





Abstract
The mutagenicity of DNA double-strand break repair in Escherichia coli is controlled by DNA-damage (SOS) and general (RpoS) stress responses, which let error-prone DNA polymerases participate, potentially accelerating evolution during stress. Either base substitutions and indels or genome rearrangements result. Here we discovered that most small basic proteins that compact the genome, nucleoid-associated proteins (NAPs), promote or inhibit mutagenic break repair (MBR) via different routes. Of 15 NAPs, H-NS, Fis, CspE, and CbpA were required for MBR; Dps inhibited MBR; StpA and Hha did neither; and five others were characterized previously. Three essential genes were not tested. Using multiple tests, we found the following: First, Dps, which reduces reactive oxygen species (ROS), inhibited MBR, implicating ROS in MBR. Second, CbpA promoted F′ plasmid maintenance, allowing MBR to be measured in an F′-based assay. Third, Fis was required for activation of the SOS DNA-damage response and could be substituted in MBR by SOS-induced levels of DinB error-prone DNA polymerase. Thus, Fis promoted MBR by allowing SOS activation. Fourth, H-NS represses ROS detoxifier sodB and was substituted in MBR by deletion of sodB, which was not otherwise mutagenic. We conclude that normal ROS levels promote MBR and that H-NS promotes MBR by maintaining ROS. CspE positively regulates RpoS, which is required for MBR. Four of five previously characterized NAPs promoted stress responses that enhance MBR. Hence, most NAPs affect MBR, the majority via regulatory functions. The data show that a total of six NAPs promote MBR by regulating stress responses, indicating the importance of nucleoid structure and function to the regulation of MBR and of coupling mutagenesis to stress, creating genetic diversity responsively.
IN Escherichia coli, the structure of the bacterial chromosome or nucleoid is governed by the action of small, abundant DNA-binding nucleoid-associated proteins (NAPs) (Ali Azam et al. 1999). NAPs bind dynamically to the genome both sequence specifically and sequence nonspecifically, causing a variety of genomic conformations (Azam et al. 2000). Through this action, several NAPs also act as global transcriptional regulators, with some of their regulons containing hundreds of genes (Oberto et al. 2009; Kahramanoglou et al. 2011; Chib and Mahadevan 2012). There are 15 NAPs: Hfq, IHF-A and IHF-B (which constitute IHF), HU-α and HU-β (which constitute HU), Fis, H-NS, CbpA, Lrp, CspC, CspE, Dps, StpA, Hha, IciA (ArgP), DnaA, and CbpB (Ali Azam et al. 1999). The NAP composition of the nucleoid changes in a growth-phase-dependent fashion, with Hfq, IHF, HU, Fis, and H-NS controlling genomic compaction primarily in actively growing cells (Ali Azam et al. 1999). Dps takes over in stationary phase, and CbpA expression peaks in late stationary phase (Ali Azam et al. 1999).
Fis, like most NAPs, is small, basic, and abundant in cells. Fis levels peak at 60,000 molecules per cell in mid-log phase (Ali Azam et al. 1999). Fis functions in site-specific recombination and also controls transcription of DNA recombination and growth genes (Ueguchi and Mizuno 1993; Esposito and Gerard 2003; Flatten and Skarstad 2013). During the transition to stationary phase, Fis drops to undetectable levels and is thought to be absent from starving cells (Ali Azam et al. 1999). H-NS functions largely as a transcription repressor, silencing especially genes recently acquired through horizontal transfer (Ueguchi and Mizuno 1993; Perez et al. 2008; Sharadamma et al. 2010; Dorman 2014). H-NS reaches 40,000 molecules per cell during growth phase but falls sharply on entry into stationary phase (Ali Azam et al. 1999). H-NS is also a global regulator controlling the transcription of a few hundred genes, including those required for protection from osmotic and acid stresses (Barth et al. 1995; Bouvier et al. 1998; Chib and Mahadevan 2012). In growing cells, Fis and H-NS work together to repress the transcription of dps (Grainger et al. 2008). Fis also represses cbpA transcription (Chintakayala et al. 2013).
When IHF, HU, H-NS, and Fis levels fall, the nucleoid becomes compacted by Dps (Akerlund et al. 1995). In stationary phase, Dps levels can reach as high as 180,000 molecules per cell. Dps binds sequences nonspecifically (Ali Azam et al. 1999) and causes the nucleoid to assume a unique state with DNA cocrystallized with Dps protein molecules (Schnetz 2008). Whereas there is no direct evidence that Dps is a global transcriptional regulator, Dps plays important roles in both protection against reactive oxygen species (ROS) and regulation of replication initiation (Chodavarapu et al. 2008b; Calhoun and Kwon 2011). Little is known about the other stationary-phase-specific NAP, CbpA. CbpA has sequence similarity to the DnaJ cochaperone and has a DNA binding motif similar to that of Dps. CbpA is not known to be a global transcription regulator.
HU and IHF are the other primary growth-phase NAPs, and they function with Fis and H-NS to compact the replicating genome (Ali Azam et al. 1999). As cells reach stationary phase, HU and IHF levels drop (Ali Azam et al. 1999). DnaA is an essential replication-initiation protein that binds to the replication origin, oriC, and promotes open-complex formation for replication initiation (Ozaki and Katayama 2009). IciA, also known as ArgP, inhibits replication initiation by binding to conserved sequences within oriC, preventing DnaA from inducing open-complex formation (Hwang and Kornberg 1990; Thony et al. 1991; Hwang et al. 1992) CbpB, also known as Rob, is similar to CbpA in that it binds preferentially to curved DNA structures (Azam et al. 2000). StpA is a homolog of H-NS and can silence horizontally acquired DNA when H-NS is absent from cells (Sonnenfield et al. 2001; Uyar et al. 2009). Unlike the other NAPs, Hha does not bind directly to DNA but rather binds a protein that binds and sequesters DNA (Madrid et al. 2007). Lrp is the closest analog to a eukaryotic histone. Lrp forms octomeric structures around which DNA winds, similarly to DNA wrapping around histones (de los Rios and Perona 2007). HU, IHF, Lrp, DnaA, and IciA are also global transcription regulators (Bouvier et al. 1998; Oberto et al. 2009; Prieto et al. 2012).
