY4859), SIR2 (LPY4860), sir2-R139K (LPY4861), sir2-G270E (LPY4862), or sir2-F296L (LPY4863). Fivefold dilutions of the transformants were plated on 5-FOA, growth on 5-FOA indicating silencing of the URA3 reporter, and on SC-leu plates, to control for growth differences. (B) Tethering Sir1p to a telomeric reporter partially suppresses the sir2eso TPE defect. A sir2Δ strain (LPY4624) containing a URA3 reporter at telomere VII, in addition to a copy of the Gal4p DNA-binding site, was cotransformed with a Gal4p DNA-binding domain (GBD)-SIR1 hybrid HIS3 2μ vector (pLP114) and an empty LEU2 CEN vector (pLP62) or the vector containing SIR2 (pLP1237), sir2-R139K (pLP1187), sir2-G270E (pLP1188), or sir2-F296L (pLP1189). The transformants were assayed for silencing as in A. (C) The sir2eso mutants demonstrate dominance by disrupting telomeric silencing even in the presence of Sir2p. A SIR2 strain (LPY1954) was transformed and assayed as in A. (D) Tethering Sir1p suppresses the dominant sir2eso phenotype. A SIR2 strain (LPY 1030) was transformed and assayed as in B.
To evaluate the effects of these mutations relative to proposed enzymatic activities, we considered the structures of Archaeoglobus fulgidus Sir2 (Sir2-Afl) complexed with NAD+ and of a human homolog, SIRT2 (Finnin et al. 2001; Min et al. 2001). The two superimposed enzymes show similarities to the regions containing the Rossman fold domain, commonly found in NAD(H)/NADP(H)-binding proteins, and to the smaller catalytic domain containing a structural zinc atom (Finnin et al. 2001) reviewed in Dutnall and Pillus (2001). The structure of the yeast Sir2 protein has not yet been determined. However, on the basis of the structural similarities between the Sir2-Afl and SIRT2, it appears that two of the sir2eso mutants contain amino acid changes in conserved residues within regions that contribute to the Rossman fold domain (Sir2-Afl in Figure 1B). The equivalent amino acid changes in Sir2-Afl are at amino acids G28 (yG270E) and A48 (yF296L). The fact that this domain is the postulated NAD+-binding portion of the enzyme suggests that these two mutants might be impaired in catalytic activity. The R139K mutation lies in a region N-terminal of the conserved core domain and a priori was not expected to influence activity. This region is not present in Sir2-Afl or SIRT2 structures and therefore the location of R139K could not be inferred (Finnin et al. 2001; Min et al. 2001). The N-terminal region is also variable in members of the yeast SIR2 family members and might be expected to influence SIR2-specific functions (Brachmann et al. 1995). Effects of the sir2eso mutants on catalysis were investigated as described below.
The sir2eso quantitative mating efficiencies
| Mating efficiencya | ||||
|---|---|---|---|---|
| Plasmid | Strain genotype: | MATa sir2Δ | MATa sir1Δ sir2Δ | MATa sir1Δ |
| SIR2 (pLP285) | 1.0 | 0.4 ± 0.07 | 0.2 ± 0.02 | |
| Vector only (pLP60) | 1 × 10-6 ± 0.04 × 10-6 | 3 × 10-6 ± 0.6 × 10-6 | 0.5 ± 0.06 | |
| sir2-R139K (pLP1102) | 0.8 ± 0.07 | 1 × 10-4 ± 0.3 × 10-4 | 0.1 ± 0.02 | |
| sir2-G270E (pLP1110) | 0.9 ± 0.07 | 3 × 10-4 ± 0.6 × 10-6 | 2 × 10-4 ± 0.4 × 10-4 | |
| sir2-F296L (pLP1112) | 0.7 ± 0.09 | 3 × 10-6 ± 0.4 × 10-6 | 2 × 10-4 ± 0.4 × 10-4 | |
| MATα sir2Δ | MATα sir1Δ sir2Δ | MATα sir1Δ | ||
| SIR2 (pLP285) | 1.0 | 0.2 ± 0.2 | 0.2 ± 0.08 | |
| Vector only (pLP60) | 7 × 10-6 ± 2 × 10-6 | 2 × 10-4 ± 0.1 × 10-4 | 0.6 ± 0.02 | |
| sir2-R139K (pLP1102) | 0.8 ± 0.04 | 2 × 10-4 ± 0.4 × 10-4 | 0.1 ± 0.01 | |
| sir2-G270E (pLP1110) | 0.9 ± 0.02 | 2 × 10-4 ± 0.3 × 10-4 | 0.01 ± 0.001 | |
| sir2-F296L (pLP1112) | 0.8 ± 0.01 | 2 × 10-4 ± 0.2 × 10-4 | 0.005 ± 0.002 | |
| Mating efficiencya | ||||
|---|---|---|---|---|
| Plasmid | Strain genotype: | MATa sir2Δ | MATa sir1Δ sir2Δ | MATa sir1Δ |
| SIR2 (pLP285) | 1.0 | 0.4 ± 0.07 | 0.2 ± 0.02 | |
| Vector only (pLP60) | 1 × 10-6 ± 0.04 × 10-6 | 3 × 10-6 ± 0.6 × 10-6 | 0.5 ± 0.06 | |
| sir2-R139K (pLP1102) | 0.8 ± 0.07 | 1 × 10-4 ± 0.3 × 10-4 | 0.1 ± 0.02 | |
| sir2-G270E (pLP1110) | 0.9 ± 0.07 | 3 × 10-4 ± 0.6 × 10-6 | 2 × 10-4 ± 0.4 × 10-4 | |
| sir2-F296L (pLP1112) | 0.7 ± 0.09 | 3 × 10-6 ± 0.4 × 10-6 | 2 × 10-4 ± 0.4 × 10-4 | |
| MATα sir2Δ | MATα sir1Δ sir2Δ | MATα sir1Δ | ||
| SIR2 (pLP285) | 1.0 | 0.2 ± 0.2 | 0.2 ± 0.08 | |
| Vector only (pLP60) | 7 × 10-6 ± 2 × 10-6 | 2 × 10-4 ± 0.1 × 10-4 | 0.6 ± 0.02 | |
| sir2-R139K (pLP1102) | 0.8 ± 0.04 | 2 × 10-4 ± 0.4 × 10-4 | 0.1 ± 0.01 | |
| sir2-G270E (pLP1110) | 0.9 ± 0.02 | 2 × 10-4 ± 0.3 × 10-4 | 0.01 ± 0.001 | |
| sir2-F296L (pLP1112) | 0.8 ± 0.01 | 2 × 10-4 ± 0.2 × 10-4 | 0.005 ± 0.002 | |
Mating efficiencies from three independent experiments are shown with standard deviations and normalized to either MATa sir2Δ or MATα sir2Δ strains transformed with a plasmid containing wild-type SIR2 (pLP285).
