





Abstract
The Spo12 protein plays a regulatory role in two of the most fundamental processes of biology, mitosis and meiosis, and yet its biochemical function remains elusive. In this study we concentrate on the genetic and biochemical analysis of its mitotic function. Since high-copy SPO12 is able to suppress a wide variety of mitotic exit mutants, all of which arrest with high Clb-Cdc28 activity, we speculated whether SPO12 is able to facilitate exit from mitosis when overexpressed by antagonizing mitotic kinase activity. We show, however, that Spo12 is not a potent regulator of Clb-Cdc28 activity and can function independently of either the cyclin-dependent kinase inhibitor (CDKi), Sic1, or the anaphase-promoting complex (APC) regulator, Hct1. Spo12 protein level is regulated by the APC and the protein is degraded in G1 by an Hct1-dependent mechanism. We also demonstrate that in addition to localizing to the nucleus Spo12 is a nucleolar protein. We propose a model where overexpression of Spo12 may lead to the delocalization of a small amount of Cdc14 from the nucleolus, resulting in a sufficient lowering of mitotic kinase levels to facilitate mitotic exit. Finally, site-directed mutagenesis of highly conserved residues in the Spo12 protein sequence abolishes both its mitotic suppressor activity as well as its meiotic function. This result is the first indication that Spo12 may carry out the same biochemical function in mitosis as it does in meiosis.
PROGRESSION through mitosis requires the precisely timed ubiquitin-dependent destruction of specific substrates. In budding yeast degradation of the anaphase inhibitor Pds1 is required for sister-chromatid separation and therefore the metaphase to anaphase transition (Cohen-Fix et al. 1996). Exit from mitosis is initiated by proteolysis of the mitotic cyclins (King et al. 1996), and destruction of the spindle mid-zone protein, Ase1, is important for the disassembly of the mitotic spindle during mitotic exit (Juang et al. 1997). Destruction of all of these proteins is mediated by a multisubunit complex known as the anaphase-promoting complex (APC; reviewed in Zachariae 1999). The APC is highly conserved in eukaryotes and acts as a ubiquitin ligase, thereby marking proteins for degradation by the proteasome. To ensure the correct order of mitotic events, the APC is controlled at the level of substrate specificity by the highly conserved accessory factors Cdc20 and Hct1/Cdh1. In yeast and human cells, both Cdc20 and Hct1 bind to the APC, but the timing and regulation of their association is controlled differently (Fang et al. 1998; Kramer et al. 1998; Zachariae et al. 1998). Thus, Cdc20 mediates the degradation of the anaphase inhibitor Pds1, whereas Hct1/Cdh1 mediates the destruction of the mitotic cyclins Clb1 and Clb2 (Schwab et al. 1997; Visintin et al. 1998) as well as Ase1 (Visintin et al. 1998).
In conjunction with the regulation of the APC, oscillations in the activities of cyclin-dependent kinases (CDKs) play a major role in governing cell cycle transitions. In the budding yeast Saccharomyces cerevisiae, Cdc28 is the major CDK and is largely responsible for controlling cell cycle progression through its association with various cyclin partners (reviewed in Deshaies 1997). The mitotic cyclin Clb2 is present from S phase until the end of mitosis when, as described above, it is rapidly degraded by APCHct1 (Schwab et al. 1997; Visintin et al. 1998). Like all mitotic cyclins, Clb2 contains a short conserved motif known as the destruction box, which serves as a signal for ubiquitination (Glotzer et al. 1991). Overproduction of Clb2 by removal of its destruction box causes cells to arrest in telophase with divided chromatin, high mitotic kinase activity, and an elongated spindle (Surana et al. 1993). Interestingly, although cells deleted for HCT1 fail to degrade Clb2, they are viable (Schwab et al. 1997; Visintin et al. 1998). This is probably due to the accumulation of the CDK inhibitor Sic1, which inactivates Clb-Cdc28 activity (Mendenhall et al. 1995). Similarly, cells deleted for SIC1are also viable. However, cells disrupted for both HCT1 and SIC1 are inviable, strongly suggesting that APCHct1-mediated degradation of Clb2 and Sic1-mediated inhibition of the Clb2-Cdc28 complex are redundant pathways for CDK inactivation during exit from mitosis (Schwab et al. 1997).
Thus, exit from mitosis requires inactivation of the mitotic kinase by proteolysis of Clb2 and/or accumulation of Sic1. Interestingly, both of these processes are regulated by a set of genes collectively referred to as the mitotic exit network (MEN). The components of the MEN are highly conserved among eukaryotes and include TEM1, LTE1, CDC15, DBF2/20, CDC5, MOB1, and CDC14 (reviewed in Morgan 1999). The proteins encoded by these genes resemble components of signaling pathways: Tem1 is a GTP-binding protein; Lte1 encodes a putative guanine nucleotide exchange factor; Dbf2/Dbf20, Cdc15, and Cdc5 are protein kinases; Mob1 is a novel protein that associates with Dbf2; and Cdc14 is a dual specificity protein phosphatase. In budding yeast, temperature-sensitive mutations in these genes cause cells to arrest in late anaphase/telophase at the restrictive temperature with a common phenotype: large buds, an elongated spindle, divided chromatin, and high Clb2-Cdc28 activity. This phenotype is similar to that observed in yeast expressing a nondegradable form of Clb2 (Surana et al. 1993) and is indicative of the fact that these mutants are defective in Clb2 proteolysis (Jaspersen et al. 1998).
Cdc14 is believed to act at the bottom of the pathway (Jaspersen et al. 1998; Morgan 1999; Jaspersen and Morgan 2000). During nuclear division Cdc14 is released from the nucleolus, leading to dephosphorylation of Hct1, which stimulates APC-dependent degradation of mitotic cyclins (Shou et al. 1999; Straight et al. 1999; Visintin et al. 1999). In addition, by dephosphorylating Sic1 and its transcription factor Swi5, Cdc14 induces accumulation of Sic1 protein, leading to further inactivation of Clb-Cdc28 kinases. The release of Cdc14 from the nucleolus is regulated by the other components of the MEN, but precisely how this is achieved is not yet known.
To identify regulators of mitotic exit, several genetic screens using budding yeast MEN mutants have been performed. Intriguingly, the SPO12 gene has been identified as a multicopy suppressor of mutations in no less than five of the MEN genes: DBF2, TEM1, LTE1, CDC5, and CDC15 (Parkes and Johnston 1992; Toyn and Johnston 1993; Shirayama et al. 1994a,b; Jaspersen et al. 1998). Even a single extra copy of SPO12 is sufficient to suppress the dbf2 defect, suggesting a stoichiometric relationship (Toyn and Johnston 1993). Further indication of a mitotic role for SPO12 is the observation that although cells deleted for SPO12 are viable (mutants exhibit a slight G2/M delay), deletion of SPO12 is synthetically lethal with deletion of DBF2 (Parkes and Johnston 1992) and also with deletion of HCT1 (Grether and Herskowitz 1999). As its name suggests, the SPO12 gene was originally identified as a null mutation causing a defect in sporulation. Mutations in SPO12 cause diploid cells to bypass meiosis I, leading to the formation of asci containing two viable diploid spores or dyads (Klapholz and Esposito 1980; Malavasic and Elder 1990). Thus, SPO12 is likely to be part of a global meiotic control mechanism as well as a key regulator of mitosis. At present, however, there are very few clues to the precise biochemical function of the Spo12 protein during either mitosis or meiosis. The SPO12 gene encodes a small protein of ~20 kD, which is novel and has no homologies to proteins of known function. Its mRNA is regulated in a cell cycle-dependent manner in both mitotic and meiotic cells (Parkes and Johnston 1992; Chu et al. 1998; Spellman et al. 1998). Spo12 protein levels are also cell cycle regulated (Grether and Herskowitz 1999), peaking in G2/M in vegetative cells, supporting a mitotic role for the protein.
Given that high-copy SPO12 is able to suppress a wide variety of mitotic exit mutants, all of which arrest with high Clb-Cdc28 activity, it is tempting to speculate that SPO12 can facilitate exit from mitosis when overexpressed, by antagonizing mitotic kinase activity. In this article, however, we show that Spo12 is not a potent regulator of Clb-Cdc28 activity but is likely to function in a pathway distinct from either Sic1 or Hct1. Spo12 itself is actively degraded in G1 by an APC- and Hct1-dependent mechanism, further emphasizing a specific role for Spo12 during mitotic exit. We also show that in addition to localizing to the nucleus, as has been previously observed (Grether and Herskowitz 1999), Spo12 is a nucleolar protein. We propose a model where overexpression of Spo12 may lead to the delocalization of a small amount of Cdc14 from the nucleolus, resulting in a sufficient lowering of mitotic kinase levels to facilitate mitotic exit.
MATERIALS AND METHODS
Yeast strains and media: Relevant yeast strains and their genotypes are indicated in Table 1. All strains are in a CG378/CG379 congenic background except for those marked with an asterisk. Yeast cell culture and genetic techniques were carried out as described in Guthrie and Fink (1991). Yeast were grown in rich broth supplemented with either 2% dextrose (YPD) or 2% galactose (YPG). Unless otherwise stated, minimal medium contained yeast nitrogen base (Difco, Detroit), the required amino acid supplements, and 2% dextrose (SD). Diploid cells were sporulated on KSM plates (1% potassium, 25 μg/ml zinc acetate, and 2% agar). Yeast transformations were carried out as described in Schiestl and Gietz (1989). Selection of ura3 cells that had lost a URA3 plasmid was carried out by plating on SD plates containing 1 mg/ml 5-fluoroorotic acid (5-FOA; Boeke et al. 1987). Cell cycle arrests were performed using 5 μg/ml α-factor, 0.1 m hydroxyurea (HU), or 15 μg/ml nocodazole (Noc). FACS analysis was carried out using a FACStar flow cytometer (Becton Dickinson, Franklin Lakes, NJ).