CspC and CspE, although belonging to the cold-shock-protein regulon, are constitutively expressed in cells even at 37° (Czapski and Trun 2014). CspC and CspE promote genomic compaction and are also required for stability of rpoS transcript levels (Cohen-Or et al. 2010) and for resistance to different environmental stressors (Cohen-Or et al. 2010; Shenhar et al. 2012), possibly by controlling levels of rpoS transcript. rpoS encodes σS (or RpoS protein), the transcriptional activator (RNA polymerase sigma factor) of the general/starvation stress response.
Here we address the roles of NAPs in mutagenesis, specifically mutations formed by mutagenic break repair (MBR), which happens in stressed, starving cells. Repair of DNA double-stranded breaks (DSBs) in E. coli is switched from high fidelity to a mutagenic mode during starvation stress under control of the RpoS general/starvation stress response (Ponder et al. 2005; Shee et al. 2011; Rosenberg et al. 2012). DSB repair switches either to a mutagenic homologous recombination (HR)–mediated repair pathway, using error-prone translesion DNA polymerases IV (DinB or Pol IV) (Ponder et al. 2005), Pol V (Petrosino et al. 2009; Shee et al. 2011), or Pol II (Frisch et al. 2010), which creates base substitution and indel mutations [single-nucleotide variations (SNVs)], or to a “micro-homologous” pathway that causes genome rearrangement [gross chromosomal rearrangements (GCRs)] using DNA Pol I [reviewed in Rosenberg et al. (2012) and Rogers et al. (2015)]. Both SNV and GCR pathways require RecA, RuvC, and RecBC HR proteins.
The general/starvation (RpoS) stress response (Layton and Foster 2003; Lombardo et al. 2004) and the unfolded periplasmic protein (RpoE) response (Gibson et al. 2010) are also required for both SNV and GCR mechanisms. RpoE promotes MBR at some genomic sites by promoting formation of DSBs by an as yet undetermined mechanism (Gibson et al. 2010). RpoS licenses the use of DinB and other error-prone DNA polymerases in break repair by an unknown mechanism (Ponder et al. 2005; Frisch et al. 2010; Shee et al. 2011). The SOS DNA-damage response is required for SNV formation (McKenzie et al. 2000) and promotes MBR by its induction of the DinB error-prone DNA polymerase (McKenzie et al. 2001; Galhardo et al. 2009). Neither SOS nor DinB plays a role in GCR formation (McKenzie et al. 2001). Formation of GCRs, but not SNVs, requires DNA Pol I (Slack et al. 2006).
Thus, there are three main hubs of stress-response regulation of MBR (Al Mamun et al. 2012; Rosenberg et al. 2012): (1) formation of a DSB, which RpoE promotes (Gibson et al. 2010), and its repair, (2) SOS induction, which upregulates DinB about 10-fold transcriptionally (Galhardo et al. 2009), and (3) activation of the RpoS general stress response, which allows DinB and other low-fidelity DNA polymerases to participate in DSB repair by as yet unknown means (Ponder et al. 2005; Frisch et al. 2010; Shee et al. 2011; Rosenberg et al. 2012).
Previous studies showed that the NAP HU was required for MBR, possibly by promoting HR (Williams and Foster 2007). Another four NAPs (Hfq, CspC, Lrp, and IhfA) were shown to promote MBR (Al Mamun et al. 2012), the majority associated with regulation of stress responses required for MBR.
Here we show that although most NAPs affect MBR, most do so via regulation, and they do so by different mechanisms.
Materials and Methods
Bacterial strains and media
E. coli K12 strains used in this work and their origins are outlined in Table 1. Mutations were introduced using standard P1-mediated transduction (Miller 1992) and/or recombineering methods (Datsenko and Wanner 2000). Genotypes were confirmed by PCR and various phenotypic tests. The media used were M9 minimal medium (Miller 1992) with 10 µg/ml vitamin B1 and 0.1% glycerol or 0.1% lactose and Luria-Bertani-Herskowitz (LBH) medium (Torkelson et al. 1997).
Escherichia coli K12 trains used in this study
| Strain/plasmid | Genotype | Source/reference |
|---|---|---|
| CAG12080 | zah281::Tn10 | Singer et al. (1989) |
| FC36 | Δ(lac proB)XIII thi ara RifR | Cairns and Foster (1991) |
| FC40 | FC36 [F’å45 = F′ proAB+ lacIq lacI33ΩlacZ] | Cairns and Foster (1991) |
| PJH18 | FC40 [F′ lac-amplified] | Hastings et al. (2000) |