The sir2eso quantitative mating efficiencies
| Mating efficiencya | ||||
|---|---|---|---|---|
| Plasmid | Strain genotype: | MATa sir2Δ | MATa sir1Δ sir2Δ | MATa sir1Δ |
| SIR2 (pLP285) | 1.0 | 0.4 ± 0.07 | 0.2 ± 0.02 | |
| Vector only (pLP60) | 1 × 10-6 ± 0.04 × 10-6 | 3 × 10-6 ± 0.6 × 10-6 | 0.5 ± 0.06 | |
| sir2-R139K (pLP1102) | 0.8 ± 0.07 | 1 × 10-4 ± 0.3 × 10-4 | 0.1 ± 0.02 | |
| sir2-G270E (pLP1110) | 0.9 ± 0.07 | 3 × 10-4 ± 0.6 × 10-6 | 2 × 10-4 ± 0.4 × 10-4 | |
| sir2-F296L (pLP1112) | 0.7 ± 0.09 | 3 × 10-6 ± 0.4 × 10-6 | 2 × 10-4 ± 0.4 × 10-4 | |
| MATα sir2Δ | MATα sir1Δ sir2Δ | MATα sir1Δ | ||
| SIR2 (pLP285) | 1.0 | 0.2 ± 0.2 | 0.2 ± 0.08 | |
| Vector only (pLP60) | 7 × 10-6 ± 2 × 10-6 | 2 × 10-4 ± 0.1 × 10-4 | 0.6 ± 0.02 | |
| sir2-R139K (pLP1102) | 0.8 ± 0.04 | 2 × 10-4 ± 0.4 × 10-4 | 0.1 ± 0.01 | |
| sir2-G270E (pLP1110) | 0.9 ± 0.02 | 2 × 10-4 ± 0.3 × 10-4 | 0.01 ± 0.001 | |
| sir2-F296L (pLP1112) | 0.8 ± 0.01 | 2 × 10-4 ± 0.2 × 10-4 | 0.005 ± 0.002 | |
| Mating efficiencya | ||||
|---|---|---|---|---|
| Plasmid | Strain genotype: | MATa sir2Δ | MATa sir1Δ sir2Δ | MATa sir1Δ |
| SIR2 (pLP285) | 1.0 | 0.4 ± 0.07 | 0.2 ± 0.02 | |
| Vector only (pLP60) | 1 × 10-6 ± 0.04 × 10-6 | 3 × 10-6 ± 0.6 × 10-6 | 0.5 ± 0.06 | |
| sir2-R139K (pLP1102) | 0.8 ± 0.07 | 1 × 10-4 ± 0.3 × 10-4 | 0.1 ± 0.02 | |
| sir2-G270E (pLP1110) | 0.9 ± 0.07 | 3 × 10-4 ± 0.6 × 10-6 | 2 × 10-4 ± 0.4 × 10-4 | |
| sir2-F296L (pLP1112) | 0.7 ± 0.09 | 3 × 10-6 ± 0.4 × 10-6 | 2 × 10-4 ± 0.4 × 10-4 | |
| MATα sir2Δ | MATα sir1Δ sir2Δ | MATα sir1Δ | ||
| SIR2 (pLP285) | 1.0 | 0.2 ± 0.2 | 0.2 ± 0.08 | |
| Vector only (pLP60) | 7 × 10-6 ± 2 × 10-6 | 2 × 10-4 ± 0.1 × 10-4 | 0.6 ± 0.02 | |
| sir2-R139K (pLP1102) | 0.8 ± 0.04 | 2 × 10-4 ± 0.4 × 10-4 | 0.1 ± 0.01 | |
| sir2-G270E (pLP1110) | 0.9 ± 0.02 | 2 × 10-4 ± 0.3 × 10-4 | 0.01 ± 0.001 | |
| sir2-F296L (pLP1112) | 0.8 ± 0.01 | 2 × 10-4 ± 0.2 × 10-4 | 0.005 ± 0.002 | |
Mating efficiencies from three independent experiments are shown with standard deviations and normalized to either MATa sir2Δ or MATα sir2Δ strains transformed with a plasmid containing wild-type SIR2 (pLP285).
By virtue of the design of the eso screen, the sir2eso mutants displayed a characteristic mating defect in the absence of Sir1p. A qualitative mating assay with one representative sir2eso mutant is shown in Figure 2A. It is notable that a sir1Δ strain mates comparably to a wild-type strain by this assay despite its known epigenetic silencing defects at the HM loci (Pillus and Rine 1989). Therefore, it remained a possibility that the sir2eso alleles displayed silencing defects at HML and HMR even in the presence of Sir1p at levels detectable only through quantitative analysis. Mating efficiencies for strains transformed with plasmids bearing the sir2eso mutations, wild-type SIR2, or vector only were calculated and normalized to wild type. The results showed that the sir2eso mutants were modestly defective in silencing in the presence of SIR1 (Table 2). Although they mated at 70-90% efficiency in comparison to wild type, the sir2eso mutants were slightly more efficient in mating than the sir1Δ SIR2 mutant strain. The sir1Δ sir2eso mutants are as defective as a sir1Δ sir2Δ mutant, with mating efficiencies four to seven orders of magnitude lower than those of wild-type strains. In addition, two lesions within the core domain, sir2-G270E and sir2-F296L, conferred dominantly derepressed phenotypes in MATa sir1Δ and MATα sir1Δ cells but not in SIR1 wild-type backgrounds (data not shown).
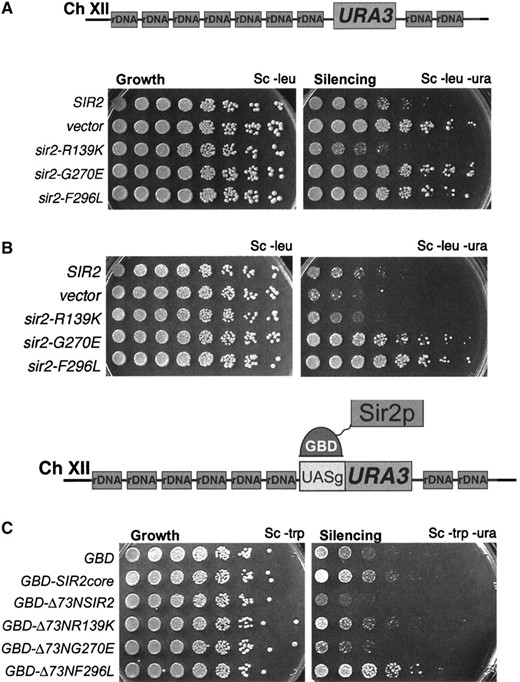
The sir2eso mutants are defective in telomeric silencing and can be partially suppressed by tethering Sir1p to telomeres: In contrast to silencing at the HM silent mating-type loci, loss of Sir1p has no effect on silencing reporter genes at telomeres (Aparicio et al. 1991). Since the sir2eso defects at the HM loci were revealed only in sir1Δ mutants, although SIR1 has no apparent role in telomeric silencing, it was possible that the sir2eso alleles would be competent in telomeric silencing. To test this hypothesis, a sir2Δ strain marked at telomere VII with URA3 was transformed with CEN plasmids containing the sir2eso alleles (pLP1187-1189), SIR2 (pLP1237), or vector only (pLP62). Telomeric silencing was assayed on 5-FOA-containing medium as described previously (Gottschling et al. 1990). All three sir2eso alleles were sensitive to 5-FOA, demonstrating that they were completely defective in silencing the telomeric reporter gene (Figure 3A).