Plasmids and DNA manipulations: The SPO12 open reading frame (ORF) was subcloned as a BglII-HindIII PCR fragment
Yeast strains
| Strain name | Relevant genotype | Source |
|---|---|---|
| YLF23 | a SPO12::SPO12-13MYC-KANr | This study |
| RS135-7C | α SPO12::SPO12-13MYC-KANr | RS132-6D × YLF23 |
| KTM208 | a apc2Δ::HIS7 papc2-8 | Kramer et al. (1998) |
| RS1-304 | a hct-Δ::LEU2 | This study |
| RS132-6D | α hct1-Δ::LEU2 | CG379 × RSI-304 |
| RS135-6B | a hct1-Δ1::LEU2 SPO12::SPO12-13 MYC-KANr | RS132-6D × YLF23 |
| MY101 | a cdc28-4 | Segal et al. (1998) |
| RSI-310* | a cdc28-4 hct1-Δ1::LEU2 | This study |
| RS142-1D* | a cdc28-4 SPO12::SPO12-13MYC-KANr | RS135-7C × RSI-310 |
| RS142-13B* | a cdc28-4 hct1-Δ1::LEU2 SPO12::SPO12-13MYC-KANr | RS135-7C × RSI-310 |
| DTY59-1CLB* | α ura3::1× pGAL-CLB2-URA3 | Toyn et al. (1997) |
| DTY59-2CLB* | α ura3::2× pGAL-CLB2-URA3 | Toyn et al. (1997) |
| JD100-1CLB | α ura3::1× pGAL-CLB2-URA3 sic1Δ::TRP1 | Toyn et al. (1977) |
| RSI-100* | α ura3::1× pGAL-CLB2-URA3 spo12Δ::TRP1 | This study |
| S2-2D | α dbf2Δ::LEU2 | Toyn et al. (1991) |
| V378 | a spo12Δ::TRP1 | Parkes and Johnston (1992) |
| V379 | α spo12Δ::TRP1 | Parkes and Johnston (1992) |
| RSI-306 | α spo12Δ::LEU2 | This study |
| JD100 | α sic1Δ::TRP1 | Donovan et al. (1994) |
| CG379sic1::HIS7 | α sic1Δ::HIS7 | K. Kramer |
| L181-6B* | α dbf2-2 | Toyn and Johnston (1993) |
| DK329-4D* | α cdc15-1 | Schweitzer and Philippsen (1991) |
| SCG15-10B | a cdc15-1 | This study |
| SCG15-3D | α cdc15-1 | This study |
| RS1-308* | α cdc15-1 dbf20Δ::KANr | This study |
| RS127-12B | a cdc15-1 hct1-Δ::LEU2 | RSI-304 × SCG15-3D |
| RS128-1C | a cdc15-1 sic1Δ::HIS7 | CG379 sic1::HIS7 × SCG15-10B |
| RS129-6B | a cdc15-1 spo12Δ::LEU2 | RSI-306 × SCG15-10B |
| YDL21 | a RNR2.13MYC::KANr | D. Liger |
| Strain name | Relevant genotype | Source |
|---|---|---|
| YLF23 | a SPO12::SPO12-13MYC-KANr | This study |
| RS135-7C | α SPO12::SPO12-13MYC-KANr | RS132-6D × YLF23 |
| KTM208 | a apc2Δ::HIS7 papc2-8 | Kramer et al. (1998) |
| RS1-304 | a hct-Δ::LEU2 | This study |
| RS132-6D | α hct1-Δ::LEU2 | CG379 × RSI-304 |
| RS135-6B | a hct1-Δ1::LEU2 SPO12::SPO12-13 MYC-KANr | RS132-6D × YLF23 |
| MY101 | a cdc28-4 | Segal et al. (1998) |
| RSI-310* | a cdc28-4 hct1-Δ1::LEU2 | This study |
| RS142-1D* | a cdc28-4 SPO12::SPO12-13MYC-KANr | RS135-7C × RSI-310 |
| RS142-13B* | a cdc28-4 hct1-Δ1::LEU2 SPO12::SPO12-13MYC-KANr | RS135-7C × RSI-310 |
| DTY59-1CLB* | α ura3::1× pGAL-CLB2-URA3 | Toyn et al. (1997) |
| DTY59-2CLB* | α ura3::2× pGAL-CLB2-URA3 | Toyn et al. (1997) |
| JD100-1CLB | α ura3::1× pGAL-CLB2-URA3 sic1Δ::TRP1 | Toyn et al. (1977) |
| RSI-100* | α ura3::1× pGAL-CLB2-URA3 spo12Δ::TRP1 | This study |
| S2-2D | α dbf2Δ::LEU2 | Toyn et al. (1991) |
| V378 | a spo12Δ::TRP1 | Parkes and Johnston (1992) |
| V379 | α spo12Δ::TRP1 | Parkes and Johnston (1992) |
| RSI-306 | α spo12Δ::LEU2 | This study |
| JD100 | α sic1Δ::TRP1 | Donovan et al. (1994) |
| CG379sic1::HIS7 | α sic1Δ::HIS7 | K. Kramer |
| L181-6B* | α dbf2-2 | Toyn and Johnston (1993) |
| DK329-4D* | α cdc15-1 | Schweitzer and Philippsen (1991) |
| SCG15-10B | a cdc15-1 | This study |
| SCG15-3D | α cdc15-1 | This study |
| RS1-308* | α cdc15-1 dbf20Δ::KANr | This study |
| RS127-12B | a cdc15-1 hct1-Δ::LEU2 | RSI-304 × SCG15-3D |
| RS128-1C | a cdc15-1 sic1Δ::HIS7 | CG379 sic1::HIS7 × SCG15-10B |
| RS129-6B | a cdc15-1 spo12Δ::LEU2 | RSI-306 × SCG15-10B |
| YDL21 | a RNR2.13MYC::KANr | D. Liger |
Yeast strains are in a CG378/CG379 congenic background except for those marked with an asterisk.
Yeast strains
| Strain name | Relevant genotype | Source |
|---|---|---|
| YLF23 | a SPO12::SPO12-13MYC-KANr | This study |
| RS135-7C | α SPO12::SPO12-13MYC-KANr | RS132-6D × YLF23 |
| KTM208 | a apc2Δ::HIS7 papc2-8 | Kramer et al. (1998) |
| RS1-304 | a hct-Δ::LEU2 | This study |
| RS132-6D | α hct1-Δ::LEU2 | CG379 × RSI-304 |
| RS135-6B | a hct1-Δ1::LEU2 SPO12::SPO12-13 MYC-KANr | RS132-6D × YLF23 |
| MY101 | a cdc28-4 | Segal et al. (1998) |
| RSI-310* | a cdc28-4 hct1-Δ1::LEU2 | This study |
| RS142-1D* | a cdc28-4 SPO12::SPO12-13MYC-KANr | RS135-7C × RSI-310 |
| RS142-13B* | a cdc28-4 hct1-Δ1::LEU2 SPO12::SPO12-13MYC-KANr | RS135-7C × RSI-310 |
| DTY59-1CLB* | α ura3::1× pGAL-CLB2-URA3 | Toyn et al. (1997) |
| DTY59-2CLB* | α ura3::2× pGAL-CLB2-URA3 | Toyn et al. (1997) |
| JD100-1CLB | α ura3::1× pGAL-CLB2-URA3 sic1Δ::TRP1 | Toyn et al. (1977) |
| RSI-100* | α ura3::1× pGAL-CLB2-URA3 spo12Δ::TRP1 | This study |
| S2-2D | α dbf2Δ::LEU2 | Toyn et al. (1991) |
| V378 | a spo12Δ::TRP1 | Parkes and Johnston (1992) |
| V379 | α spo12Δ::TRP1 | Parkes and Johnston (1992) |
| RSI-306 | α spo12Δ::LEU2 | This study |
| JD100 | α sic1Δ::TRP1 | Donovan et al. (1994) |
| CG379sic1::HIS7 | α sic1Δ::HIS7 | K. Kramer |
| L181-6B* | α dbf2-2 | Toyn and Johnston (1993) |
| DK329-4D* | α cdc15-1 | Schweitzer and Philippsen (1991) |
| SCG15-10B | a cdc15-1 | This study |
| SCG15-3D | α cdc15-1 | This study |
| RS1-308* | α cdc15-1 dbf20Δ::KANr | This study |
| RS127-12B | a cdc15-1 hct1-Δ::LEU2 | RSI-304 × SCG15-3D |
| RS128-1C | a cdc15-1 sic1Δ::HIS7 | CG379 sic1::HIS7 × SCG15-10B |
| RS129-6B | a cdc15-1 spo12Δ::LEU2 | RSI-306 × SCG15-10B |
| YDL21 | a RNR2.13MYC::KANr | D. Liger |
| Strain name | Relevant genotype | Source |
|---|---|---|
| YLF23 | a SPO12::SPO12-13MYC-KANr | This study |
| RS135-7C | α SPO12::SPO12-13MYC-KANr | RS132-6D × YLF23 |
| KTM208 | a apc2Δ::HIS7 papc2-8 | Kramer et al. (1998) |
| RS1-304 | a hct-Δ::LEU2 | This study |
| RS132-6D | α hct1-Δ::LEU2 | CG379 × RSI-304 |
| RS135-6B | a hct1-Δ1::LEU2 SPO12::SPO12-13 MYC-KANr | RS132-6D × YLF23 |
| MY101 | a cdc28-4 | Segal et al. (1998) |
| RSI-310* | a cdc28-4 hct1-Δ1::LEU2 | This study |
| RS142-1D* | a cdc28-4 SPO12::SPO12-13MYC-KANr | RS135-7C × RSI-310 |
| RS142-13B* | a cdc28-4 hct1-Δ1::LEU2 SPO12::SPO12-13MYC-KANr | RS135-7C × RSI-310 |
| DTY59-1CLB* | α ura3::1× pGAL-CLB2-URA3 | Toyn et al. (1997) |
| DTY59-2CLB* | α ura3::2× pGAL-CLB2-URA3 | Toyn et al. (1997) |
| JD100-1CLB | α ura3::1× pGAL-CLB2-URA3 sic1Δ::TRP1 | Toyn et al. (1977) |
| RSI-100* | α ura3::1× pGAL-CLB2-URA3 spo12Δ::TRP1 | This study |
| S2-2D | α dbf2Δ::LEU2 | Toyn et al. (1991) |
| V378 | a spo12Δ::TRP1 | Parkes and Johnston (1992) |
| V379 | α spo12Δ::TRP1 | Parkes and Johnston (1992) |
| RSI-306 | α spo12Δ::LEU2 | This study |
| JD100 | α sic1Δ::TRP1 | Donovan et al. (1994) |
| CG379sic1::HIS7 | α sic1Δ::HIS7 | K. Kramer |
| L181-6B* | α dbf2-2 | Toyn and Johnston (1993) |
| DK329-4D* | α cdc15-1 | Schweitzer and Philippsen (1991) |
| SCG15-10B | a cdc15-1 | This study |
| SCG15-3D | α cdc15-1 | This study |
| RS1-308* | α cdc15-1 dbf20Δ::KANr | This study |
| RS127-12B | a cdc15-1 hct1-Δ::LEU2 | RSI-304 × SCG15-3D |
| RS128-1C | a cdc15-1 sic1Δ::HIS7 | CG379 sic1::HIS7 × SCG15-10B |
| RS129-6B | a cdc15-1 spo12Δ::LEU2 | RSI-306 × SCG15-10B |
| YDL21 | a RNR2.13MYC::KANr | D. Liger |
Yeast strains are in a CG378/CG379 congenic background except for those marked with an asterisk.
into the BamHI/HindIII sites of pEMBLYex4.6HIS (pGAL, 2μ, URA3). This generated plasmid pGAL6HIS-SPO12, which contains SPO12 N-terminally tagged with a 6HIS epitope, under the control of the inducible GAL promoter. For high-copy-number expression of SPO12 under the control of its own promoter, a 1.7-kb PvuII genomic restriction fragment (containing the entire SPO12 gene and >1 kb of upstream promoter sequence) was cloned into the PvuII site of YEplac-195 and YEplac181 (2μ; Gietz and Sugino 1988) to generate plasmids YEpSPO12(URA3) and YEpSPO12(LEU2), respectively. Multicopy expression of SIC1 was achieved by plasmids YEpSDB25 (2μ, URA3) and YEplacSDB25 (2μ, LEU2), which have been described previously (Donovan et al. 1994).