| PJH33 | FC40 [F′ lac-amplified] | Hastings et al. (2000) |
| PJH51 | FC40 [F′ lac-amplified] | Hastings et al. (2000) |
| PJH1242 | SMR4562 Δlrp::KanFRT | SMR4562 × P1(JW0802) |
| PJH1390 | SMR4562 ΔrpoS::KanFRT | SMR4562 × P1(SMR10336) |
| PJH1399 | SMR4562 ΔrpoS::FRT | PJH1390 × pCP20 |
| PJH1428 | SMR4562 ΔleuO::KanFRT | SMR4562 × P1(JW0075)a |
| PJH1438 | SMR4562 ∆hns::KanFRT | SMR4562 × P1(JW1225)a |
| PJH1440 | SMR4562 ∆hha::KanFRT | SMR4562 × P1(JW0449)a |
| PJH1442 | SMR4562 ∆hfq::KanFRT | SMR4562 × P1(JW4130)a |
| PJH1444 | SMR4562 ∆stpA::KanFRT | SMR4562 × P1(JW2644)a |
| PJH1446 | SMR4562 ∆ihfB::KanFRT | SMR4562 × P1(JW0895)a |
| PJH1448 | SMR4562 ∆fis::KanFRT | SMR4562 × P1(JW3229a |
| PJH1948 | SMR4562 ∆cbpA::KanFRT | SMR4562 × P1(JW0985)a |
| PJH1952 | SMR4562 ∆dps::KanFRT | SMR4562 × P1(JW0797)a |
| PJH1966 | SMR4562 ∆cspE::KanFRT | SMR4562 × P1(JW618)a |
| PJH2602 | SMR4562 ∆dps::KanFRT ∆rpoS::FRT | PJH1399 × P1(JW0797)a |
| PJH2608 | SMR4562 ∆dps::FRT | PJH1952 × pCP20 |
| PJH2610 | SMR4562 Δhns::FRT | PJH1438 × pCP20 |
| PJH2673 | SMR4562 [F′ zah281::Tn10 Lac+] ∆fis::KanFRT | PJH1448 × P1(SMR8847) |
| PJH2677 | SMR4562 [F’ zah281::Tn10 Lac+] ∆cbpA::KanFRT | PJH1948 × P1(SMR8847) |
| PJH2715 | FC36 ∆hns::KanFRT | FC36 × P1(JW1225)a |
| PJH2716 | FC36 ∆fis::KanFRT | FC36 × P1(JW3229)a |
| PJH2732 | FC36 ∆cbpA::KanFRT | FC36 × P1(JW0985)a |
| PJH2790 | SMR4562 ∆fis::KanFRT [F′ lac-amplified] | PJH2716 × PH18 |
| PJH2792 | SMR4562 ∆fis::KanFRT [F′ lac-amplified] | PJH2716 × PJH33 |
| PJH2794 | SMR4562 ∆fis::KanFRT [F′ lac-amplified] | PJH2716 × PJH51 |
| PJH2804 | SMR4562 ∆cbpA::KanFRT [F′ lac-amplified] | PJH2732 × PH18 |
| PJH2806 | SMR4562 ∆cbpA::KanFRT [F′ lac-amplified] | PJH2732 × PJH33 |
| PJH2841 | SMR4562 ∆hns::KanFRT[F′ lac-amplified] | PJH2715 × PJH18 |
| PJH2843 | SMR4562 ∆hns::KanFRT [F′ lac-amplified] | PJH2715 × PJH33 |
| PJH2845 | SMR4562 ∆hns::KanFRT [F′ lac-amplified] | PJH2715 × PJH51 |
| PJH2863 | SMR4562 ∆hns::KanFRT [F′ zah281::Tn10 Lac+] | PJH1438 × P1(SMR8847) |
| PJH2867 | SMR4562 ∆dps::FRT recA::Tn10dCam | PJH2608 × P1(SMR4610) |
| PJH2874 | SMR4562 ∆cbpA::KanFRT [F′ lac-amplified] | PJH2732 × PJH51 |
| PJH2947 | SMR4562 soxR104 zjc2206::Tn10Kan | SMR4562 × P1(JTG1048) (Nunoshiba and Demple 1994) |
| PJH2961-4 | SMR4562 ∆attλ::PsulA-gfp ∆hns::KanFRT | SMR6039 × P1(JW1225)a |
| PJH2971-2974 | SMR4562 ∆attλ::PsulA-gfp ∆cbpA::KanFRT | SMR6039 × P1(JW0985)a |
| PJH2977 | SMR4562 ∆dps::KanFRT ∆dinB50::FRT [F′ ∆dinB50::FRT] | SMR4562 × P1(JTG1048) (Nunoshiba and Demple 1994) |
| PJH3019 | SMR4562 ∆rssB::tet ∆cbpA::KanFRT | SMR6039 × P1(JW1225)a |
| PJH3092 | SMR4562 ∆rssB::tet ∆fis::KanFRT | SMR6039 × P1(JW0985)a |
| PJH3114 | SMR4562 [F′ lafU2::FRTcatFRT dinB(oc)] ∆fis::KanFRT | SMR5889 × P1(JW0797)a |
| PJH3122 | SMR4562 [F′’ lafU2::FRTcatFRT dinB(oc)] ∆cbpA::KanFRT | SMR12566 × P1(JW0985)a |
| PJH3215 | SMR4562 ∆sodB::KanFRT | SMR12566 × P1(JW3229)a |
| PJH3217 | SMR4562 ∆hns::FRT ∆sodB::KanFRT | PJH2610 × P1(JW1648)a |
| PJH3219-3222 | SMR4562 ∆attλ::PsulA-gfp ∆fis::KanFRT | SMR6039 × P1(JW3229)a |
| PJH3245 | SMR4562 ∆dps::FRT ruvC53 eda::Tn10dCam | PJH2608 × P1(SMR6906) |
| PJH3267 | SMR4562 ∆dps::FRT soxR104 zjc2206::Tn10Kan | PJH2608 × P1(JTG1048) |
| SMR601 | ruvC53 eda51::Tn10 | Lopez et al. (2005) |
| SMR789 | FC40 ruvC53 eda51::Tn10 | FC40 × P1(SMR601) |
| SMR4562 | Independent construction of FC40 | McKenzie et al. (2000) |
| SMR4610 | SMR4562 recA::Tn10dCam | Bull et al. (2001) |
| SMR5889 | SMR4562 ∆dinB50::FRT [F’ ∆dinB50::FRT] | Galhardo et al. (2009) |
| SMR6039 | SMR4562 ∆attλ::PsulA-gfp | Hastings et al. (2004) |
| SMR6178 | SMR4562 ∆attλ::PsulA-gfp sulA::Tn5 lexA3(Ind−) malB::Tn9 | Al Mamun et al. (2012) |
| SMR6263 | MG1655 leuB::Tn10 | Gibson et al. (2010) |
| SMR6906 | SMR4562 ruvC53 eda::Tn10dCam | By linear replacement (Datsenko and Wanner 2000) into SMR789 |
| SMR8847 | SMR4562 [F′ zah281::Tn10 Lac+] | SMR4562 × P1(CAG12080) (Singer et al. 1989) |
| SMR10308 | SMR4562 [F’ lafU2::FRTcatFRT dinB21(oc)] | Galhardo et al. (2009) |
| SMR12566 | SMR4562 ∆rssB::tet | Al Mamun et al. (2012) |
| Strain/plasmid | Genotype | Source/reference |
|---|---|---|
| CAG12080 | zah281::Tn10 | Singer et al. (1989) |
| FC36 | Δ(lac proB)XIII thi ara RifR | Cairns and Foster (1991) |