Recently, it was determined that there are only ∼30 molecules of Sir1p per cell (Gardner and Fox 2001), suggesting that Sir1p may be limiting in the cell. Therefore, we tested whether increased SIR1 gene dosage suppressed the sir2eso telomeric silencing defects. Expression of SIR1 using a high-copy 2μ plasmid did not suppress their defects; thus this simple possibility does not explain the sir2eso phenotype (data not shown). Next, we directed Sir1p to the telomeres using a GBD-Sir1p fusion protein and a modified telomeric reporter gene containing an adjacent Gal4p DNA-binding site (UASg). Previous results demonstrated improvement in silencing upon tethering GBD-Sir1p at a telomeric reporter. However, such improved silencing is dependent on the other Sir proteins, including Sir2p (Chien et al. 1993). A sir2Δ strain marked at telomere VII with a UASg-URA3 marker was cotransformed with GBD-Sir1p (pLP114) and with the same set of sir2eso plasmids used for the telomeric assay shown in Figure 3A. The telomeric defects for all three alleles were suppressed by tethering Sir1p, albeit to differing degrees (Figure 3B). The sir2-R139K defect was fully suppressed whereas the two core mutants were only partially suppressed. Therefore, the sir2eso mutants require Sir1p for silencing at the HM loci and can function if Sir1p is directed to the telomeres. This suggested that at some level, the sir2eso mutant proteins had the capacity to function in telomeric silencing, although this function appeared limited.
To evaluate further the nature of sir2eso function at telomeres, we performed a dominance test. In this experiment, the sir2eso mutant genes on centromeric plasmids were transformed into a telomere-marked strain containing wild-type Sir2p. Somewhat surprisingly, the sir2eso mutants disrupted silencing in the presence of SIR2 (Figure 3C). However, tethering Sir1p to the telomeres overcame this dominant phenotype (Figure 3D). A potential molecular explanation for the dominance observed could be that the sir2eso mutant proteins were mislocalized in the cell and thus titrated Sir4p or wild-type Sir2p away from the telomeres. To test this possibility sir2eso mutant proteins were localized using immunofluorescence analysis.
Localization of sir2-G270Ep at telomeres and nucleolus
| Total no. of cells | Telomeric staining | ||
|---|---|---|---|
| Strain | >3 foci (%) | 0-3 foci (%) | |
| SIR1 SIR2 | 114 | 44 (38) | 70 (62) |
| sir1Δ SIR2 | 87 | 35 (38) | 54 (62) |
| SIR1 sir2Δ | 69 | 0 (0) | 69 (100) |
| sir1Δ G270E | 106 | 2 (2) | 104 (98) |
| SIR1 G270E | 138 | 57 (41) | 75 (54) |
| Nucleolar staining | |||
| Strain | Total no. of cells | Colocalized with Nop1p (%) | No colocalization (%) |
| SIR1 SIR2 | 114 | 94 (83) | 20 (17) |
| sir1Δ SIR2 | 87 | 78 (90) | 9 (10) |
| SIR1 sir2Δ | 69 | 0 (0) | 69 (100) |
| sir1Δ G270E | 106 | 23 (22) | 79 (78) |
| SIR1 G270E | 138 | 124 (90) | 14 (10) |
| Total no. of cells | Telomeric staining | ||
|---|---|---|---|
| Strain | >3 foci (%) | 0-3 foci (%) | |
| SIR1 SIR2 | 114 | 44 (38) | 70 (62) |
| sir1Δ SIR2 | 87 | 35 (38) | 54 (62) |
| SIR1 sir2Δ | 69 | 0 (0) | 69 (100) |
| sir1Δ G270E | 106 | 2 (2) | 104 (98) |
| SIR1 G270E | 138 | 57 (41) | 75 (54) |
| Nucleolar staining | |||
| Strain | Total no. of cells | Colocalized with Nop1p (%) | No colocalization (%) |
| SIR1 SIR2 | 114 | 94 (83) | 20 (17) |
| sir1Δ SIR2 | 87 | 78 (90) | 9 (10) |
| SIR1 sir2Δ | 69 | 0 (0) | 69 (100) |
| sir1Δ G270E | 106 | 23 (22) | 79 (78) |
| SIR1 G270E | 138 | 124 (90) | 14 (10) |
The sir2eso mutant strains were analyzed by immunofluorescence in sir1Δ and SIR1 strain backgrounds. The total cell number was derived from four independent experiments. Images were blinded and the number of telomeric foci counted and represented as >3 foci and 0-3 foci. Percentages are the number of cells with a specific pattern divided by the total number of cells analyzed for that sample.
Localization of sir2-G270Ep at telomeres and nucleolus
| Total no. of cells | Telomeric staining | ||
|---|---|---|---|
| Strain | >3 foci (%) | 0-3 foci (%) | |
| SIR1 SIR2 | 114 | 44 (38) | 70 (62) |
| sir1Δ SIR2 | 87 | 35 (38) | 54 (62) |
| SIR1 sir2Δ | 69 | 0 (0) | 69 (100) |
| sir1Δ G270E | 106 | 2 (2) | 104 (98) |
| SIR1 G270E | 138 | 57 (41) | 75 (54) |
| Nucleolar staining | |||
| Strain | Total no. of cells | Colocalized with Nop1p (%) | No colocalization (%) |
| SIR1 SIR2 | 114 | 94 (83) | 20 (17) |
| sir1Δ SIR2 | 87 | 78 (90) | 9 (10) |
| SIR1 sir2Δ | 69 | 0 (0) | 69 (100) |
| sir1Δ G270E | 106 | 23 (22) | 79 (78) |
| SIR1 G270E | 138 | 124 (90) | 14 (10) |
| Total no. of cells | Telomeric staining | ||
|---|---|---|---|
| Strain | >3 foci (%) | 0-3 foci (%) | |
| SIR1 SIR2 | 114 | 44 (38) | 70 (62) |
| sir1Δ SIR2 | 87 | 35 (38) | 54 (62) |
| SIR1 sir2Δ | 69 | 0 (0) | 69 (100) |
| sir1Δ G270E | 106 | 2 (2) | 104 (98) |
| SIR1 G270E | 138 | 57 (41) | 75 (54) |
| Nucleolar staining | |||
| Strain | Total no. of cells | Colocalized with Nop1p (%) | No colocalization (%) |
| SIR1 SIR2 | 114 | 94 (83) | 20 (17) |
| sir1Δ SIR2 | 87 | 78 (90) | 9 (10) |
| SIR1 sir2Δ | 69 | 0 (0) | 69 (100) |
| sir1Δ G270E | 106 | 23 (22) | 79 (78) |
| SIR1 G270E | 138 | 124 (90) | 14 (10) |
The sir2eso mutant strains were analyzed by immunofluorescence in sir1Δ and SIR1 strain backgrounds. The total cell number was derived from four independent experiments. Images were blinded and the number of telomeric foci counted and represented as >3 foci and 0-3 foci. Percentages are the number of cells with a specific pattern divided by the total number of cells analyzed for that sample.