C-terminal tagging of SPO12 with 13MYC (strain YLF23) was achieved by the direct chromosomal integration of a PCR fragment (Longtine et al. 1998). The PCR fragment was generated by primers SPO12-F2 (5′-GAT TCA GAG GAT GTA GAA ATC GAT GAA GAT GAG GAG TAT TTC TAC CGG ATC CCC GGG TTA ATT AA-3′) and SPO12-R1 (5′-GGT TTA GTG TAG CAT TTG GCT ATT TTT GGA TGA CTA GAA AGG CAG GAA TTC GAG CTC GTT TAA AC-3′), with plasmid pFA6a-13Myc-kanMX6 as template (Longtine et al. 1998). Underlined regions represent homology with the vector template. Correct integration was confirmed by diagnostic colony PCR analysis and expression of Spo12-13Myc protein was verified by Western analysis. The functionality of Spo12-13Myc was tested by two independent methods: First, in a cross between YLF23 and S2-2D (a dbf2Δ strain; Toyn et al. 1991), spores containing both SPO12-13MYC and dbf2Δ were viable, indicating that the epitope-tagged Spo12 protein was functional (deletion of SPO12 is synthetically lethal with deletion of DBF2; Parkes and Johnston 1992). Second, strain YLF23 was crossed with V379 (a spo12Δ strain). The resulting diploid cells were sporulated on KSM plates and produced normal asci, i.e., containing four haploid spores, indicating that Spo12-13Myc was functioning normally in meiosis.
The SPO12 gene was deleted from strain DTY-1CLB by transformation with PvuII-digested pBSSPO12Δ::TRP1 plasmid (Parkes and Johnston 1992) to generate strain RSI-100. To produce strain RSI-306, a PCR-based gene disruption technique was used to delete the SPO12 gene from CG379 cells, with the YDpLEU plasmid as template (Berben et al. 1991). Primers used were SPO12KO5 (5′-AGT CTA AAC CGC CAT TTC AAA CTA GTT TTT AGG AGG TCA GCC GAA TTC CCG GGG ATC CGG TGA-3′) and SPO12KO3 (5′-GGT TTA GTG TAG CAT TTG GCA ATT TTT GGA TGA CTA GAA AGG CAG GCA GG TCG ACG GAT CCG GTG A-3′). Underlined regions indicate homology with plasmid YDpLEU. Disruption of SPO12 by either method was confirmed by diagnostic colony PCR. In addition, transformants, which appeared to have lost SPO12, were further tested by mating with the spo12Δ strain V378 to generate diploids. Subsequent sporulation of these diploids gave rise to asci containing only two diploid spores, a phenotype that is displayed only when both chromosomal copies of SPO12 are absent.
The HCT1 ORF was removed from CG378 cells, resulting in strain RSI-304, via transformation with PvuII-digested pWS176 plasmid (Schwab et al. 1997). HCT1 was deleted from MY101 (Segal et al. 1998), with the same method, to generate strain RSI-310. Strain CG379 sic1Δ::HIS7 (courtesy of K. Kramer) was constructed by transformation of CG379 with AatII/SphI-digested YIpΔsic1::HIS7 plasmid. Deletion of the DBF20 ORF from DK329-4D cells (Schweitzer and Philippsen 1991) to produce strain RSI-308 was carried out, with direct chromosomal integration of a PCR fragment generated using primers DBF20-F1 (5′-TGA TTA GCA ACC CAG AAT ATC GTA TTC AAC AAT AAT TC-3′) and DBF20-R1 (5′-TCA AAA TGG AAA ATG TTA GCA CAT CTT GTT GCT GTT GT-3′) and using plasmid pFA6a-kanMX6 as template (Longtine et al. 1998). In all cases, disruption of the desired gene was confirmed by diagnostic colony PCR analysis of transformants.
Site-directed mutagenesis of SPO12: Mutagenesis was carried out using the ExSite PCR-based site-directed mutagenesis kit (Stratagene, La Jolla, CA). Plasmid pBSSPO12-MCS (Parkes and Johnston 1992) containing the entire SPO12 ORF was used as template together with specifically designed mutagenic primers. The position and nature of the mutations introduced into the SPO12 ORF are indicated in Figure 5. Mutants MutAA and MutEE were generated by replacement of S118 and S125 with alanine residues and glutamic acid residues, respectively, and MutC127A was produced by changing C127 to an alanine. Mutations in the putative cyclin destruction box of SPO12, to produce MutDB, consisted of the replacement of R99 and L102 with alanine residues. Mutant MutKEN was generated by changing K58, E59, and N60 (of the putative KEN-box sequence KENXXXD/N) to alanine residues. Correct introduction of each of the desired mutations was verified by DNA sequence analysis. For multicopy expression, BamHI-HindII fragments from mutants MutAA, MutEE, and MutC127A were used to replace the wild-type BamHI-HindII segment from plasmid YEpSPO12(LEU2). For experiments requiring inducible expression, the mutated SPO12 ORFs were subcloned as BglII-HindIII PCR fragments into the BamHI/HindIII sites of vector pEMBLYex4.6HIS (pGAL, 2μ, URA3). This generated plasmids that contained 6HIS-epitope-tagged fusions of the mutated SPO12 sequences under the control of the inducible GAL promoter.
Preparation of crude yeast extracts and protein analysis: Unless otherwise stated, cells were grown to midlog phase, harvested, and cell pellets (2 × 108 cells) were resuspended in 100 μl breaking buffer containing 50 mm Tris-HCl pH 7.5, 150 mm NaCl, 15 mm MgCl2, 5 mm EDTA, 1% NP-40, 10 μg/ml pepstatin, 87 μg/ml phenylmethylsulfonyl fluoride, 1 μm DTT, and 2× complete protease inhibitor cocktail (Boehringer Mannheim, Mannheim, Germany). Two volumes of 0.5-mm glass beads were added to cell suspensions and cells were lysed using a RiboLyser cell disrupter (Hybaid, Ashford, Middlesex, United Kingdom). Lysates were separated from beads by brief centrifugation, and protein concentrations were determined using the Bio-Rad (Richmond, CA) Bradford assay. For Western blot analysis, 50 μg of protein in urea sample buffer (Printen and Sprague 1994) was resolved by SDS-PAGE and transferred to Protran nitrocellulose membrane (Schleicher & Schuell, Dassel, Germany). Proteins were detected using chemiluminescence detection (ECL; Amersham, Arlington Heights, IL), after probing with primary antibodies RGS-HIS (mouse; QIAGEN, Valencia, CA), 9E10 (mouse anti-c-myc; BAbCO, Richmond CA), anti-Clb2 (rabbit; a gift from C. Mann, CEA/Saclay), or TAT-1 (mouse antitubulin; a gift from K. Gull).
Clb2-Cdc28 kinase assays: Protein extracts (200 μg) were immunoprecipitated with 1 μg of anti-Clb2 (described above) on a rotating wheel for 1 hr at 4°. Protein A Sepharose beads (Pharmacia, Piscataway, NJ) were added and incubation was continued at 4° with rotation for a further 1 hr. The protein A beads immune complex was washed three times with breaking buffer and twice with kinase buffer (25 mm MOPS pH 7.2, 10 mm MgCl2) and was incubated for 20 min at room temperature with 10 μl kinase buffer containing 5 μg histone HI (Sigma, St. Louis), 50 μm ATP, and 0.1 μl [γ-32P]ATP (10 mCi/ml). The reaction was stopped by adding Laemmli buffer, and phosphorylated proteins were analyzed by SDS-PAGE and autoradiography.
Indirect immunofluorescence: Immunolocalization of Spo12-13Myc, Rnr2-13Myc, and Nop2 was performed essentially as detailed in Pringle et al. (1991). Spo12-13Myc and Rnr2-13Myc were detected using the 9E10 primary antibody (1:50 dilution) and a donkey FITC-conjugated anti-mouse IgG secondary antibody (1:50; Jackson ImmunoResearch Laboratories, West Grove, PA). Nop2 was detected with an affinity-purified rabbit anti-Nop2p antibody (1:100 dilution; de Beus et al. 1994), followed by an AlexaFluor 568-conjugated goat anti-rabbit IgG secondary antibody (1:400 dilution; Molecular Probes, Eugene, OR). Cells were mounted in Vectashield mounting medium containing 1.5 μg/ml 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories, Burlingame, CA) and observed using a Photometrics CH350L liquid-cooled CCD camera on an Olympus IX70 inverted microscope, with a ×100 magnification U-PLAN-APO 1.35 NA oil objective lens. Images were captured and manipulated using SoftWoRx software (Applied Precision, Issaquah, WA) and Adobe PhotoShop version 5.5 (Adobe Systems, Mountain View, CA).
RESULTS
Spo12 protein degradation in G1 is dependent on the APC: During mitosis the SPO12 gene is expressed under cell cycle control coordinately with the DBF2 gene (Parkes and Johnston 1992; Chu et al. 1998; Spellman et al. 1998). To examine Spo12 protein levels, we constructed a strain expressing a 13Myc epitope-tagged version of Spo12 from the endogenous promoter. This version of Spo12 was found to be fully functional by the following criteria. First, the growth rate and FACS profile of strains containing Spo12-13Myc was no different from that of an isogenic wild type (data not shown), whereas spo12Δ strains grow more slowly and show an accumulation of 2C cells (Parkes and Johnston 1992). Second, spore clones containing both a DBF2 deletion and SPO12-13Myc were found to be viable (data not shown). Since dbf2Δ spo12Δ double mutants are inviable, this result strongly suggests that the presence of the 13Myc tag was not deleterious to the normal function of Spo12 in mitosis. Third, the meiotic function of Spo12 was also unaffected since diploid strains carrying SPO12-13MYC not only sporulated at wild-type frequencies (data not shown), but a diploid strain deleted for one of its copies of the SPO12 gene and carrying SPO12-13MYC in place of the other produced normal four-spored asci on sporulation.
We next examined Spo12 protein levels and found these to be cell cycle regulated (data not shown), as previously reported by Grether and Herskowitz (1999). Protein levels are low in G1, rise to a peak in mitosis, and decrease as cells enter the next G1 phase. Thus, the pattern of Spo12 protein expression parallels the cell cycle regulation of SPO12 mRNA (Parkes and Johnston 1992), suggesting that the protein is highly unstable or is actively degraded in late anaphase/G1. Consistent with this, Spo12 is undetectable in G1-arrested cells and readily detectable in cells arrested in mitosis (Figure 1A). Interestingly, degradation of the mitotic proteins Clb2, Cdc5, and Cdc20, which exhibit similar expression profiles to Spo12, is carried out by the APC in anaphase/G1 (Irniger and Nasmyth 1997; Charles et al. 1998; Shirayama et al. 1998). We therefore investigated the possibility that Spo12 stability is regulated in G1 by the APC.