| FC40 | FC36 [F’å45 = F′ proAB+ lacIq lacI33ΩlacZ] | Cairns and Foster (1991) |
| PJH18 | FC40 [F′ lac-amplified] | Hastings et al. (2000) |
| PJH33 | FC40 [F′ lac-amplified] | Hastings et al. (2000) |
| PJH51 | FC40 [F′ lac-amplified] | Hastings et al. (2000) |
| PJH1242 | SMR4562 Δlrp::KanFRT | SMR4562 × P1(JW0802) |
| PJH1390 | SMR4562 ΔrpoS::KanFRT | SMR4562 × P1(SMR10336) |
| PJH1399 | SMR4562 ΔrpoS::FRT | PJH1390 × pCP20 |
| PJH1428 | SMR4562 ΔleuO::KanFRT | SMR4562 × P1(JW0075)a |
| PJH1438 | SMR4562 ∆hns::KanFRT | SMR4562 × P1(JW1225)a |
| PJH1440 | SMR4562 ∆hha::KanFRT | SMR4562 × P1(JW0449)a |
| PJH1442 | SMR4562 ∆hfq::KanFRT | SMR4562 × P1(JW4130)a |
| PJH1444 | SMR4562 ∆stpA::KanFRT | SMR4562 × P1(JW2644)a |
| PJH1446 | SMR4562 ∆ihfB::KanFRT | SMR4562 × P1(JW0895)a |
| PJH1448 | SMR4562 ∆fis::KanFRT | SMR4562 × P1(JW3229a |
| PJH1948 | SMR4562 ∆cbpA::KanFRT | SMR4562 × P1(JW0985)a |
| PJH1952 | SMR4562 ∆dps::KanFRT | SMR4562 × P1(JW0797)a |
| PJH1966 | SMR4562 ∆cspE::KanFRT | SMR4562 × P1(JW618)a |
| PJH2602 | SMR4562 ∆dps::KanFRT ∆rpoS::FRT | PJH1399 × P1(JW0797)a |
| PJH2608 | SMR4562 ∆dps::FRT | PJH1952 × pCP20 |
| PJH2610 | SMR4562 Δhns::FRT | PJH1438 × pCP20 |
| PJH2673 | SMR4562 [F′ zah281::Tn10 Lac+] ∆fis::KanFRT | PJH1448 × P1(SMR8847) |
| PJH2677 | SMR4562 [F’ zah281::Tn10 Lac+] ∆cbpA::KanFRT | PJH1948 × P1(SMR8847) |
| PJH2715 | FC36 ∆hns::KanFRT | FC36 × P1(JW1225)a |
| PJH2716 | FC36 ∆fis::KanFRT | FC36 × P1(JW3229)a |
| PJH2732 | FC36 ∆cbpA::KanFRT | FC36 × P1(JW0985)a |
| PJH2790 | SMR4562 ∆fis::KanFRT [F′ lac-amplified] | PJH2716 × PH18 |
| PJH2792 | SMR4562 ∆fis::KanFRT [F′ lac-amplified] | PJH2716 × PJH33 |
| PJH2794 | SMR4562 ∆fis::KanFRT [F′ lac-amplified] | PJH2716 × PJH51 |
| PJH2804 | SMR4562 ∆cbpA::KanFRT [F′ lac-amplified] | PJH2732 × PH18 |
| PJH2806 | SMR4562 ∆cbpA::KanFRT [F′ lac-amplified] | PJH2732 × PJH33 |
| PJH2841 | SMR4562 ∆hns::KanFRT[F′ lac-amplified] | PJH2715 × PJH18 |
| PJH2843 | SMR4562 ∆hns::KanFRT [F′ lac-amplified] | PJH2715 × PJH33 |
| PJH2845 | SMR4562 ∆hns::KanFRT [F′ lac-amplified] | PJH2715 × PJH51 |
| PJH2863 | SMR4562 ∆hns::KanFRT [F′ zah281::Tn10 Lac+] | PJH1438 × P1(SMR8847) |
| PJH2867 | SMR4562 ∆dps::FRT recA::Tn10dCam | PJH2608 × P1(SMR4610) |
| PJH2874 | SMR4562 ∆cbpA::KanFRT [F′ lac-amplified] | PJH2732 × PJH51 |
| PJH2947 | SMR4562 soxR104 zjc2206::Tn10Kan | SMR4562 × P1(JTG1048) (Nunoshiba and Demple 1994) |
| PJH2961-4 | SMR4562 ∆attλ::PsulA-gfp ∆hns::KanFRT | SMR6039 × P1(JW1225)a |
| PJH2971-2974 | SMR4562 ∆attλ::PsulA-gfp ∆cbpA::KanFRT | SMR6039 × P1(JW0985)a |
| PJH2977 | SMR4562 ∆dps::KanFRT ∆dinB50::FRT [F′ ∆dinB50::FRT] | SMR4562 × P1(JTG1048) (Nunoshiba and Demple 1994) |
| PJH3019 | SMR4562 ∆rssB::tet ∆cbpA::KanFRT | SMR6039 × P1(JW1225)a |
| PJH3092 | SMR4562 ∆rssB::tet ∆fis::KanFRT | SMR6039 × P1(JW0985)a |
| PJH3114 | SMR4562 [F′ lafU2::FRTcatFRT dinB(oc)] ∆fis::KanFRT | SMR5889 × P1(JW0797)a |
| PJH3122 | SMR4562 [F′’ lafU2::FRTcatFRT dinB(oc)] ∆cbpA::KanFRT | SMR12566 × P1(JW0985)a |
| PJH3215 | SMR4562 ∆sodB::KanFRT | SMR12566 × P1(JW3229)a |
| PJH3217 | SMR4562 ∆hns::FRT ∆sodB::KanFRT | PJH2610 × P1(JW1648)a |
| PJH3219-3222 | SMR4562 ∆attλ::PsulA-gfp ∆fis::KanFRT | SMR6039 × P1(JW3229)a |
| PJH3245 | SMR4562 ∆dps::FRT ruvC53 eda::Tn10dCam | PJH2608 × P1(SMR6906) |
| PJH3267 | SMR4562 ∆dps::FRT soxR104 zjc2206::Tn10Kan | PJH2608 × P1(JTG1048) |
| SMR601 | ruvC53 eda51::Tn10 | Lopez et al. (2005) |
| SMR789 | FC40 ruvC53 eda51::Tn10 | FC40 × P1(SMR601) |
| SMR4562 | Independent construction of FC40 | McKenzie et al. (2000) |
| SMR4610 | SMR4562 recA::Tn10dCam | Bull et al. (2001) |
| SMR5889 | SMR4562 ∆dinB50::FRT [F’ ∆dinB50::FRT] | Galhardo et al. (2009) |
| SMR6039 | SMR4562 ∆attλ::PsulA-gfp | Hastings et al. (2004) |
| SMR6178 | SMR4562 ∆attλ::PsulA-gfp sulA::Tn5 lexA3(Ind−) malB::Tn9 | Al Mamun et al. (2012) |
| SMR6263 | MG1655 leuB::Tn10 | Gibson et al. (2010) |
| SMR6906 | SMR4562 ruvC53 eda::Tn10dCam | By linear replacement (Datsenko and Wanner 2000) into SMR789 |
| SMR8847 | SMR4562 [F′ zah281::Tn10 Lac+] | SMR4562 × P1(CAG12080) (Singer et al. 1989) |
| SMR10308 | SMR4562 [F’ lafU2::FRTcatFRT dinB21(oc)] | Galhardo et al. (2009) |
| SMR12566 | SMR4562 ∆rssB::tet | Al Mamun et al. (2012) |
All JW strains are from the Keio Collection, described in Baba et al. (2006).