Localization of the sir2eso mutant proteins: Genetic and biochemical studies place Sir2p at the HM loci, the telomeres, and the rDNA repeats that serve as a nucleolar organizer. Consistent with this, by indirect immunofluorescence Sir2p localizes in a crescent or cup-shaped form in the nucleolus and as clustered spots at telomeric foci found at the nuclear periphery directly opposite the nucleolus (Gotta et al. 1997). Using a polyclonal antibody raised to a peptide specific for the C terminus of Sir2p (2916/8; Smith et al. 1998), we determined the localization of the sir2eso mutant proteins in sir1Δ and SIR1 strains. To assess nucleolar localization, we evaluated colocalization with the nucleolar marker Nop1p, the yeast homolog of fibrillarin (Aris and Blobel 1988). To assess telomeric localization, we quantitated the number of telomeric foci in the various strain backgrounds.
Two of the mutant proteins, sir2-R139Kp and sir2-F296Lp, showed localization indistinguishable from wild-type Sir2p in both SIR1 and sir1Δ backgrounds (data not shown). In contrast, proper localization of sir2-G270Ep appeared to depend on the status of SIR1. The sir2-G270E mutant protein localized to telomeres in only ∼5% of sir1Δ cells relative to wild-type Sir2p localization. In addition, on the basis of failure to colocalize with Nop1p, this mutant protein showed no localization to the nucleolus in ∼30% of the cells analyzed relative to wild-type Sir2p (Table 3). Furthermore, the few cells with wild-type localization showed decreased staining intensity, represented in Figure 4. In marked contrast, in SIR1 strains, the sir2-G270Ep localization was restored to telomeres and the nucleolus in a manner indistinguishable from wild type. Therefore, the localization pattern of this mutant does not account for its defects in telomeric silencing since comparable defects are observed in both SIR1 (Figure 3) and sir1Δ (data not shown) backgrounds. Perhaps the sir2eso mutant proteins do reach the telomeres and the nucleolus but their association to the chromatin is functionally inadequate at these silenced regions.
Sir1p has been shown to function in the establishment of silencing (Pillus and Rine 1989), serving as a recruitment factor for the other Sir proteins at the silent mating-type loci (Fox et al. 1997; Gardner and Fox 2001). Because Sir1p does not ordinarily serve as a recruitment protein at the telomeres, we hypothesized that Sir2p might also function in recruiting Sir3p and Sir4p to the HM loci and the telomeres. The sir2eso mutants may be defective in this hypothesized recruitment function, rendering them fully dependent on other recruitment proteins. This is consistent with the sir2eso dependence on Sir1p at the HM loci and GBD-Sir1p at the telomeres. To test this hypothesis, we fused the sir2eso mutant proteins to the GBD to tether the mutants directly through engineered binding sites on the chromosome. If the sir2eso mutants are defective in a recruitment function, then their defects might be suppressed if targeted to a reporter gene at the telomeres via a GBD domain. A sir2Δ strain marked at telomere VII with a URA3 reporter and an adjacent Gal4p DNA-binding site was transformed with a vector expressing GBD, GBD-Sir2pcore(210-440), GBD-Δ73NSir2p(73-562), GBD-Δ73Nsir2-R139Kp, GBD-Δ73Nsir2-G270Ep, or GBD-Δ73Nsir2-F296Lp and the transformants were tested for silencing. The GBD and GBD-Sir2pcore constructs served as negative controls since it has been shown that the core domain of Sir2p is necessary but not sufficient for silencing even when tethered to a reporter (Cockell et al. 2000).
In all cases, tethering the sir2eso mutants restored telomeric position effect (TPE) to wild-type levels (Figure 5). This restoration was fully dependent on tethering since the constructs were unable to silence at telomeres in strains lacking the Gal4p DNA-binding site (UASg) adjacent to the URA3 reporter (data not shown). Therefore, the sir2eso mutants are properly localized to the telomeres by immunofluorescence, yet can fully function only if targeted to the locus either directly via a Gal4p DNA-binding domain or indirectly via GBD-Sir1p.
The sir2eso mutants show distinct phenotypes at the rDNA: PolII-transcribed reporter genes engineered within the rDNA are subject to SIR2-dependent silencing. To determine whether the sir2eso mutants function in silencing at the rDNA locus, a sir2Δ strain containing a URA3 cassette inserted in the rDNA was transformed with the various sir2eso plasmids as described above. Silencing was assayed by evaluating growth on plates lacking uracil with less growth indicating more silencing (Smith and Boeke 1997). The two sir2eso mutants that carry a mutation within the conserved core domain of SIR2 were defective in silencing the URA3 rDNA reporter (Figure 6A). However, the mutant sir2-R139K functioned fully at this locus, silencing as well as or better than wild-type SIR2.
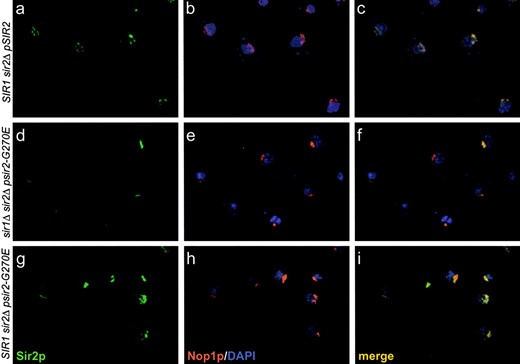
—The sir2-G270E mutant protein is mislocalized in cells lacking Sir1p. (Top: a, b, and c) SIR1 sir2Δ transformed with SIR2 on a HIS3 CEN plasmid (LPY4595). (Middle: d, e, and f) sir1Δ sir2Δ strain transformed with sir2-G270E on a HIS3 CEN plasmid (LPY4602). (Bottom: g, h, and i) SIR1 sir2Δ transformed with sir2-G270E on a HIS3 CEN plasmid (LPY4597). Strains (a, d, and g) were stained with anti-Sir2p affinity-purified antisera (2916/8) and detected by FITC-conjugated secondary antibodies (b, e, and h) with anti-Nop1p antibodies detected by a Texas-red-conjugated secondary antibody and (c, f, and i) the merge of Nop1p, Sir2p, and DNA staining with 4′,6-diamidino-2-phenylindole.