For this purpose we analyzed the stability of Spo12
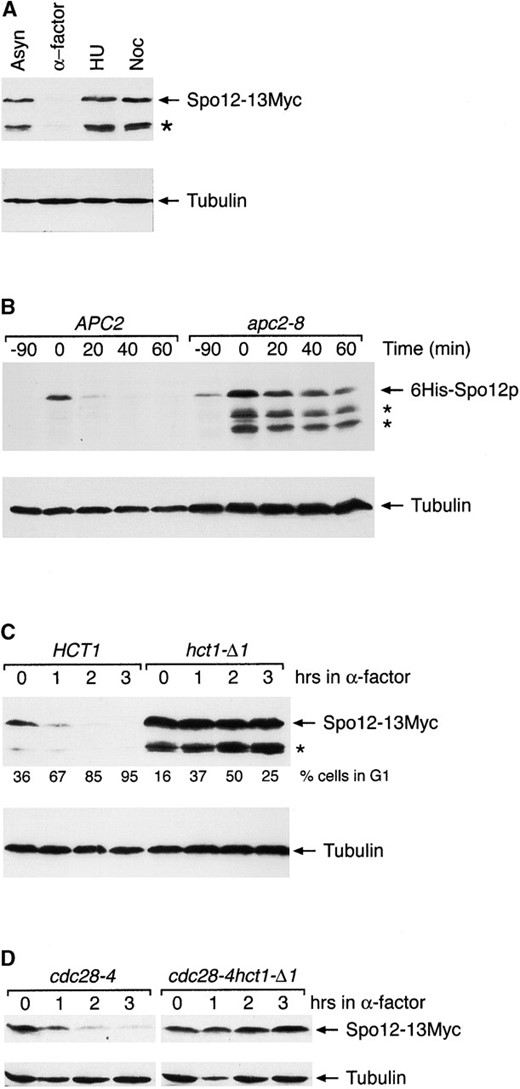
Spo12 degradation in G1 is dependent on the APC and Hct1. (A) Spo12 is absent in G1-arrested cells. YLF23 cells expressing Spo12-13Myc were arrested at specific stages of the cell cycle using chemicals as follows: G1 with α-factor (α-factor), S-phase with hydroxyurea (HU), or mitotic arrest with nocadazole (Noc). Samples for extract preparation were taken when >95% of cells were appropriately arrested. A sample from an asynchronous (Asyn) culture was included for reference. Spo12-13Myc (top) and tubulin (bottom) were detected by immunoblotting with anti-Myc 9E10 and anti-tubulin TAT-1 antibodies, respectively. Degradation products are indicated by an asterisk. (B) Spo12 degradation in G1 is dependent on the APC. Wild-type (CG378) and apc2-8 cells (KTM208) containing plasmid pGAL6HIS-SPO12 were grown in selective medium containing sucrose at 25°. Cells were arrested in G1 with 5 μg/ml α-factor and then shifted to 37° for 1 hr. 6HIS-SPO12 was induced with 4% galactose for 90 min, followed by addition of dextrose (4%) and cycloheximide (10 μg/ml), to repress transcription and translation, respectively. Samples for extract preparation were taken at the indicated times after repression. 6His-Spo12 (top) and tubulin (bottom) were detected by immunoblotting with anti-RGS.His and TAT-1 antibodies, respectively. Degradation products are indicated by asterisks. (C) Spo12 degradation is Hct1 dependent in α-factor-arrested cells. Wild-type and hct11-Δ1 cells (RS135-6B) containing integrated Spo12-13Myc were grown in YPD at 25°. Cells were arrested with 5 μg/ml α-factor, and samples for extract preparation were taken at the indicated times after α-factor addition. Samples for FACS analysis were also taken, and the percentage of cells in G1 at each time point is indicated. Spo12-13Myc (top) and tubulin (bottom) were detected as in A. Degradation products are indicated by an asterisk. (D) Spo12 degradation is Hct1 dependent in cdc28-arrested cells. Strains RS142-1D (cdc28-4) and RS142-13B (cdc28-4 hct1-Δ1) containing integrated Spo12-13Myc were grown in YPD at 25°. Cells were shifted to 35° to induce a G1 arrest, and samples were taken at the indicated times for extract preparation. At the 3-hr time point, >95% of cells in both cultures were arrested. Spo12-13Myc (top) and tubulin (bottom) were detected as in A.
after transient induction of 6HIS-SPO12 from the GAL promoter, in α-factor-arrested cells (Figure 1B). Spo12 was rapidly destroyed in wild-type cells, with the bulk of protein disappearing within 20 min of repression of transcription and translation by addition of dextrose and cycloheximide, respectively. In contrast, Spo12 was stable in cells carrying an apc2-8 mutation, which inactivates APC function at 37°. Thus Spo12 degradation in G1 is dependent on the APC.
Hct1/Cdh1 controls the degradation of Spo12: The specificity of the two forms of APC in mitosis is controlled by two related accessory factors, Cdc20 and Hct1/Cdh1 (Schwab et al. 1997; Visintin et al. 1997; Lim et al. 1998; Shirayama et al. 1998). We next investigated whether Spo12 degradation is dependent on Cdc20 or Hct1. The stability of Spo12, like that of Clb2, was unaffected during G1 arrest in a cdc20-1 strain held at the restrictive temperature when compared to wild-type cells (data not shown). Cultures of wild-type and hct1-Δ1 mutant cells were treated with α-factor and Spo12 protein levels were monitored as cells arrested in G1 (Figure 1C). In wild-type cells, the amount of Spo12 was dramatically reduced within 1 hr of α-factor addition (67% of cells were by then in G1). At later time points, Spo12 was undetectable in these cells. In contrast, Spo12 protein levels remained constant in hct1-Δ1 cells. At the 2-hr time point, where 50% of cells were in G1, no loss of Spo12 protein had occurred. This strongly suggests that Spo12 degradation is regulated by Hct1.
Cells containing the hct1-Δ1 mutation arrest only transiently in G1 following α-factor treatment and then re-replicate their DNA (Schwab et al. 1997). To circumvent this problem and to confirm our data, instead of using α-factor, we arrested cells in G1 by use of the cdc28-4 mutation (Figure 1D). As in the α-factor-arrested cells, Spo12 was quickly destroyed in cdc28-4-arrested cells. In cdc28-4 hct1-Δ1 double mutants, however, Spo12 protein was greatly stabilized. We conclude, therefore, that Spo12 degradation in G1 is dependent on both the APC and Hct1.
Almost all APC substrates identified so far contain sequences known as destruction boxes (Glotzer et al. 1991; King et al. 1996). Mutation of a conserved arginine and leucine in this sequence can prevent APC-mediated proteolysis. A single putative destruction box was identified in Spo12 (see Figure 5). However, mutation of the relevant arginine and leucine residues to alanine resulted in the Spo12 protein being turned over more rapidly in G1 than the wild-type protein (data not shown). This result suggests that APCHct1 may recognize Spo12 through other sequences. A candidate is the KEN-box sequence, which was recently shown to be responsible for targeting the human Cdc20 protein for destruction by APCHct1 (Pfleger and Kirschner 2000). KEN-box-mediated destruction of yeast proteins has not yet been reported but Spo12 contains a possible KEN-box sequence (see Figure 5). All three residues of this were mutated to alanines but, once again, this greatly destabilized the protein (data not shown). It is likely that the introduced mutations lead to deleterious changes in the tertiary structure of Spo12 and result in its rapid nonspecific turnover. Therefore, at present we cannot exclude the possibility that the destruction box and/or KEN-box sequences are involved in the APCHct1-mediated degradation of Spo12.
Spo12 does not control Clb-Cdc28 kinase activity: SPO12 has been isolated as a high-copy suppressor of a number of mutations affecting cell cycle progression such as dbf2-2, cdc15-1, cdc5-1, and tem1-3 (Parkes and Johnston 1992; Shirayama et al. 1996; Jaspersen et al. 1998). All of these mutants are defective in the degradation of Clb2 and arrest late in mitosis with high Clb-Cdc28 activity. These observations suggest that SPO12 could be facilitating exit from mitosis by lowering mitotic kinase activity. Sic1 is a known CDK inhibitor and inactivates the Clb-Cdc28 kinase by direct interaction (Mendenhall et al. 1995). Interestingly, almost all the mitotic exit mutants that are rescued by multicopy SPO12 are also rescued by multicopy SIC1 (Toyn et al. 1997; Jaspersen et al. 1998), further suggesting that overexpression of Spo12 facilitates the inactivation of the mitotic kinase.
Cells deleted for either SIC1 or HCT1 are hypersensitive to elevated levels of Clb2, which is consistent with their known functions (Schwab et al. 1997; Toyn et al. 1997). We investigated whether spo12Δ mutants share this phenotype, by using strains containing integrated copies of CLB2 under the control of the inducible GAL promoter (Figure 2A). In agreement with previous published data, we observed that two copies of GAL-CLB2 are toxic to wild-type cells grown on galactose. A single copy of GAL-CLB2 is tolerated under these growth conditions but in the absence of SIC1 cells is hypersensitive to elevated levels of Clb2 (Toyn et al. 1997 and Figure 2A). This was not the case with SPO12, as spo12Δ cells carrying a single copy of GAL-CLB2 were viable on galactose. Thus, in the absence of SPO12, cells are not hypersensitive to Clb2 overproduction, indicating that spo12Δ mutants are unlikely to be defective in deactivation of the mitotic kinase.
To investigate this further, we looked at the effect of SPO12 overexpression on the lethality incurred by ectopic expression of Clb2. Multicopy plasmids carrying either SIC1 or SPO12 were introduced into a strain carrying two integrated copies of the CLB2 gene under the control of the inducible GAL promoter (Figure 2B). The toxic effect of expressing two copies of GAL-CLB2 is clearly prevented by multicopy SIC1, consistent with its known function as a CDKi. However, multicopy SPO12 was unable to rescue this phenotype. This is a surprising result and indicates that SPO12 overexpression is unable to significantly reduce Clb2-Cdc28 kinase activity.
This result was confirmed biochemically (Figure 2C). Here, cells were transformed with a multicopy plasmid containing the SPO12 gene under the control of the GAL promoter. Overproduction of Spo12 was induced by the addition of galactose to the media, and samples were taken for Western analysis and kinase assays. Both the endogenous Clb2 protein levels and Clb2-Cdc28 kinase levels remained constant, despite the large accumulation of Spo12 protein. The result was the same even after prolonged periods of galactose induction (5–24 hr; data not shown). Moreover, this overexpression of Spo12 had no detectable effect on the viability, growth rate, or morphology of the cells (our unpublished results). This is in stark contrast to overproduction of Sic1, which results in G1/G2 cell cycle delays, elongated multibudded cells (Nishizawa et al. 1998; Sanchez-Diaz et al. 1998), and overproduction of Hct1, which is lethal, causing cells to arrest in G2 (Schwab et al. 1997; Visintin et al. 1997). Our data, therefore, strongly indicate that Spo12 does not play a significant role in the normal regulation of Clb-Cdc28 kinase activity.