| Strain/plasmid | Genotype | Source/reference |
|---|---|---|
| CAG12080 | zah281::Tn10 | Singer et al. (1989) |
| FC36 | Δ(lac proB)XIII thi ara RifR | Cairns and Foster (1991) |
| FC40 | FC36 [F’å45 = F′ proAB+ lacIq lacI33ΩlacZ] | Cairns and Foster (1991) |
| PJH18 | FC40 [F′ lac-amplified] | Hastings et al. (2000) |
| PJH33 | FC40 [F′ lac-amplified] | Hastings et al. (2000) |
| PJH51 | FC40 [F′ lac-amplified] | Hastings et al. (2000) |
| PJH1242 | SMR4562 Δlrp::KanFRT | SMR4562 × P1(JW0802) |
| PJH1390 | SMR4562 ΔrpoS::KanFRT | SMR4562 × P1(SMR10336) |
| PJH1399 | SMR4562 ΔrpoS::FRT | PJH1390 × pCP20 |
| PJH1428 | SMR4562 ΔleuO::KanFRT | SMR4562 × P1(JW0075)a |
| PJH1438 | SMR4562 ∆hns::KanFRT | SMR4562 × P1(JW1225)a |
| PJH1440 | SMR4562 ∆hha::KanFRT | SMR4562 × P1(JW0449)a |
| PJH1442 | SMR4562 ∆hfq::KanFRT | SMR4562 × P1(JW4130)a |
| PJH1444 | SMR4562 ∆stpA::KanFRT | SMR4562 × P1(JW2644)a |
| PJH1446 | SMR4562 ∆ihfB::KanFRT | SMR4562 × P1(JW0895)a |
| PJH1448 | SMR4562 ∆fis::KanFRT | SMR4562 × P1(JW3229a |
| PJH1948 | SMR4562 ∆cbpA::KanFRT | SMR4562 × P1(JW0985)a |
| PJH1952 | SMR4562 ∆dps::KanFRT | SMR4562 × P1(JW0797)a |
| PJH1966 | SMR4562 ∆cspE::KanFRT | SMR4562 × P1(JW618)a |
| PJH2602 | SMR4562 ∆dps::KanFRT ∆rpoS::FRT | PJH1399 × P1(JW0797)a |
| PJH2608 | SMR4562 ∆dps::FRT | PJH1952 × pCP20 |
| PJH2610 | SMR4562 Δhns::FRT | PJH1438 × pCP20 |
| PJH2673 | SMR4562 [F′ zah281::Tn10 Lac+] ∆fis::KanFRT | PJH1448 × P1(SMR8847) |
| PJH2677 | SMR4562 [F’ zah281::Tn10 Lac+] ∆cbpA::KanFRT | PJH1948 × P1(SMR8847) |
| PJH2715 | FC36 ∆hns::KanFRT | FC36 × P1(JW1225)a |
| PJH2716 | FC36 ∆fis::KanFRT | FC36 × P1(JW3229)a |
| PJH2732 | FC36 ∆cbpA::KanFRT | FC36 × P1(JW0985)a |
| PJH2790 | SMR4562 ∆fis::KanFRT [F′ lac-amplified] | PJH2716 × PH18 |
| PJH2792 | SMR4562 ∆fis::KanFRT [F′ lac-amplified] | PJH2716 × PJH33 |
| PJH2794 | SMR4562 ∆fis::KanFRT [F′ lac-amplified] | PJH2716 × PJH51 |
| PJH2804 | SMR4562 ∆cbpA::KanFRT [F′ lac-amplified] | PJH2732 × PH18 |
| PJH2806 | SMR4562 ∆cbpA::KanFRT [F′ lac-amplified] | PJH2732 × PJH33 |
| PJH2841 | SMR4562 ∆hns::KanFRT[F′ lac-amplified] | PJH2715 × PJH18 |
| PJH2843 | SMR4562 ∆hns::KanFRT [F′ lac-amplified] | PJH2715 × PJH33 |
| PJH2845 | SMR4562 ∆hns::KanFRT [F′ lac-amplified] | PJH2715 × PJH51 |
| PJH2863 | SMR4562 ∆hns::KanFRT [F′ zah281::Tn10 Lac+] | PJH1438 × P1(SMR8847) |
| PJH2867 | SMR4562 ∆dps::FRT recA::Tn10dCam | PJH2608 × P1(SMR4610) |
| PJH2874 | SMR4562 ∆cbpA::KanFRT [F′ lac-amplified] | PJH2732 × PJH51 |
| PJH2947 | SMR4562 soxR104 zjc2206::Tn10Kan | SMR4562 × P1(JTG1048) (Nunoshiba and Demple 1994) |
| PJH2961-4 | SMR4562 ∆attλ::PsulA-gfp ∆hns::KanFRT | SMR6039 × P1(JW1225)a |
| PJH2971-2974 | SMR4562 ∆attλ::PsulA-gfp ∆cbpA::KanFRT | SMR6039 × P1(JW0985)a |
| PJH2977 | SMR4562 ∆dps::KanFRT ∆dinB50::FRT [F′ ∆dinB50::FRT] | SMR4562 × P1(JTG1048) (Nunoshiba and Demple 1994) |
| PJH3019 | SMR4562 ∆rssB::tet ∆cbpA::KanFRT | SMR6039 × P1(JW1225)a |
| PJH3092 | SMR4562 ∆rssB::tet ∆fis::KanFRT | SMR6039 × P1(JW0985)a |
| PJH3114 | SMR4562 [F′ lafU2::FRTcatFRT dinB(oc)] ∆fis::KanFRT | SMR5889 × P1(JW0797)a |
| PJH3122 | SMR4562 [F′’ lafU2::FRTcatFRT dinB(oc)] ∆cbpA::KanFRT | SMR12566 × P1(JW0985)a |
| PJH3215 | SMR4562 ∆sodB::KanFRT | SMR12566 × P1(JW3229)a |
| PJH3217 | SMR4562 ∆hns::FRT ∆sodB::KanFRT | PJH2610 × P1(JW1648)a |
| PJH3219-3222 | SMR4562 ∆attλ::PsulA-gfp ∆fis::KanFRT | SMR6039 × P1(JW3229)a |
| PJH3245 | SMR4562 ∆dps::FRT ruvC53 eda::Tn10dCam | PJH2608 × P1(SMR6906) |
| PJH3267 | SMR4562 ∆dps::FRT soxR104 zjc2206::Tn10Kan | PJH2608 × P1(JTG1048) |
| SMR601 | ruvC53 eda51::Tn10 | Lopez et al. (2005) |
| SMR789 | FC40 ruvC53 eda51::Tn10 | FC40 × P1(SMR601) |
| SMR4562 | Independent construction of FC40 | McKenzie et al. (2000) |
| SMR4610 | SMR4562 recA::Tn10dCam | Bull et al. (2001) |