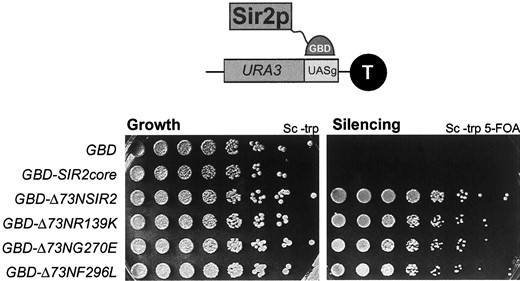
—Tethering the sir2eso alleles directly to the telomeres rescued their telomeric silencing defects. A sir2Δ strain (LPY5611) marked at telomere VII with aURA3 reportergene with anadjacent Gal4p DNA-binding site was transformed with GBD(1-147) (LPY5777), GBD-Sir2-pcore(210-440) (LPY5778), GBD-Δ73NSir2-p(73-562) (LPY5779), GBD-Δ73NSir2-R139Kp (LPY5780), GBD-Δ73NSir2-G270Ep (LPY-5781), or GBD-Δ73NSir2-F296Lp (LPY-5782). Fivefold dilutions were plated and assayed for growth on Sc-trp or silencing on 5-FOA.
Transcriptional and recombinational silencing within the rDNA is distinct because it is fully dependent on SIR2, yet is independent of the other SIR genes (Gottlieb and Esposito 1989; Bryk et al. 1997; Fritze et al. 1997; Smith and Boeke 1997). Because the sir2eso alleles had a conditional dependence on SIR1 at the HM loci, we tested whether this dependence might also exist at the rDNA. The absence of Sir1p had no effect on the sir2eso phenotypes observed at the rDNA (data not shown). Since the sir2-G270E and sir2-F296L mutations caused dominant derepression at the HM loci and the telomeres, the dominance test was repeated at the rDNA locus. The sir2eso genes on plasmids were transformed into a SIR2 strain marked within the rDNA and, as before, transformants were assayed for growth on plates lacking uracil. The two mutants defective in rDNA silencing, sir2-G270E and sir2-F296L, also showed dominant effects at this locus (Figure 6B).
—The sir2eso mutants have distinct silencing phenotypes within the rDNA locus. (A) A sir2Δ strain (LPY2447) marked at the rDNA with a URA3 cassette was transformed with wild-type SIR2 on a LEU2 CEN plasmid (pLP1237), empty vector (pLP62), sir2-R139K (pLP1187), sir2-G270E (pLP1188), or sir2-F296L (pLP1189), and fivefold dilutions of the transformants were assayed for growth on Sc-leu or silencing on Sc-ura. The sir2eso mutants with mutations in the conserved domain of Sir2p are defective in rDNA silencing. (B) A SIR2 strain (LPY2446) transformed and assayed as in A. The sir2-G270E and sir2-F296L mutants are dominantly defective in rDNA silencing. (C) A sir2Δ strain (LPY5378) containing four Gal4p DNA-binding sites adjacent to the rDNA URA3 reporter was transformed with GBD (LPY5637), GBD-Sir2pcore210-440 (LPY5638), GBD-Δ73NSir2p73-562 (LPY5639), GBD-Δ73NSir2-R139Kp (LPY5640), GBD-Δ73N-Sir2-G270Ep (LPY5641), or GBD-Δ73NSir2-F296Lp (LPY5642). Fivefold dilutions were assayed for growth and silencing. GBD alone or fused to the conserved core domain of Sir2p failed to silence the reporter. The sir2-G270E mutant is rescued when tethered to the rDNA; however, the sir2-F296L mutant remains defective. Note that mutant sir2-R139K becomes defective in silencing at the rDNA when tethered to the locus.
We proposed in the section above that the sir2eso mutants could be defective in a type of recruitment function at the telomeres. To extend this hypothesis to the rDNA, we tested the ability of the sir2eso strains to silence the rDNA if targeted directly to this locus. The GBD-Δ73NSIR2 constructs were transformed into a sir2Δ strain containing a modified URA3 reporter gene with four adjacent Gal4p DNA-binding sites (4X-UASg) within the rDNA locus (Cuperus et al. 2000). Because of inherent variability in silencing assays, in each case multiple independent transformants were evaluated. We observed, as expected, that the GBD-Δ73NSir2p consistently silenced when tethered. Neither GBD-Sir2pcore nor GBD constructs alone silenced. The modest differences between these controls (Figure 6) demonstrate the occasional variability noted above. The results with the sir2eso alleles were somewhat surprising. One of the two rDNA silencing-defective sir2eso mutants, sir2-G270E, was rescued when tethered, whereas the other mutant, sir2-F296L, was not. Further, the rDNA silencing-competent sir2eso mutant, sir2-R139K, became impaired for silencing when tethered to the rDNA reporter (Figure 6C). We observed that simply expressing these constructs in a strain with a silencing reporter but no UAS had no effect (data not shown); thus the effects observed are completely dependent on the tethering site.
Therefore, all three mutants showed a different spectrum of phenotypes with respect to rDNA silencing, implying that they had different silencing defects at this locus. Although inadequate association with chromatin might explain the silencing phenotypes of the sir2eso mutants at the HM loci and the telomeres, this model seemed inadequate to explain the diversity of phenotypes of the sir2eso mutants at the rDNA. A significant element of Sir2p silencing function is its NAD+-dependent deacetylase activity. We therefore asked if the differences between the sir2eso silencing abilities were reflected in their catalytic activities.
The sir2eso mutants are defective in NADase activity
| GST fusion protein | Normalized NADase activity |
|---|---|
| GST-Sir2p | 1.00 ± 0.08 |
| GST-sir2-R139Kp | 0.37 ± 0.01 |
| GST-sir2-G270Ep | 0.59 ± 0.04 |
| GST-sir2-F296Lp | 0.34 ± 0.07 |
| GST | 0.05 ± 0.02 |
| GST fusion protein | Normalized NADase activity |
|---|---|
| GST-Sir2p | 1.00 ± 0.08 |
| GST-sir2-R139Kp | 0.37 ± 0.01 |
| GST-sir2-G270Ep | 0.59 ± 0.04 |
| GST-sir2-F296Lp | 0.34 ± 0.07 |
| GST | 0.05 ± 0.02 |
Purified proteins GST-Sir2p (pLP1275), GST-sir2R139K (pLP1335), GST-sir2G270E (pLP1336), GST-sir2F296L (pLP1337), or GST (pLP1334) were tested for their ability to convert NAD+ to nicotinamide and ADP-ribose in a histone-dependent manner. Reactions were performed in the presence of [3H]NAD+ and 3.7 μg of purified enzymes. Radiolabeled [3H]nicotinamide was detected as described by Landry et al. (2000a; see materials and methods). The rate of change over five time points (10 min and 3, 7, 24, and 33 hr) was calculated and normalized to wild-type Sir2p. Multiple experiments were performed. The experiment shown was performed in triplicate and is shown with standard deviations.