Spo12 can function independently of either Sic1 or
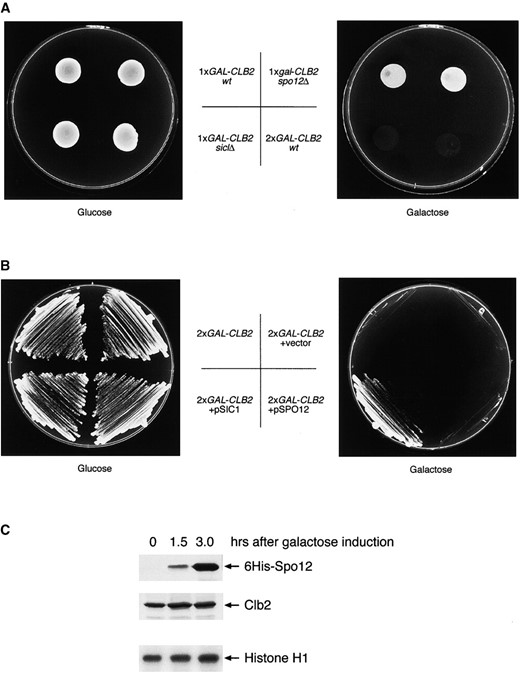
Clb2-Cdc28 kinase activity is unaffected by SPO12 deletion or overexpression. (A) Cells deleted for SPO12 are not sensitive to CLB2 overexpression. The SPO12 gene was deleted from a strain containing one integrated copy of GAL-CLB2, to create strain RSI-100. Strain RSI-100 and control strains DTY59-1CLB (1 × GAL-CLB2), DTY59-2CLB (2 × GAL-CLB2), and JD100-1CLB (1 × GAL-CLB2 sic1Δ) were grown in YPD and 5-μl aliquots were spotted onto YPD (glucose) and YPG (galactose) plates. Plates were incubated at 25° for 1 (YPD) or 2 (YPG) days. (B) Multicopy SIC1, but not SPO12, rescues mitotic arrest caused by CLB2 overexpression. Strain DTY59-2CLB, containing two integrated copies of the CLB2 gene under the control of the GAL promoter, was transformed with empty vector (YEplac181; LEU2) or multicopy plasmids containing either SPO12 (YEpSPO12; LEU2) or SIC1 (YEplacSDB25; LEU2). Transformants were streaked for single colonies on YPD and YPG plates and grown at 25° for 1 (YPD) or 2 (YPG) days. (C) Clb2 protein and associated kinase levels are unaffected by SPO12 overexpression. Wild-type cells transformed with plasmid pGAL6HIS - SPO12 were grown in selective medium containing sucrose at 25°. Production of 6His-Spo12 was induced with 2% galactose, and samples were taken at the indicated times for extract preparation. 6His-Spo12 (top) and Clb2 protein levels (middle) were detected by immunoblotting with anti-RGS.His and anti-Clb2 antibodies, respectively. Cdc28-Clb2 kinase assays (bottom) are described in materials and methods, with histone HI as substrate.
Hct1: If Spo12 does not regulate Clb-Cdc28 kinase activity, how is its overproduction able to suppress such a wide range of mitotic exit mutants? One possibility is that SPO12 overexpression leads to a limited activation of one of the known pathways required for inactivation of mitotic kinase and only at a specific stage in the cell cycle, i.e., late anaphase/telophase where MEN mutants arrest. Obvious candidates are the redundant Sic1 and Hct1 pathways. A prediction of this hypothesis is that if the correct pathway is inactivated, then multicopy SPO12 would no longer be able to rescue the arrest of a mitotic exit mutant. To examine this, we introduced various gene disruptions into a cdc15-1 background and investigated whether 2μ SPO12 was still able to overcome the cdc15-1-mediated arrest at the restrictive temperature. Empty vector and 2μ SIC1 served as negative and positive controls, respectively. SPO12 was able to rescue the cdc15-1 mutation in the absence of either SIC1 or HCT1; i.e., overexpression of SPO12 rescued growth of both the cdc15-1 sic1Δ and cdc15-1 hct1-Δ1 double mutants at (and above) the restrictive temperature (Table 2). This suggests that Spo12 can function independently of either Sic1 or Hct1. 2μ SIC1 is also able to suppress the lethality of the cdc15-1 spo12Δ double mutant at 37°.
Multicopy SPO12 is able to suppress the cdc15-1 mutation in combination with other gene disruptions
| Rescue of growth at restrictive temperature by multicopy plasmida | ||||
|---|---|---|---|---|
| Strain genotype | Minimum restrictive temperature | Vector | SPO12 | SIC1 |
| cdc 15-1 | 35° | − | + | + |
| cdc15-1 sic1Δ::HIS7 | 30° | − | + (33.5) | + (35) |
| cdc15-1 hct1-Δ1::LEU2 | 30° | − | + (35) | + (35) |
| cdc15-1 spol2Δ::LEU2 | 30° | − | + (35) | + |
| cdc15-1 dbf20Δ::KANr | 33.5° | − | + (35) | + |
| Rescue of growth at restrictive temperature by multicopy plasmida | ||||
|---|---|---|---|---|
| Strain genotype | Minimum restrictive temperature | Vector | SPO12 | SIC1 |
| cdc 15-1 | 35° | − | + | + |
| cdc15-1 sic1Δ::HIS7 | 30° | − | + (33.5) | + (35) |
| cdc15-1 hct1-Δ1::LEU2 | 30° | − | + (35) | + (35) |
| cdc15-1 spol2Δ::LEU2 | 30° | − | + (35) | + |
| cdc15-1 dbf20Δ::KANr | 33.5° | − | + (35) | + |
Mutant strains were transformed at 25° with the indicated gene on a 2μ plasmid. LEU2 or URA3 plasmids were used, depending on the genotype of the strain being transformed.
Suppression of each mutant arrest was analyzed by streaking for single colonies on SD selective plates at 25°, 30°, 33.5° and 35°. Failure (−) or ability (+) of multicopy plasmids to rescue growth of strains at their respective minimum restrictive temperatures is indicated. The maximum temperature of rescue, if above the minimum restrictive temperature, is indicated in parentheses. In a control experiment, all mutant strains showed robust growth at 35° when transformed with a 2μ plasmid carrying the CDC15 gene (data not shown).
Multicopy SPO12 is able to suppress the cdc15-1 mutation in combination with other gene disruptions
| Rescue of growth at restrictive temperature by multicopy plasmida | ||||
|---|---|---|---|---|
| Strain genotype | Minimum restrictive temperature | Vector | SPO12 | SIC1 |
| cdc 15-1 | 35° | − | + | + |
| cdc15-1 sic1Δ::HIS7 | 30° | − | + (33.5) | + (35) |
| cdc15-1 hct1-Δ1::LEU2 | 30° | − | + (35) | + (35) |
| cdc15-1 spol2Δ::LEU2 | 30° | − | + (35) | + |
| cdc15-1 dbf20Δ::KANr | 33.5° | − | + (35) | + |
| Rescue of growth at restrictive temperature by multicopy plasmida | ||||
|---|---|---|---|---|
| Strain genotype | Minimum restrictive temperature | Vector | SPO12 | SIC1 |
| cdc 15-1 | 35° | − | + | + |
| cdc15-1 sic1Δ::HIS7 | 30° | − | + (33.5) | + (35) |
| cdc15-1 hct1-Δ1::LEU2 | 30° | − | + (35) | + (35) |
| cdc15-1 spol2Δ::LEU2 | 30° | − | + (35) | + |
| cdc15-1 dbf20Δ::KANr | 33.5° | − | + (35) | + |
Mutant strains were transformed at 25° with the indicated gene on a 2μ plasmid. LEU2 or URA3 plasmids were used, depending on the genotype of the strain being transformed.
Suppression of each mutant arrest was analyzed by streaking for single colonies on SD selective plates at 25°, 30°, 33.5° and 35°. Failure (−) or ability (+) of multicopy plasmids to rescue growth of strains at their respective minimum restrictive temperatures is indicated. The maximum temperature of rescue, if above the minimum restrictive temperature, is indicated in parentheses. In a control experiment, all mutant strains showed robust growth at 35° when transformed with a 2μ plasmid carrying the CDC15 gene (data not shown).
Thus, Spo12 and Sic1 are not only able to function in different pathways, but they are clearly not dependent on each other for their suppression of mitotic exit mutants. These conclusions contrast with those of Grether and Herskowitz (1999), who were unable to detect rescue of either cdc15-1 sic1Δ by overexpressed SPO12 or of cdc15-1 spo12Δ by overexpressed SIC1. This discrepancy may reflect strain differences but, on the other hand, these workers carried out all their suppression studies at 37°. It is possible that this temperature may have been too stringent for the mutant strains, and therefore rescue of growth was not observed. In our experiments, we often found that the restrictive temperatures of the double mutants were much lower than that of the cdc15-1 single mutation (for example, 30° compared to 35°; see Table 2). For this reason our suppressor studies were carried out at a range of temperatures from 25° to 35°.
Further proof that Spo12 can function independently of either Sic1 or Hct1 comes from the synthetic interactions between the relevant genes. A strain containing a deletion of either HCT1 or SIC1 is viable but deletion of both is lethal (Schwab et al. 1997). Moreover, hct1-Δ1 and spo12Δ are also a lethal combination (Grether and Herskowitz 1999; our unpublished observations). On the other hand, spo12Δ sic1Δ mutants are viable (Grether and Herskowitz 1999) and without a synthetic phenotype (our unpublished observations). This might imply that Spo12 and Sic1 could be acting in the same pathway. However, our suppression data (Table 2) suggest otherwise. In addition, multicopy SIC1 is able to rescue the lethality of the spo12Δ hct1-Δ1 double mutation (Figure 3A). hct1-Δ1 and spo12Δ mutant strains were crossed in the presence of a 2μ SIC1 plasmid. Tetrad analysis showed that >95% of spores deleted for both HCT1 and SPO12 were now viable in the presence of multicopy SIC1. We found that ~60% of these double mutants were unable to survive in the absence of the plasmid (examples are shown in Figure 3A). A similar ratio of lethality was observed in crosses of spo12Δ and hct1-Δ1 in CG378/9 and W303 backgrounds (not shown). In contrast to this result, we found that multicopy SPO12 is unable to rescue the synthetic lethality of hct1-Δ1 sic1Δ double mutants (data not shown). So, although Spo12 can act in the absence of either Sic1 or Hct1, it is still dependent on at least one of these pathways being available for it to carry out its function.
Other lethal combinations of late mitotic gene disruptions include dbf2Δ sic1Δ (Donovan et al. 1994) and dbf2Δ spo12Δ (Parkes and Johnston 1992). The arrest phenotype of these cells as dumbbells indicates a failure in completion of mitosis and suggests that Dbf2 and Sic1 between them perform an essential function, as do Dbf2 and Spo12. By genetic analysis, we found that multicopy SPO12 was able to rescue the lethality of dbf2Δ sic1Δ, and, conversely, multicopy SIC1 rescued the lethality of dbf2Δ spo12Δ (Figure 3B).
While all of the genetic data so far presented could be interpreted as Spo12 functioning in a mitotic exit pathway distinct from either Sic1 or Hct1, they are also consistent with Spo12 impinging on part of the MEN regulatory pathway upstream of both Sic1 and Hct1.