| SMR5889 | SMR4562 ∆dinB50::FRT [F’ ∆dinB50::FRT] | Galhardo et al. (2009) |
| SMR6039 | SMR4562 ∆attλ::PsulA-gfp | Hastings et al. (2004) |
| SMR6178 | SMR4562 ∆attλ::PsulA-gfp sulA::Tn5 lexA3(Ind−) malB::Tn9 | Al Mamun et al. (2012) |
| SMR6263 | MG1655 leuB::Tn10 | Gibson et al. (2010) |
| SMR6906 | SMR4562 ruvC53 eda::Tn10dCam | By linear replacement (Datsenko and Wanner 2000) into SMR789 |
| SMR8847 | SMR4562 [F′ zah281::Tn10 Lac+] | SMR4562 × P1(CAG12080) (Singer et al. 1989) |
| SMR10308 | SMR4562 [F’ lafU2::FRTcatFRT dinB21(oc)] | Galhardo et al. (2009) |
| SMR12566 | SMR4562 ∆rssB::tet | Al Mamun et al. (2012) |
| Strain/plasmid | Genotype | Source/reference |
|---|---|---|
| CAG12080 | zah281::Tn10 | Singer et al. (1989) |
| FC36 | Δ(lac proB)XIII thi ara RifR | Cairns and Foster (1991) |
| FC40 | FC36 [F’å45 = F′ proAB+ lacIq lacI33ΩlacZ] | Cairns and Foster (1991) |
| PJH18 | FC40 [F′ lac-amplified] | Hastings et al. (2000) |
| PJH33 | FC40 [F′ lac-amplified] | Hastings et al. (2000) |
| PJH51 | FC40 [F′ lac-amplified] | Hastings et al. (2000) |
| PJH1242 | SMR4562 Δlrp::KanFRT | SMR4562 × P1(JW0802) |
| PJH1390 | SMR4562 ΔrpoS::KanFRT | SMR4562 × P1(SMR10336) |
| PJH1399 | SMR4562 ΔrpoS::FRT | PJH1390 × pCP20 |
| PJH1428 | SMR4562 ΔleuO::KanFRT | SMR4562 × P1(JW0075)a |
| PJH1438 | SMR4562 ∆hns::KanFRT | SMR4562 × P1(JW1225)a |
| PJH1440 | SMR4562 ∆hha::KanFRT | SMR4562 × P1(JW0449)a |
| PJH1442 | SMR4562 ∆hfq::KanFRT | SMR4562 × P1(JW4130)a |
| PJH1444 | SMR4562 ∆stpA::KanFRT | SMR4562 × P1(JW2644)a |
| PJH1446 | SMR4562 ∆ihfB::KanFRT | SMR4562 × P1(JW0895)a |
| PJH1448 | SMR4562 ∆fis::KanFRT | SMR4562 × P1(JW3229a |
| PJH1948 | SMR4562 ∆cbpA::KanFRT | SMR4562 × P1(JW0985)a |
| PJH1952 | SMR4562 ∆dps::KanFRT | SMR4562 × P1(JW0797)a |
| PJH1966 | SMR4562 ∆cspE::KanFRT | SMR4562 × P1(JW618)a |
| PJH2602 | SMR4562 ∆dps::KanFRT ∆rpoS::FRT | PJH1399 × P1(JW0797)a |
| PJH2608 | SMR4562 ∆dps::FRT | PJH1952 × pCP20 |
| PJH2610 | SMR4562 Δhns::FRT | PJH1438 × pCP20 |
| PJH2673 | SMR4562 [F′ zah281::Tn10 Lac+] ∆fis::KanFRT | PJH1448 × P1(SMR8847) |
| PJH2677 | SMR4562 [F’ zah281::Tn10 Lac+] ∆cbpA::KanFRT | PJH1948 × P1(SMR8847) |
| PJH2715 | FC36 ∆hns::KanFRT | FC36 × P1(JW1225)a |
| PJH2716 | FC36 ∆fis::KanFRT | FC36 × P1(JW3229)a |
| PJH2732 | FC36 ∆cbpA::KanFRT | FC36 × P1(JW0985)a |
| PJH2790 | SMR4562 ∆fis::KanFRT [F′ lac-amplified] | PJH2716 × PH18 |
| PJH2792 | SMR4562 ∆fis::KanFRT [F′ lac-amplified] | PJH2716 × PJH33 |
| PJH2794 | SMR4562 ∆fis::KanFRT [F′ lac-amplified] | PJH2716 × PJH51 |
| PJH2804 | SMR4562 ∆cbpA::KanFRT [F′ lac-amplified] | PJH2732 × PH18 |
| PJH2806 | SMR4562 ∆cbpA::KanFRT [F′ lac-amplified] | PJH2732 × PJH33 |
| PJH2841 | SMR4562 ∆hns::KanFRT[F′ lac-amplified] | PJH2715 × PJH18 |
| PJH2843 | SMR4562 ∆hns::KanFRT [F′ lac-amplified] | PJH2715 × PJH33 |
| PJH2845 | SMR4562 ∆hns::KanFRT [F′ lac-amplified] | PJH2715 × PJH51 |
| PJH2863 | SMR4562 ∆hns::KanFRT [F′ zah281::Tn10 Lac+] | PJH1438 × P1(SMR8847) |
| PJH2867 | SMR4562 ∆dps::FRT recA::Tn10dCam | PJH2608 × P1(SMR4610) |
| PJH2874 | SMR4562 ∆cbpA::KanFRT [F′ lac-amplified] | PJH2732 × PJH51 |
| PJH2947 | SMR4562 soxR104 zjc2206::Tn10Kan | SMR4562 × P1(JTG1048) (Nunoshiba and Demple 1994) |
| PJH2961-4 | SMR4562 ∆attλ::PsulA-gfp ∆hns::KanFRT | SMR6039 × P1(JW1225)a |
| PJH2971-2974 | SMR4562 ∆attλ::PsulA-gfp ∆cbpA::KanFRT | SMR6039 × P1(JW0985)a |
| PJH2977 | SMR4562 ∆dps::KanFRT ∆dinB50::FRT [F′ ∆dinB50::FRT] | SMR4562 × P1(JTG1048) (Nunoshiba and Demple 1994) |
| PJH3019 | SMR4562 ∆rssB::tet ∆cbpA::KanFRT | SMR6039 × P1(JW1225)a |
| PJH3092 | SMR4562 ∆rssB::tet ∆fis::KanFRT | SMR6039 × P1(JW0985)a |
| PJH3114 | SMR4562 [F′ lafU2::FRTcatFRT dinB(oc)] ∆fis::KanFRT | SMR5889 × P1(JW0797)a |