The sir2eso mutants are defective in NADase activity
| GST fusion protein | Normalized NADase activity |
|---|---|
| GST-Sir2p | 1.00 ± 0.08 |
| GST-sir2-R139Kp | 0.37 ± 0.01 |
| GST-sir2-G270Ep | 0.59 ± 0.04 |
| GST-sir2-F296Lp | 0.34 ± 0.07 |
| GST | 0.05 ± 0.02 |
| GST fusion protein | Normalized NADase activity |
|---|---|
| GST-Sir2p | 1.00 ± 0.08 |
| GST-sir2-R139Kp | 0.37 ± 0.01 |
| GST-sir2-G270Ep | 0.59 ± 0.04 |
| GST-sir2-F296Lp | 0.34 ± 0.07 |
| GST | 0.05 ± 0.02 |
Purified proteins GST-Sir2p (pLP1275), GST-sir2R139K (pLP1335), GST-sir2G270E (pLP1336), GST-sir2F296L (pLP1337), or GST (pLP1334) were tested for their ability to convert NAD+ to nicotinamide and ADP-ribose in a histone-dependent manner. Reactions were performed in the presence of [3H]NAD+ and 3.7 μg of purified enzymes. Radiolabeled [3H]nicotinamide was detected as described by Landry et al. (2000a; see materials and methods). The rate of change over five time points (10 min and 3, 7, 24, and 33 hr) was calculated and normalized to wild-type Sir2p. Multiple experiments were performed. The experiment shown was performed in triplicate and is shown with standard deviations.
The sir2eso mutants are impaired in NADase (deacetylase) activity: Previous reports showed that the Sir2p family of proteins functions as NAD+-dependent protein deacetylases and that decreases in deacetylase activity correlate with loss of silencing (Imai et al. 2000; Landry et al. 2000b; Smith et al. 2000). The role of NAD+ in the deacetylation reaction has been investigated and has led to a deeper understanding of the mechanism of catalysis (Imai et al. 2000; Landry et al. 2000a; Borra et al. 2002). On the basis of the products released, the reaction is described as the hydrolysis of one NAD+ to form nicotinamide and the novel product acetyl-ADP-ribose (AADPR), for each acetyl group removed. This results in a 1:1:1 molar ratio of acetyl-ADP-ribose (AADPR), nicotinamide, and a deacetylated peptide substrate with an enzyme-ADP-ribose intermediate (Landry et al. 2000a). This finding allows for a direct correlation between NADase activity and histone deacetylation for the Sir2 family members.
To determine whether the sir2eso proteins retained wild-type deacetylase activity, recombinant GST-sir2eso mutant fusion proteins were expressed in E. coli, purified, and tested for NADase activity in vitro (Landry et al. 2000a). The results of three independent NADase assays showed that the mutants were all partially impaired in enzymatic activity (Table 4 and data not shown). The GST-sir2-R139Kp and GST-sir2-F296Lp mutant proteins consistently had <50% activity when compared to wild-type GST-Sir2p. The GST-sir2-G270E mutant protein was slightly more active than the other two mutant enzymes but never achieved activities >70% of wild-type activity. The activities shown in Table 4 reflect consistent differences of mutant activity relative to wild type. Further distinctions between the GST-sir2eso mutant proteins may become apparent with extensive kinetic analyses or with comparisons between NADase and deacetylase activities. Since NADase and deacetylase activities are coupled, however, the decreases in activity for the sir2eso mutant proteins suggest that they are not significant enough to completely abolish silencing because these mutants were capable of silencing when tethered to a reporter or when tethered by Sir1p. Since the sir2eso mutants are not catalytically dead but have only partially impaired enzymatic activity, we asked whether the silencing defects observed in vivo might also reflect inefficient complex formation or protein interactions.
The sir2eso mutant proteins interact with Sir4p and Net1p: Sir2p is known to interact with a number of other proteins. Among these are Sir4p (Moazed et al. 1997) and Net1p (Shou et al. 1999; Straight et al. 1999), associations with which are correlated with telomeric and rDNA silencing, respectively. We considered the possibility that the sir2eso phenotype was caused by impaired interactions with these proteins and tested their associations by immunoprecipitation experiments. Immunoprecipitation was performed with either anti-Sir2p or anti-Sir4p reagents. The immunoprecipitated samples were analyzed by protein immunoblotting using the relevant antisera as noted (Figure 7). The results indicated that the sir2eso mutant proteins interacted with both Sir4p and Net1p, even in the absence of Sir1p (Figure 7 and data not shown). This suggests that Sir and RENT complex formation is not grossly disrupted in the sir2eso mutants. It is also clear that Sir1p does not stabilize the components of the complexes since interactions with Sir4p and Net1p are found in both SIR1 and sir1Δ backgrounds (sir1Δ background for Sir4p, Figure 7A; both backgrounds for Net1p, Figure 7B). One difference observed in the sir1Δ background was the consistent loss of an additional α-myc-reactive band that commonly migrates with the Net1p in immunoblot analyses (Straight et al. 1999; Figure 7B). The band could represent a protein modification of Net1p that, either directly or indirectly, requires Sir1p, although SIR1 has not been previously implicated in NET1 function. Further experiments will be necessary to explore these ideas, but together they suggest that the sir2eso phenotypes do not result from gross disruptions of Sir2p-containing complexes or activity.
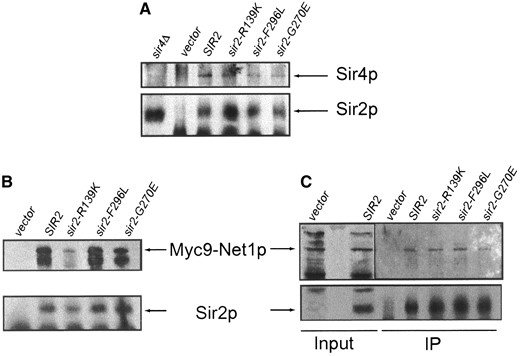
—The sir2eso mutant proteins interact with Sir4p and Net1p. Extracts were prepared from SIR1 sir2Δ net1Δ::Myc9-NET1-LEU2 and sir1Δ sir2Δ net1Δ::Myc9-NET1-LEU2 strains transformed with vector (LPY6402), SIR2 (LPY6403), sir2-R139K (LPY6404), sir2-G270E (LPY6405), or sir2-F296L (LPY6406). (A) Strain LPY6400 sir1Δ sir2Δ net1Δ::Myc9-NET1-LEU2. Sir4p was immunoprecipitated and tested for coimmunoprecipitation of Sir2p and sir2eso mutant proteins by immunoblot analysis. Immunoprecipitations were performed with anti-Sir4p (7795) and immunoblots were probed with antisera against Sir4p (2913/8; top) and Sir2p (2916/8; bottom). (B) Strains LPY5615 SIR1 sir2Δ net1Δ::Myc9-NET1-LEU2 (left) and LPY6400 sir1Δ sir2Δ net1Δ::Myc9-NET1-LEU2 (right). Immunoblot analysis of Net1-myc9 immunoprecipitated with anti-Sir2p (2916/8) and probed with anti-myc monoclonal antibody 9E10 (top) and against Sir2p (2916/8, bottom) is shown; it is also shown in a SIR1-sir2Δ background (left).