Spo12 function is dependent on either Dbf2 or Dbf20: Since Spo12 may act in a novel late mitotic pathway, it is important to determine what other genes, if any, might lie in the same pathway. On the basis of genetic arguments, it was previously suggested that Spo12 may be acting as a regulatory subunit for the protein kinase Dbf2 and its homolog Dbf20, both of which function as part of the mitotic exit network. This is based on the observation that in the absence of DBF20, multicopy SPO12 is no longer able to rescue dbf2 mutants (Toyn and Johnston 1993). However, this is not the case with the cdc15-1 mutation, as we found that 2μ
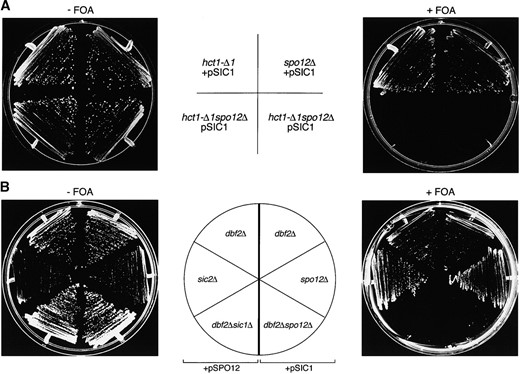
Evidence that SPO12 can function independently of SIC1. (A) Multicopy SIC1 can rescue the lethality of hct1-Δ1 spo12Δ mutants. Isogenic strains deleted for HCT1 (RSI-304) and SPO12 (V379) were crossed, and the resulting diploid was transformed with a multicopy plasmid containing the SIC1 gene (YEpSDB25; URA3). Tetrad analysis was performed on cells sporulated on KSM plates. Colonies derived from two independent hct1-Δ1 spo12Δ spores, containing the pSIC1 plasmid, were selected and streaked for growth on SD − ura plates (−FOA; growth in presence of plasmid) and FOA plates (+FOA; growth in absence of plasmid). Mutant hct1-Δ1 and spo12Δ cells, derived from the same cross and also containing the pSIC1 plasmid, were streaked as controls. Plates were incubated at 25° for 2–3 days. (B) Multicopy rescue of dbf2Δ sic1Δ and dbf2Δ spo12Δ mutants. In two independent crosses, a dbf2Δ strain (S2-2D) was mated with isogenic strains JD-100 (sic1Δ) and V378 (spo12Δ). The resulting diploid strains were transformed with multicopy plasmids containing SPO12 (YEpSPO12; URA3) or SIC1 (YEpSDB25; URA3), respectively. Cells were sporulated and tetrad analysis was performed. Colonies containing the appropriate URA3+ plasmid, representing both single and double mutations from each cross, were selected and streaked for growth on SD − ura plates (−FOA; growth in presence of plasmid) and FOA plates (+FOA; growth in absence of plasmid). Plates were incubated at 25° for 2–3 days.
SPO12 was able to rescue the arrest of a cdc15-1 dbf20Δ double mutant (Table 2). This result suggests that Spo12 is not dependent on Dbf20 for its suppressor function. However, in this instance, it is possible that Spo12 is acting through Dbf2. Deletion of DBF2 is lethal in a cdc15-1 background (our unpublished data). To test whether high-copy SPO12 can suppress cdc15-1 in the absence of Dbf2 activity, we constructed a cdc15-1 dbf2Δ double mutant kept alive by providing DBF2 on a URA3-based plasmid. We found that transformation with multicopy SPO12 allowed the DBF2 plasmid to be lost and rescued the growth of the cdc15-1 dbf2Δ strain at the restrictive temperature of 37° (data not shown). Therefore, the ability of Spo12 to suppress cdc15-1 depends on the presence of either functional Dbf2 or Dbf20.
Spo12 is a nuclear and nucleolar protein: To look for additional clues to Spo12 function, we investigated the subcellular localization of the protein. We were unable to detect Spo12 fused to green fluorescent protein (GFP) in live cells microscopically and therefore employed indirect immunofluorescence. Asynchronous cells expressing Spo12-13Myc showed the protein to be located predominantly in the nucleus at most stages in the cell cycle (Figure 4A). The exception was in G1-arrested cells where the fluorescence signal was absent due to the fact that Spo12 is not expressed at this stage of the cell cycle. These results are consistent with the previously reported nuclear location of Spo12 (Grether and Herskowitz 1999). However, the anti-myc fluorescence did not strictly correlate with bulk DNA, as determined by DAPI staining. As shown in Figure 4A, a significant concentration of Spo12-13Myc was often located in a region adjacent to the bulk of chromatin (white arrows). This pattern of staining is similar to that of nucleolar antigens (de Beus et al. 1994). To determine whether Spo12 was indeed locating to the nucleolus, Nop2, an essential nucleolar protein, was visualized in SPO12-13MYC cells, with the anti-Nop2 mAb (de Beus et al. 1994). In budded cells, Spo12 clearly located to two spheres, one corresponding to the DNA and the other to the Nop2 signal, and hence the nucleolus (Figure 4B). As the cells separated their chromosomes, the nucleolus also appeared to segregate asymmetrically between mother and daughter cells, with the majority in the mother. This became apparent toward the end of anaphase B and in telophase. Throughout these latter stages of the cell cycle, Spo12 staining persistently co-localized with the DAPI and Nop2 signals. We conclude, therefore, that Spo12 is located to both the nucleus and nucleolus throughout mitosis.
As a control for the nucleolar localization of Spo12, the localization of Rnr2 was examined. Rnr2 is the small subunit of ribonucleotide reductase and is a nuclear
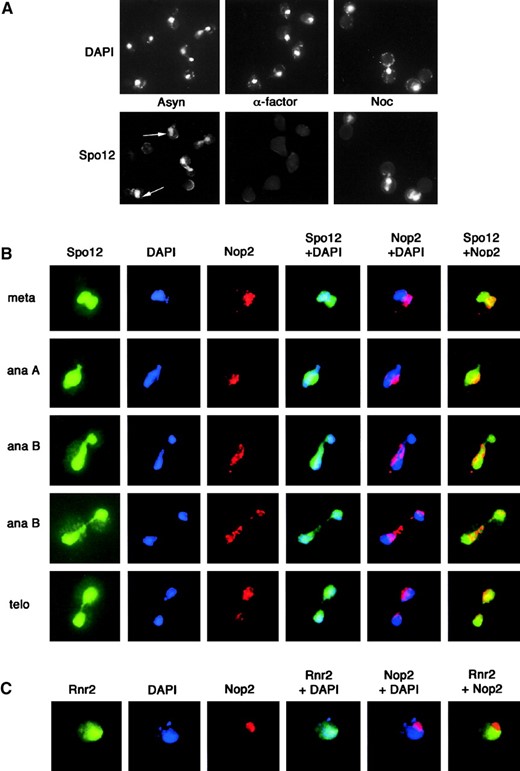
Subcellular localization of Spo12 protein. (A) Strain YLF23, expressing Spo12-13Myc, was grown to midlog phase (Asyn), arrested in G1 with α-factor (α-factor), or arrested in metaphase with nocodazole (Noc). Following fixation, cells were stained with the anti-Myc mAb 9E10 to visualize Spo12 and with DAPI to visualize nuclei. In some cells, localization of Spo12 was seen clearly outside the bulk of the DNA indicated by white arrows. (B) Spo12 is a nuclear and nucleolar protein. Asynchronous cells expressing Spo12-13Myc (strain YLF23) were grown to midlog phase and fixed for indirect immunofluorescence. Samples were stained with anti-Myc mAb 9E10 (to detect Spo12), anti-Nop2 mouse antibody (to detect the exclusively nucleolar Nop2 protein), and DAPI (to visualize nuclei). Cells representative of progressive stages of mitosis are shown, i.e., metaphase (meta), anaphase (ana), and telophase (telo). The merged images clearly show that Spo12 is present both in the nucleus and the nucleolus. (C) Rnr2 does not co-localize with Nop2. Asynchronous cells expressing Rnr2-13Myc (strain YDL21) were grown to midlog phase and fixed for indirect immunofluorescence. Samples were stained for localization of Rnr2 (anti-Myc mAb 9E10), Nop2 (anti-Nop2 antibody), and chromatin (DAPI).
protein (D. Liger and L. H. Johnston, unpublished observations). Localization of Rnr2-13Myc was then compared with Nop2, with similar fixation and immunofluorescence techniques. No coincidence of Rnr2 localization with that of Nop2 was observed (Figure 4C). At no stage of the cell cycle did the Nop2 stain coincide with DAPI or Rnr2. Thus, the Spo12 nucleolar localization is likely to be of physiological significance.
In view of this nucleolar localization of Spo12, we investigated the possibility that overexpression of SPO12 might activate release of the Cdc14 protein phosphatase from the nucleolus. Cdc14 plays a key role in promoting inactivation of mitotic kinase by dephosphorylating Hct1, Sic1, and Swi5 (Visintin et al. 1998; Jaspersen et al. 1999). In turn, Cdc14 activity is regulated by Cfi1/Net1 (Shou et al. 1999; Straight et al. 1999; Visintin et al. 1999), which sequesters and inhibits Cdc14 in the nucleolus during G1, S phase, and early mitosis. During nuclear division, Cdc14 is released from the nucleolus, a process regulated by the mitotic exit network of proteins by an as yet unknown mechanism. Interestingly, although SPO12 overexpression rescues a wide range of mitotic exit mutants, it cannot rescue a mutation in the CDC14 gene (Jaspersen et al. 1998; our unpublished data). SPO12 overexpression could therefore rescue mitotic exit mutants by stimulating the release of Cdc14 from the nucleolus, thereby bypassing the need for a functional MEN.
To investigate this, we looked at the localization of Cdc14-18Myc in cells carrying multicopy SPO12. However, we were unable to detect any significant differences in the cell cycle-dependent localization of Cdc14 in either
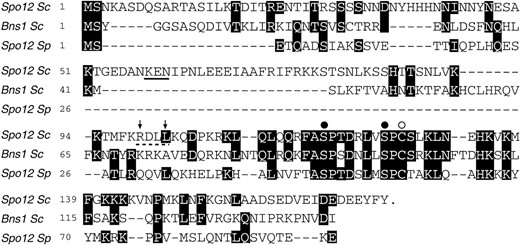
Comparison of Spo12 homologs and mutagenesis of Spo12 consensus sequence. Sequence alignment of the S. cerevisiae (Sc) Spo12 and Bns1 proteins and the S. pombe (Sp) Spo12 protein is shown. Numbers correspond to the amino acid positions in each protein. Amino acids identical to those found in S. cerevisiae Spo12 are shaded black. Substitutions in the S. cerevisiae Spo12 sequence were made by site-directed mutagenesis as follows: The highly conserved amino acids S118 and S125 (solid circles) were mutated to alanine or glutamic acid residues to produce MutAA and MutEE, respectively. Cysteine 127 (open circle) was mutated to alanine in MutC127A. A putative cyclin destruction box is marked with a dashed line, and residues R99 and L102 within this sequence (indicated by arrows) were mutated to alanines to generate MutDB. Alanine substitutions were also made to replace the three residues of a putative KEN-box motif (underlined) to make MutKEN.
the absence or the presence of the plasmid (data not shown). This does not rule out the possibility, however, that a small but undetectable fraction of Cdc14 is released on SPO12 overexpression, which may be sufficient to allow cells to exit mitosis.
Site-directed mutagenesis indicates a similar function for Spo12 in mitosis and meiosis: Proteins related to Spo12 have been identified in both S. cerevisiae (Bns1) and in Schizosaccharomyces pombe (Spo12 s.p.). The precise functional relationship between these proteins is not clear (Jaspersen et al. 1998; Grether and Herskowitz 1999; Samuel et al. 2000). Strains deleted for BNS1 have no obvious vegetative phenotype and there is no synthetic interaction between spo12Δ and bns1Δ (Grether and Herskowitz 1999; our unpublished data). The double mutant has the same growth rate and FACS profile as spo12Δ alone (data not shown). In addition, there is no synthetic interaction between bns1Δ and dbf20Δ or dbf2Δ (data not shown), whereas dfb2Δ and spo12Δ are a lethal combination. Constitutively overexpressed BNS1 does, however, rescue some late mitotic mutants (Jaspersen et al. 1998). These data indicate little, if any, overlap in vegetative function between Bns1 and Spo12. Grether and Herskowitz (1999) came to the same conclusion, these authors also showing that high-copy BNS1 only partially suppresses the absence of Spo12 function in meiosis.