| PJH3122 | SMR4562 [F′’ lafU2::FRTcatFRT dinB(oc)] ∆cbpA::KanFRT | SMR12566 × P1(JW0985)a |
| PJH3215 | SMR4562 ∆sodB::KanFRT | SMR12566 × P1(JW3229)a |
| PJH3217 | SMR4562 ∆hns::FRT ∆sodB::KanFRT | PJH2610 × P1(JW1648)a |
| PJH3219-3222 | SMR4562 ∆attλ::PsulA-gfp ∆fis::KanFRT | SMR6039 × P1(JW3229)a |
| PJH3245 | SMR4562 ∆dps::FRT ruvC53 eda::Tn10dCam | PJH2608 × P1(SMR6906) |
| PJH3267 | SMR4562 ∆dps::FRT soxR104 zjc2206::Tn10Kan | PJH2608 × P1(JTG1048) |
| SMR601 | ruvC53 eda51::Tn10 | Lopez et al. (2005) |
| SMR789 | FC40 ruvC53 eda51::Tn10 | FC40 × P1(SMR601) |
| SMR4562 | Independent construction of FC40 | McKenzie et al. (2000) |
| SMR4610 | SMR4562 recA::Tn10dCam | Bull et al. (2001) |
| SMR5889 | SMR4562 ∆dinB50::FRT [F’ ∆dinB50::FRT] | Galhardo et al. (2009) |
| SMR6039 | SMR4562 ∆attλ::PsulA-gfp | Hastings et al. (2004) |
| SMR6178 | SMR4562 ∆attλ::PsulA-gfp sulA::Tn5 lexA3(Ind−) malB::Tn9 | Al Mamun et al. (2012) |
| SMR6263 | MG1655 leuB::Tn10 | Gibson et al. (2010) |
| SMR6906 | SMR4562 ruvC53 eda::Tn10dCam | By linear replacement (Datsenko and Wanner 2000) into SMR789 |
| SMR8847 | SMR4562 [F′ zah281::Tn10 Lac+] | SMR4562 × P1(CAG12080) (Singer et al. 1989) |
| SMR10308 | SMR4562 [F’ lafU2::FRTcatFRT dinB21(oc)] | Galhardo et al. (2009) |
| SMR12566 | SMR4562 ∆rssB::tet | Al Mamun et al. (2012) |
All JW strains are from the Keio Collection, described in Baba et al. (2006).
Determination of Lac+ mutation rates
Experimental procedures are described in Harris et al. (1996) and Hastings et al. (2000). In all experiments reported, Lac− viable cell counts did not vary significantly over the course of the experiment, and mutations introduced by transduction did not affect colony formation time, measured in reconstruction experiments [reviewed by Rosenberg (2001)]. The proportion of lac-amplified and Lac+ point mutants (carrying −1-bp indels) was determined based on Hastings et al. (2000) by replating samples of colonies on rich medium plates containing 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (x-gal) dye and scoring for stable blue phenotype (Lac+ indels) vs. sectored-colony morphology (lac amplification). Absolute values for mutation rates (Lac+ colonies per cell per day) vary from day to day, but relative values remain constant (Lombardo et al. 2004). Errors bars shown are 1 SEM of three or more experiments.
Chromosome and F′ plasmid copy-number analyses
DAPI staining and flow cytometry were used to determine numbers of chromosomes per cell. Aliquots of saturated cultures grown for 3 days at 32° in M9 glycerol medium were fixed in ethanol and stained with 1 mg/ml DAPI in PBS. The average number of chromosomes per cell was calculated by measuring the DAPI fluorescence per cell and normalizing to the fluorescence value of the single-chromosome peak in the flow cytometry DAPI histogram for each sample. F′ plasmid copy number relative to the chromosome was calculated in the same cells by quantitative PCR (qPCR). Portions of cell cultures were collected, and genomic DNA was isolated using a modified cetyltrimethyl ammonium bromide (CTAB) method (Wilson 2001). Reactions contained 10 ng of genomic DNA and 350 µM primer in 1× KAPA SYBR FAST ABI Prism qPCR mix and were run on an Applied Biosystems 7900 HT. F′ copy number relative to the chromosome was determined using the ΔΔCt method (Livak and Schmittgen 2001), and from these measurements, F′ plasmid content per cell was calculated. The F′-specific primer sequences used were (5′–3′): F1: GATGGGACTTGATGTCTGTTAGG/CAGAGGAAGCAGAGGAGATAAATG; F2: GCTATAGCTGGCTCAAGTTAGG/CATGCAGACCTCACGAGTTAAT; and F3: GATGGGACTTGATGTCTGTTAGG/CAGAGGAAGCAGAGGAGATAAAT. oriC-specific primers were TTCGATCACCCCTGCGTACA/CGCAACAGCATGGCGATAAC. ter-specific primers were AATGATGCCGGTTACCCAAAGC/AGTTGCGTTTCGACGGTCATTC.