DISCUSSION
Sir2p is an NAD+-dependent deacetylase whose association with Sir4p and Net1p correlates with transcriptional silencing (reviewed in Gartenberg 2000; Moazed 2001). This study describes three new sir2 mutants isolated as enhancers of sir-oneΔ, the sir2eso mutants. These mutants retain interactions with Sir4p and Net1p and are only partially compromised for catalytic activity. They do, however, display distinct phenotypes at three distinct silenced loci, including dominant effects at telomeres and within the rDNA. Many of these defects can be ameliorated by molecular targeting strategies. Thus a range of catalytic activity may be compatible with silencing functions, as long as that activity is appropriately directed to its required sites of action.
The sir2eso mutants show mating defects in the absence of SIR1: The sir2eso mutants are defective in silencing HML and HMR only in the absence of Sir1p and encode mutations in residues that are conserved in the Sir2 protein family. In quantitative mating analyses, the sir2eso mutants were only slightly impaired in their mating ability in the presence of SIR1. In contrast, in a sir1Δ background, the sir2eso mutants were as defective as sir2Δ strains. Although the mutants were not dominant in the presence of SIR1 (data not shown), sir2-G270E and sir2-F296L displayed moderate mating defects in a sir1Δ SIR2 background. Considering that Sir1p is required for targeting the Sir2/4 complex to modified synthetic silencers (Fox et al. 1997; Gardner and Fox 2001), sir2eso silencing defects may arise from unstable targeting to the HM loci. Such instability, when coupled with the sir1Δ establishment defect, may synergistically lead to the complete loss of silencing at HML and HMR.
The sir2eso mutants are impaired, yet not totally defective, in enzymatic activity. Therefore, it is possible that in the absence of Sir1p, wild-type activity levels are required to maintain a silenced state at the HM loci or to initiate a stable silenced chromatin structure that can then be propagated. Our data do not yet distinguish these possibilities.
Telomeric silencing is disrupted in the sir2eso mutants: Although the sir2eso mutants were identified through a screen for silent mating-type defects, the mutants were also dominantly defective in telomeric silencing in both sir1Δ (data not shown) and SIR1 backgrounds. Tethering directly to the telomeres, or indirectly through GBD-Sir1p, rescued these silencing defects and reversed the dominant effects. This raised the possibility that the mutant proteins were not properly localized in the cell. However, immunofluorescence analysis revealed that the sir2eso proteins were localized indistinguishably to telomeric foci from wild-type localization in SIR1 cells. Recent studies have also evaluated silencing protein localization by the independent technique of chromatin immunoprecipitation. In these studies, it was observed that Sir2p’s enzymatic activity not only is necessary for the spread of the Sir proteins but also may influence efficient association of the Sir proteins with chromatin at the telomeres (Armstrong et al. 2002; Hoppe et al. 2002; Luo et al. 2002). Subtle quantitative distinctions in association or dynamic occupancy may result from the decreased activity of sir2eso mutant proteins. Indeed, that the mutant proteins are somehow impaired in functional association with chromatin is supported by the observation that the sir2eso mutants became silencing competent when tethered via GBD or GBD-Sir1p, showing that their weakened enzymatic activity did not render these mutants incapable of promoting silencing.
An activity-dependent model for function at the silent mating-type loci and telomeres: Long-standing models for the establishment of silent chromatin at the telomeres involve the recruitment of the Sir2/4 complex to DNA, followed by propagation of condensed chromatin through interactions among Sir3p, Sir4p, and deacetylated histone tails (Ghidelli et al. 2001; Carmen et al. 2002; Hoppe et al. 2002; Luo et al. 2002; and reviewed in Moazed 2001). The recruitment of Sir3p to the telomeres appears to be a regulated step since stable Sir3p-Sir4p interactions are detected only when the N terminus of Sir4p is removed or under conditions that strengthen Sir-nucleosome interactions (Moretti et al. 1994; Moazed et al. 1997; Strahl-Bolsinger et al. 1997; Ghidelli et al. 2001). We propose that if Sir3p is indeed also recruited through strengthened Sir-nucleosome interactions, its recruitment could become progressively less dependent on Sir2p enzymatic activity as the number of associated Sir complexes increases and spreads. This idea is consistent with the observation that overexpression of Sir3p can extend telomeric silencing into regions of chromatin that do not contain Sir2p and Sir4p (Renauld et al. 1993; Hecht et al. 1996) and provides an explanation for our observation that silencing of a reporter gene is restored by targeting enzymatically impaired sir2eso mutant proteins to a single Gal4p DNA-binding site.
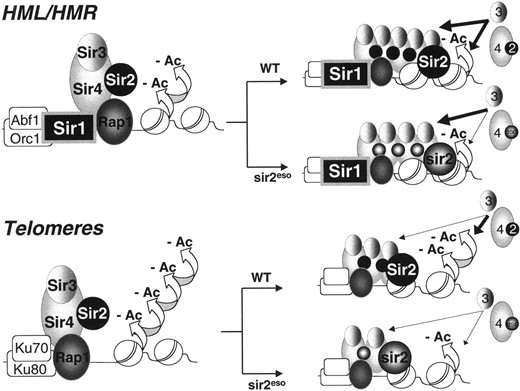
—An activity-sensitive model for HML/HMR and telomeric silencing. The Sir2/4 complex is recruited to the silent mating-type loci and telomeres via Rap1-Sir4p interactions (Sir1p also participates in recruitment at HML/HMR and Ku70p and Ku80p do so at telomeres). After initial recruitment and assembly, Sir2p activity is required to deacetylate (-Ac) histones H3 and H4, thereby recruiting additional Sir3 and Sir4 proteins leading to the spread of condensed chromatin. As the Sir proteins accumulate, subsequent spreading becomes less dependent on Sir2p activity. Although many aspects of sir2esop function support previous models of HM and telomeric chromatin, distinct from these models, sir2esop functions suggest that there may be locus-specific threshold requirements for NAD+-dependent catalytic activity. Thus, robust NAD+-dependent deacetylase activity is not necessary in all circumstances for nucleating stable silenced chromatin. For example, high levels of Sir2p enzymatic activity may not be critical when Sir1p or another targeting molecule such as GBD ensures that the Sir proteins remain associated with the locus. The sir2eso mutants, impaired in enzymatic activity, rely heavily on a targeting factor such as Sir1p or GBD for stable retention of the Sir proteins at the locus and for propagation of silencing.
We suggest that at the HM loci, Sir1p recruits the Sir complex, thereby strengthening Sir-nucleosome interactions and serving as one method of Sir3p recruitment even when Sir2p activity is limiting. Since Sir1p is not present to strengthen Sir-nucleosome interactions at the telomeres, stable spreading of the Sir proteins may rely solely on robust Sir2p enzymatic activity. An occasional successful deacetylation event, which can then be propagated, may underlie the variegation of silencing that is the hallmark of telomeric position effects.
Our model, outlined in Figure 8, may also explain why npt1Δ mutants, required for the nuclear NAD+ salvage pathway, are selectively defective in silencing, only slightly affecting the HM loci (Smith et al. 2000). Perhaps even modest decreases in enzymatic activity have amplified effects at telomeres that would be masked at HML and HMR in the presence of Sir1p.