Despite this lack of a clear functional relationship between the proteins, there is a striking region of identity corresponding to a 22-amino-acid segment in the C-terminal half of the Spo12 protein (Figure 5). To investigate whether this region is important for Spo12 function, we performed site-directed mutagenesis of a few highly conserved amino acids (indicated in Figure 5). Serines S118 and S125 are potential CDK phosphorylation sites and these were mutated to either alanine or glutamic acid residues to generate MutAA and MutEE, respectively, and cysteine 127 was mutated to an alanine, to generate MutC127A. Like the wild-type protein, overexpression of these mutants either as multicopy plasmids or as GAL constructs had no detectable effect on the cells (data not shown). However, there were marked differences when these mutants were tested for their ability to rescue mitotic exit mutants. Neither multicopy MutEE nor MutAA was able to rescue the mitotic arrest phenotype of dbf2-2 or cdc15-1 cells at the restrictive temperature (Table 3). MutC127A, on the other hand, rescued both dbf2-2 and cdc15-1 cells with the same efficiency as wild-type Spo12.
Interestingly, when we investigated the effect of these mutations on meiotic function, we found that MutAA and MutEE were unable to rescue the meiotic defect of a spo12Δ/spo12Δ diploid strain, unlike MutC127A, which rescued the defect, again with the same efficiency as the wild-type construct (Table 4). Western analysis of epitope-tagged versions of these mutants indicated that they were being expressed to similar levels and were also cell cycle regulated in the same way as wild-type protein (data not shown). This suggests that the suppression differences observed between the various mutants are not due to differences in their relative protein levels. We conclude, therefore, that amino acids S118 and S125 play a crucial role in Spo12 function and that mutation of these residues abolishes Spo12 activity. These results suggest for the first time that Spo12 may carry out the same biochemical function in mitosis as it does in meiosis.
DISCUSSION
The Spo12 protein is a key regulatory protein in two of the most fundamental processes of biology, mitosis and meiosis, and yet its biochemical function remains elusive. In this study we concentrated on the genetic
Effect of mutagenesis of SPO12 on its ability to suppress mitotic exit mutations
| Effect of multicopy plasmid on colony growth at 35°a | |||||
|---|---|---|---|---|---|
| Vector | SPO12 (WT) | MutAA | MutEE | MutC127A | |
| dbf2-2 | − | +b | − | − | + |
| cdc15-1 | − | +b | − | − | + |
| Effect of multicopy plasmid on colony growth at 35°a | |||||
|---|---|---|---|---|---|
| Vector | SPO12 (WT) | MutAA | MutEE | MutC127A | |
| dbf2-2 | − | +b | − | − | + |
| cdc15-1 | − | +b | − | − | + |
Mutant strains dbf2-2 (L181-6B) and cdc15-1 (SCG15-3D) were transformed at 25° with the indicated gene on a 2μ plasmid. Suppression of each mutant arrest was analyzed by streaking for single colonies on selective SD plates at 35°. Robust growth (+) or no growth (−) is indicated.
Similar results were obtained previously (Parkes and Johnston 1992; Shirayama et al. 1996).
Effect of mutagenesis of SPO12 on its ability to suppress mitotic exit mutations
| Effect of multicopy plasmid on colony growth at 35°a | |||||
|---|---|---|---|---|---|
| Vector | SPO12 (WT) | MutAA | MutEE | MutC127A | |
| dbf2-2 | − | +b | − | − | + |
| cdc15-1 | − | +b | − | − | + |
| Effect of multicopy plasmid on colony growth at 35°a | |||||
|---|---|---|---|---|---|
| Vector | SPO12 (WT) | MutAA | MutEE | MutC127A | |
| dbf2-2 | − | +b | − | − | + |
| cdc15-1 | − | +b | − | − | + |
Mutant strains dbf2-2 (L181-6B) and cdc15-1 (SCG15-3D) were transformed at 25° with the indicated gene on a 2μ plasmid. Suppression of each mutant arrest was analyzed by streaking for single colonies on selective SD plates at 35°. Robust growth (+) or no growth (−) is indicated.
Similar results were obtained previously (Parkes and Johnston 1992; Shirayama et al. 1996).
and biochemical analysis of its mitotic function. We present findings that, for the first time, give insights into its mitotic role. These observations may also provide clues to its meiotic function.
Spo12 proteolysis is mediated by APCHct1: The regulation of Spo12 is under tight cell cycle control, both at the level of mRNA and protein. During mitosis, Spo12 protein levels are low in G1, rise to a peak in mitosis, and decrease as cells enter the next G1 phase (Grether and Herskowitz 1999; our unpublished data). This periodicity of the protein levels mirrors that of SPO12 mRNA (Parkes and Johnston 1992). In this study we observed that the rapid turnover of Spo12 protein during G1 is mediated by the APC and the APC regulator, Hct1 (Figure 1). The fact that the Spo12 protein peaks during mitosis and is then actively degraded under control of the APC in the next G1 phase is consistent with genetic data (outlined below), which strongly suggest that this protein has a regulatory role during mitosis and mitotic exit.
During our attempts to construct a nondestructible version of Spo12, mutagenesis of a putative cyclin destruction box and a putative KEN box within the Spo12
Multicopy suppression of the meiotic phenotype of a spo12Δ/spo12Δ diploid strain
| Plasmid | Tetrads (%) | Dyads (%) | Sporulation (%) |
|---|---|---|---|
| Vector | 0 | 100 | 16 |
| SPO12 (WT) | 88 | 12 | 30 |
| MutAA | 0 | 100 | 15 |
| MutEE | 0 | 100 | 18 |
| MutC127A | 87 | 13 | 32 |
| Plasmid | Tetrads (%) | Dyads (%) | Sporulation (%) |
|---|---|---|---|
| Vector | 0 | 100 | 16 |
| SPO12 (WT) | 88 | 12 | 30 |
| MutAA | 0 | 100 | 15 |
| MutEE | 0 | 100 | 18 |
| MutC127A | 87 | 13 | 32 |
Plasmids containing the indicated wild-type (WT) or mutated SPO12 gene (as for Table 3) were transformed into an a/α spo12Δ/spo12Δ strain (V378 × V379), sporulated on KSM plates, and analyzed for efficiency of sporulation. Tetrad values are the number of tetrads divided by the number of tetrads plus dyads, and dyad values are the number of dyads divided by the number of tetrads plus the number of dyads. Sporulation values are the number of tetrads plus dyads divided by the total number of cells counted in each case (400).
Multicopy suppression of the meiotic phenotype of a spo12Δ/spo12Δ diploid strain
| Plasmid | Tetrads (%) | Dyads (%) | Sporulation (%) |
|---|---|---|---|
| Vector | 0 | 100 | 16 |
| SPO12 (WT) | 88 | 12 | 30 |
| MutAA | 0 | 100 | 15 |
| MutEE | 0 | 100 | 18 |
| MutC127A | 87 | 13 | 32 |
| Plasmid | Tetrads (%) | Dyads (%) | Sporulation (%) |
|---|---|---|---|
| Vector | 0 | 100 | 16 |
| SPO12 (WT) | 88 | 12 | 30 |
| MutAA | 0 | 100 | 15 |
| MutEE | 0 | 100 | 18 |
| MutC127A | 87 | 13 | 32 |
Plasmids containing the indicated wild-type (WT) or mutated SPO12 gene (as for Table 3) were transformed into an a/α spo12Δ/spo12Δ strain (V378 × V379), sporulated on KSM plates, and analyzed for efficiency of sporulation. Tetrad values are the number of tetrads divided by the number of tetrads plus dyads, and dyad values are the number of dyads divided by the number of tetrads plus the number of dyads. Sporulation values are the number of tetrads plus dyads divided by the total number of cells counted in each case (400).
sequence (indicated in Figure 5) appeared to destabilize rather than stabilize the protein. The introduced mutations presumably led to deleterious changes in the tertiary structure of Spo12 and resulted in its rapid nonspecific turnover. Therefore, it is unclear at present whether DB- or KEN-box sequences or perhaps other sequences as have been reported for Cdc5 (Charles et al. 1998) are important for the APC-dependent degradation of Spo12. Also, as we have not shown biochemically that Spo12 is an APC substrate, it is possible that its degradation is being controlled by the APC in an indirect fashion.
Spo12 is not a major regulator of Clb2-Cdc28 activity: Given that high-copy SPO12 is able to suppress the late mitotic arrest phenotype of a number of mutants that arrest with high Clb2-Cdc28 kinase activity (Parkes and Johnston 1992; Shirayama et al. 1994a,b; Jaspersen et al. 1998), we speculated that Spo12 may be able to facilitate mitotic exit by antagonizing Clb2-Cdc28 kinase activity either by acting as a CDKi, like Sic1, or as an APC activator, like Hct1. However, four lines of evidence presented in this study strongly argue against this. First, cells deleted for SPO12 are not compromised by Clb2 overproduction (Figure 2A). This is in contrast to cells deleted for either SIC1 or HCT1, which are defective in deactivation of the mitotic kinase and are therefore hypersensitive to elevated levels of Clb2 (Schwab et al. 1997; Toyn et al. 1997). Second, overexpression of Spo12 fails to rescue the lethality induced by the ectopic overproduction of Clb2 (Figure 2B). In contrast, multicopy SIC1 is able to rescue this phenotype, which is consistent with its known function as a CDKi. Third, during the overproduction and accumulation of Spo12 in vegetative cells, endogenous levels of Clb2 protein and Clb2-Cdc28 kinase levels remained constant (Figure 2C), further indicating that Spo12 is not an antagonist of the mitotic kinase. In addition, we found that this overexpression of Spo12 has no detectable effect on the viability, growth rate, or morphology of yeast cells (our unpublished results). This is in stark contrast to overproduction of Hct1, which results in the rapid degradation of Clb2 and Ase1 and causes cells to arrest in G2 (Schwab et al. 1997; Visintin et al. 1997), and overproduction
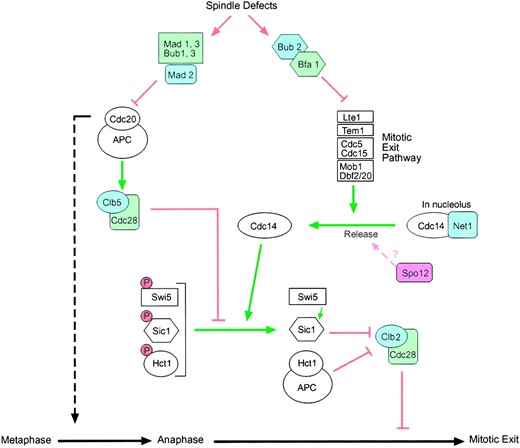
Schematic diagram of proteins that control anaphase and exit from mitosis. Arrows indicate activation and bars indicate inhibition. Negative regulators are shaded and positive regulators are white. The mitotic exit pathway triggers the release of the Cdc14 phosphatase from its inhibitor Cfi1/Net1, which sequesters Cdc14 in the nucleolus. Once released from Cfi1/Net1, Cdc14 dephosphorylates the three key substrates Hct1, Sic1, and the Swi5 transcription factor. The combined activity of APCHct1 and Sic1 leads to the collapse of the Clb2-Cdc28 kinase activity and allows exit from mitosis. Release of Cdc14 from the nucleolus is not sufficient for it to trigger inactivation of mitotic kinases. Clb5-Cdc28, a potent antagonist of Cdc14, also needs to be inactivated for Cdc14 to efficiently dephosphorylate its targets. This is achieved by APCCdc20, which degrades Clb5 during anaphase. APCCdc20 is also responsible for the degradation of the Pds1 protein (not shown), which ultimately leads to separation of sister chromatids and therefore passage into anaphase (dashed line). The spindle checkpoint impinges on both anaphase and mitotic exit as shown. Mad2 inhibits APCCdc20 in response to kinetochore defects, thereby blocking metaphase. Bub2 and Bfa1 form a putative GAP complex that probably blocks the late mitotic pathway by converting the Tem1 GTPase into an inactive GDP-bound form. The mechanism by which the mitotic exit pathway is activated during a normal cell cycle is unknown and the ordering of its components is still speculative. The proposed pathway by which overexpression of Spo12 is able to suppress defects in the late mitotic exit genes is indicated by a question mark.
of Sic1, which not only greatly reduces Clb-Cdc28 kinase levels but also results in G1/G2 cell cycle delays and elongated multibudded cells (Nishizawa et al. 1998; Sanchez-Diaz et al. 1998). Taken together, therefore, our data strongly indicate that Spo12 does not play a role in the normal regulation of Clb-Cdc28 kinase activity during mitosis. This also appears to be the case in meiosis, as reported by Grether and Herskowitz (1999). These workers observed that there were no significant differences in the timing or regulation of specific Clb-Cdc28 kinases between wild-type or spo12 mutant diploid cells undergoing a meiotic cell cycle.