Is dominance a threshold effect? If it is, this might explain why CEN-plasmid dosage of the sir2eso mutants has dominant phenotypes. In these cases of marginally increased amounts of mutant proteins, a critical threshold of wild-type activity may not be met. One possibility is that even slightly increased amounts of mutant proteins are sufficient to limit availability of Sir2p interacting molecules such as NAD+, acetylated substrates, or Sir4p, thereby interfering with proper Sir2p function. Therefore, although the sir2eso mutants are impaired in enzymatic activity, they may be sufficiently active to initiate stable silencing if a targeting factor such as Sir1p or GBD ensures that the Sir complex remains associated with the locus for the silenced state to be propagated. In the absence of the targeting factor, the sir2eso enzymatic activity may become limiting and unable to overcome a threshold required for stable initiation of chromatin decondensation.
Distinct sir2eso phenotypes in the rDNA underscore mechanistic differences at this locus: Although otherwise similar, the sir2eso mutants differ from one another in their rDNA phenotypic profiles. For instance, although the sir2-G270E and sir2-F296L mutant strains were dominantly defective in rDNA silencing, they differed in their localization and their ability to rescue silencing when tethered to the rDNA array.
The sir2-G270E mutant was defective in rDNA silencing in the presence or absence of Sir1p but was rescued if tethered to the locus via a GBD. In addition, in the absence of Sir1p this mutant protein did not localize normally to the nucleolus. Therefore, this mutant is the only sir2eso mutant that displays similar phenotypes at the HM loci, the telomeres, and the rDNA and appears to be impaired in its ability to associate with chromatin at all three silenced loci. In contrast, the sir2-F296L mutant protein localized properly to the nucleolus in the presence or absence of Sir1p and had decreased levels of silencing and increased levels of recombination (data not shown) in both backgrounds. However, it did not function in silencing at the rDNA when tethered directly to the locus. Therefore, the cause for the sir2-F296L defects in the rDNA likely differs from the impaired associations postulated for it at the telomeres.
In further contrast, sir2-R139K repressed recombination normally at the rDNA (data not shown) in the presence or absence of Sir1p and, within the limits of the silencing bioassay, was even more efficient than wild-type Sir2p at silencing the URA3 reporter. However, synthetically tethering this mutant to the rDNA array via a GBD abrogates its function. These paradoxical effects in rDNA silencing by sir2-R139K may be explained by inefficient interaction with Net1p. Coimmunoprecipitation analyses showed that sir2-R139Kp consistently immunoprecipitated Net1p less efficiently than did wild-type Sir2p. Perhaps the decreased sir2-R139Kp-Net1p interaction allows function of the RENT complex at the rDNA only when the sir2-R139K mutant protein is targeted to the nucleolus exclusively through Net1p. Targeting via a GBD may abolish the already weakened interaction with Net1p and/or interactions with other RENT complex members, thereby disrupting silencing.
Together, these observations underscore and extend the growing view that there are fundamental differences between Sir2p function within the rDNA compared to the HM loci and telomeres. First, rDNA silencing requires a distinct (RENT) complex including Sir2p, Net1p, and Cdc14p (Shou et al. 1999; Straight et al. 1999). Also silenced rDNA repeats are in a region interspersed with highly transcribed rDNA units (reviewed in Hsieh and Fire 2000). It is not clear whether components of the RENT complex, or other proteins as yet unidentified, directly bind histones and thereby target and promote the spread of silenced chromatin at particular rDNA repeats. Such additional targeting might explain why sir2-R139Kp, which is enzymatically impaired and fails to initiate silencing at the telomeres, might still function slightly more efficiently than SIR2 in silencing at the rDNA. The enzymatically inactive mutant sir2-H364Yp did not immunoprecipitate telomeric chromatin efficiently, yet it was able to immunoprecipitate rDNA chromatin (Tanny et al. 1999; Armstrong et al. 2002; Hoppe et al. 2002). Together, these mutant analyses support the idea that optimal levels of NAD+-dependent activity appear to be required for the initiation steps of telomeric silencing but may be of less importance in initiating stable silencing within the rDNA array.
When catalysis is not enough: Other non-null sir2 mutant alleles with locus-specific defects that raise intriguing possibilities, but leave some key unanswered questions, have been described (Sherman et al. 1999; Tanny et al. 1999; Cuperus et al. 2000; Imai et al. 2000; Perrod et al. 2001). One engineered mutant protein in particular, sir2-G270Ap, was shown to have enzymatic activities almost identical to those reported in this study for sir2-G270Ep (Imai et al. 2000; Tanny and Moazed 2001). Despite their enzymatic similarities, there are phenotypic differences between sir2-G270E and sir2-G270A mutants. Although sir2-G270E was defective at all silenced loci unless tethered via a GBD or Sir1p, sir2-G270A was mating proficient, showed partial silencing defects at the telomeres, and silenced as well as wild-type SIR2 at the rDNA (Imai et al. 2000). The amino acid difference at this residue does not appear to cause major changes in relative enzymatic activity and both mutants share an isogenic background. One possible explanation for the phenotypic differences is that sir2-G270A is also an eso mutant. Thus, it might display mating defects only in a sir1Δ background, a condition not tested in the original report (Imai et al. 2000). However, this explanation does not account for the differences observed in silencing within the rDNA array and at the telomeres. Further exploration of the sir2-G270A mutant and its molecular associations may provide additional clues to the mechanism of silencing at the rDNA.
In summary, the sir2eso phenotypes highlight the requirement for a targeting molecule such as Sir1p or GBD to initiate stable silenced chromatin at the HM loci and the telomeres when Sir2p enzymatic activity is limiting. Our studies also identified a residue outside the conserved core domain of Sir2p (R139), which, when mutated, results in impaired enzymatic activity yet wild-type levels of silencing within the rDNA array. The phenotypic differences observed among the sir2eso mutants suggest that wild-type levels of activity, although essential for stable initiation of silencing at the telomeres, are not critical for rDNA silencing and confirm that there are mechanistic differences for Sir2p’s nucleolar functions that require further investigation. Analysis of other sir2 mutants for potential eso phenotypes may reveal additional mutations outside the core domain of SIR2 that can have impaired enzymatic activity yet function in silencing when tethered to the HM loci or to a telomere. Such studies may help further understanding of the unique dependence on Sir1p in establishing silencing and the intricate relationships among enzymatic targeting, chromatin modification, and gene regulation.
Acknowledgement
We thank C. Reifsnyder, M. McVey, and J. Sherman for early contributions to this work; J. Wilson, P. Laurenson, and R. Dutnall for constructive criticism on this manuscript; G. Cuperus, D. Shore, J. Smith, J. Boeke, R. Deshaies, J. Aris, and S. Gasser for reagents; E. Stone and M. Sharp for help with immunofluorescence analysis; J. Landry for advice on NAD+ hydrolysis assays; and the entire Pillus lab for countless contributions. This work was carried out with the support of the National Institutes of Health.
Footnotes
Communicating editor: M. Hampsey
LITERATURE CITED

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}