Spo12—a possible regulator of Cdc14 release: If Spo12 does not directly regulate Clb-Cdc28 kinase activity, how is its overproduction able to suppress such a wide range of mitotic exit mutants? One possibility is that overexpression of Spo12 leads to the release of a small but significant amount of Cdc14 from the nucleolus that is sufficient to initiate deactivation of mitotic kinase (through dephosphorylation of both Hct1 and Sic1), thereby facilitating mitotic exit (Figure 6). There are several observations that are consistent with this theory. First, localization of Spo12 to the nucleolus during mitosis (Figure 4) places the protein in the right place at the right time to carry out this function. Second, although multicopy SPO12 can rescue a wide range of mitotic exit mutants, it cannot overcome the arrest of cdc14 mutants at the restrictive temperature (Jaspersen et al. 1998; our unpublished results). Thus, the suppression phenotype of SPO12 is dependent on functional Cdc14 but not on the other members of the MEN. Third, if SPO12 overexpression leads to a limited delocalization of Cdc14 from the nucleolus, then mitotic kinase levels will be lowered through the dephosphorylation and subsequent activation of Hct1 and accumulation of Sic1. It is likely, therefore, that in the absence of one of these redundant pathways, the presence of the other would be sufficient to adequately lower mitotic kinase levels during SPO12 overproduction. As indicated in Table 2, we have shown this to be the case; i.e., SPO12 can carry out its suppressor function in the absence of either Sic1 or Hct1. The notion that SPO12 acts upstream of both HCT1 and SIC1 is also consistent with the observation that although the synthetic lethality of spo12Δ hct1-Δ1 mutants is rescued by multicopy SIC1 (Figure 3A), multicopy SPO12 is unable to rescue the lethality of hct1-Δ1 sic1Δ mutants (data not shown).
We have been unable to demonstrate a gross delocalization of Cdc14 from the nucleolus on SPO12 overexpression (data not shown). However, it has been previously shown that even a tiny, almost undetectable, release of Cdc14 is sufficient to facilitate mitotic exit. Shou et al. (1999) recently described a mutation, net1-1, which, despite being able to rescue the lethality of tem1Δ mutants by allowing both Clb2 degradation and Sic1 accumulation, demonstrated an apparently normal wild-type localization of Cdc14. On closer examination, it was observed that the net1-1 mutation did indeed lead to a small delocalization of Cdc14 in a fraction of Tem1-depleted net1-1 cells but, significantly, only during late anaphase/telophase. This led these researchers to propose a model whereby the release of Cdc14 from the nucleolus requires the action of two independent signals: an unknown signal and the Tem1-dependent (and therefore MEN-dependent) signal, both of which are active only during mitosis. They suggested that net1-1 relieves the requirement for the latter but not the former. In this article, we propose that overexpression of SPO12 may fulfill a similar role to net1-1. That is, in the absence of a functional MEN, it may facilitate a limited release of Cdc14 during late mitosis. In addition, a temporally limited (rather than constitutive) release of Cdc14 by multicopy SPO12 confined to anaphase would also explain the anomaly that we were unable to detect gross effects on Clb-Cdc28 kinase levels. Unfortunately, we have been unable to assess whether overexpression of Spo12 can lead to a small and temporally limited release of Cdc14 from the nucleolus, as our attempts to achieve highly synchronous arrest-release of MEN mutant strains in the presence of high-copy Spo12 have so far failed. However, we have observed that the level of Cdc14 is not affected in spo12Δ mutants or in cells overexpressing Spo12, suggesting that the Spo12 does not regulate the expression/stability of Cdc14 (data not shown).
To identify proteins that may interact with Spo12, we performed extensive two-hybrid screens using a Gal4 activation domain-Spo12 fusion as bait (data not shown). This construct was able to carry out the suppressor function of the wild-type Spo12 protein (not shown). Only one potent interactor, Fob1, was identified multiple times. Fob1 is a nucleolar protein required for mitotic recombination hotspot activity and replication fork blocking (Kobayashi and Horiuchi 1996). Intriguingly, it is also thought to act in the same pathway as the regulator of nucleolar silencing and telophase (RENT) component Sir2 in determination of life span (Defossez et al. 1999; Kaeberlein et al. 1999). However, the significance of this two-hybrid result is unclear at present, as an in vivo interaction between tagged versions of Spo12 and Fob1 has yet to be established.
What is the mitotic function of single-copy Spo12? There are now a number of lines of evidence that clearly suggest a regulatory role for Spo12 in mitosis. First, its protein levels are tightly cell cycle regulated, peaking in G2/M and decreasing in G1 via destruction by the APC (Grether and Herskowitz 1999; this study). Second, although cells deleted for SPO12 are viable, spo12Δ mutant cells exhibit a slight but significant G2/M delay (Parkes and Johnston 1992). Third, spo12 is synthetically lethal with dbf2 and hct1 mutations, both of which are important regulators of mitotic exit (Parkes and Johnston 1992; Grether and Herskowitz 1999; this study). Fourth, multicopy SPO12 not only suppresses the mitotic arrest phenotype of a wide range of mitotic exit mutants (reviewed by Morgan 1999) but it can also rescue the lethality of dbf2Δ sic1Δ double mutants (Figure 3B). So exactly what is the mitotic function of Spo12? Unfortunately, analysis of BNS1 does not help resolve this issue. Despite the homology of Bns1 with Spo12, bns1Δ strains display no obvious mitotic defects and no synthetic interaction was evident in a spo12Δ bns1Δ double mutant (Grether and Herskowitz 1999; our unpublished observations). Given the unusual localization of Spo12 to both the nucleus and nucleolus, it may have more than one role. To explore possible functions, we investigated the basis of the G2/M delay exhibited by spo12Δ mutants. Cells deleted for SPO12 do not demonstrate any defects in regulation of Clb2 protein or mitotic kinase levels, nor do they show any abnormalities in spindle assembly/disassembly or spindle orientation (our unpublished data). In addition, spo12Δ cells exhibit wild-type levels of benomyl sensitivity (data not shown), indicating that Spo12 is unlikely to be part of a signaling mechanism for the spindle assembly checkpoint (SAC), of which the MEN is a target (Fesquet et al. 1999). Further proof of this comes from genetic studies, which indicate that there are no synthetic interactions between spo12 and mutations in genes representing either branch of the SAC, i.e., MAD2 or BUB2 (data not shown).
In this study we demonstrate that when overexpressed, Spo12 is unable to directly regulate Clb-Cdc28 kinase levels. However, we propose that it may do so indirectly through its possible action on the Cdc14 phosphatase. A role for Spo12 in the normal regulation of Cdc14 release is at present unclear, as we have been unable to detect any defects in the timing and pattern of Cdc14 localization in spo12Δ mutants (data not shown). However, this does not rule out a redundant and therefore nonessential role for Spo12 in Cdc14 regulation, which may account for the subtle mitotic defect of spo12Δ mutants.
Spo12 may have a similar function in mitosis and meiosis: The SPO12 gene was first identified as a null mutation causing a defect in sporulation. During meiosis, mutations in SPO12 cause diploid cells to bypass meiosis I, leading to the formation of asci containing two viable diploid spores or dyads. The biochemical function of Spo12 during meiosis is not understood. In this study, we present, for the first time, direct evidence to indicate that Spo12 may carry out the same biochemical function during both mitosis and meiosis. We observed that mutagenesis of two highly conserved serine residues within the Spo12 protein sequence abolishes not only its mitotic suppressor function but also its meiotic function (Table 4). It is tempting to speculate that if Spo12 does indeed play a minor role in the regulation of Cdc14 activity during mitosis, this function could become more important during meiosis. Very little is known about the meiotic role of Cdc14 or any of the MEN components. However, it has been shown that CDC14 is expressed to significant levels during meiosis (Chu et al. 1998). In addition, cdc14 mutants not only exhibit a defect in vegetative growth but also are defective in meiosis. Intriguingly, homozygous diploid cdc14 mutant cells produce dyads on sporulation at the restrictive temperature, as do spo12Δ diploid cells (Schild and Byers 1980).
Spo12 is clearly a global regulator of meiosis, controlling an entire meiotic division. Our previous work and that described here show it to play an additional important, albeit redundant, role in mitotic exit. Indeed, our data highlight the redundancy of late mitotic events, emphasizing the biological complexity of this late cell cycle transition. While it seems likely that Spo12 impinges upon the core regulatory pathway of mitotic exit, which is Cdc14 release from the nucleolus, further work is necessary to prove this notion.
Acknowledgement
We are very grateful to the following for providing reagents: M. Segal (Scripps Research Institute, California) for strain MY101, P. Philippsen (University of North Carolina) for strain DK329-4D, W. Seufert (Universtity of Stuttgart, Germany) for plasmid pWS176, K. Kramer (National Institute for Medical Research, UK) for strain CG379sic1::HIS7, C. Mann (CEA/Saclay, France) for anti-Clb2 antibody, K. Gull (University of Manchester, United Kingdom) for antitubulin antibody, J. Aris (University of Florida) for anti-Nop2 antibody, and D. Liger (NIMR) for strain DLY4. We are also indebted to D. Liger for providing the Rnr2-13Myc-tagged strain and to other members of the Yeast Genetics Division for valuable discussions. This work was supported by the Medical Research Council (MRC) and MRC Fellowship Training Grant no. G81/141 (R.S.). S.J. was supported by a fellowship from the Danish Medical Research Council.
Footnotes
Communicating editor: J. Rine
LITERATURE CITED
Author notes
Present address: Imperial Cancer Research Fund, Clare Hall Laboratories, Herts, EN6 3LD, United Kingdom.
Present address: Cyclacel/Polgen, CRC Cell Cycle Research Group, Department of Genetics, University of Cambridge, Cambridge, CB2 3EH, United Kingdom.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}