Quantitative P1 phage transduction assay for HR
Five-milliliter cultures of SMR4562 (Rec+), isogenic NAP deletion mutants, and an isogenic ∆recA control strain were grown in triplicate to saturation at 37° in LBH medium, as indicated by Gibson et al. (2010). Dilutions of each culture were plated on LBH medium to determine viable cell titers. P1 transduction (Miller 1992) was performed with 50 µl of either a 10−8, 10−7 dilution of P1 lysate from E. coli strain SMR6263 of known titer containing the mutation leuB::Tn10 (TetR) or 50 ml of LBH medium and 100 µl of each saturated culture. Then 1.5 × 107 phage particles and 1.5 to 2.1 × 109 cells were used in each reaction to ensure a multiplicity of infection of <0.1. Entire reaction mixtures were plated on solid LBH medium containing 3.3 µg/ml tetracycline and 20 mM sodium citrate. Using the titer of TetR transductant colonies and calculated phage particles added, frequencies of transductant colony-forming units (cfu) per phage were calculated.
UV-sensitivity assays
Five-milliliter cultures of repair-proficient SMR4562, isogenic NAP deletion mutants, and ∆recA control strain were grown in triplicate to saturation at 37° in LBH medium. Serial dilutions in M9 salts were plated in duplicate on LBH medium to determine viable cell titer, and appropriate dilutions were plated on LBH solid medium and exposed to UV light at 20, 40, 60, and 80 mJ/cm2. Irradiated plates were incubated in the dark at 37° overnight. Colonies were counted the next day, and survival was calculated.
Flow cytometric assay of spontaneous DNA damage and SOS response
All strains used for SOS induction determination carried the SOS-regulated PsulA-gfp allele (Pennington and Rosenberg 2007). PsulA-gfp is inserted in a nongenic chromosomal site in sulA+ cells (Pennington and Rosenberg 2007). Five-milliliter cultures of isogenic SMR6039 (SOS-proficient control), SMR6178 [lexA3(Ind) SOS-defective], and an NAP deletion mutant were grown for 3 days at 32° in M9 minimal glycerol medium as in MBR starvation assay conditions. Controls were grown in duplicate with four independently isolated strains of each NAP deletion derivative. Gates for GFP fluorescence in flow cytometry were set by using the parent strain (SMR6039) and the SOS induction-deficient control (SMR6178). Then 105 cells were measured per determination and the fraction of GFP+ cells reported. UV induction of the SOS response was measured at 80 mJ/cm2.
Results
We tested NAPs for possible roles in MBR. Only two, StpA and Hha, had no effect, and one, Dps, inhibited MBR (Figure 1A). Of the 10 that influenced MBR, 5 had been shown previously to be wholly or partly required for MBR, and their functions were partly characterized (Williams and Foster 2007; Al Mamun et al. 2012). We investigated the roles of the others.
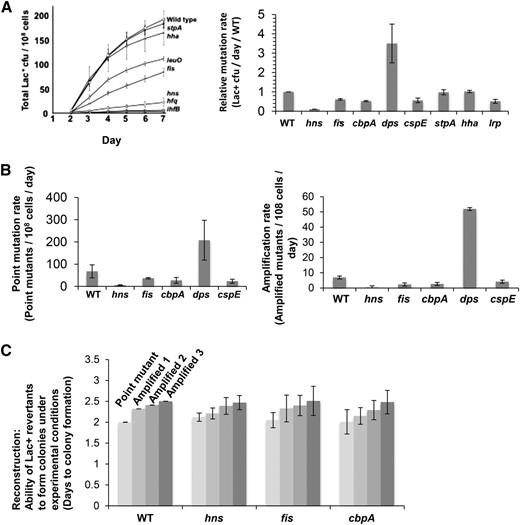
NAPs are required for mutagenic break repair. (A) Representative Lac assay experiment (left) showing requirements for NAPs for MBR and summary of MBR rates from three experiments (right) (mean rates ± SEM) compared with the wild-type protein. Deletion of hns, fis, cbpA, and cspE reduced MBR rates (P = 0.005, 0.03, 0.03, and 0.049, respectively, Student’s two-tailed t-test), whereas ∆dps increased the mutation rate. (B) Both point mutation and amplification are affected. Relative rates of point mutant (left) and lac-amplified colony formation (right). (C) Average time to colony formation in reconstruction experiments. These measure the time taken for strains carrying either the Lac+ point mutant or lac-amplified alleles to form colonies under MBR experimental conditions. hns, fis, and cbpA deletion strains formed colonies as rapidly as NAP+ cells and so are defective in mutagenesis, not growth of mutant colonies afterward. Strain designations relevant to the genotypes in all figures are given in Table 1.
We used the Lac assay for MBR (Cairns and Foster 1991). E. coli cells carrying a leaky lacIZ +1-bp frameshift allele can revert either by a single-base deletion compensatory frameshift mutation (SNV) or by amplification of the leaky lac allele to give a tandem array of 20 or more copies (GCR) (Hastings et al. 2000, 2004; Hersh et al. 2004; Rogers et al. 2015). TraI single-strand endonuclease makes nicks in the F′ that become DSBs (Ponder et al. 2005; Wimberly et al. 2013), which are required for both SNV and GCR (Harris et al. 1994; Wimberly et al. 2013).
Strategy
We used various assays that dissect known components of MBR under stress to distinguish how various NAPs affect MBR: (1) a quantitative P1 phage–mediated transduction test of HR proficiency at DSB ends, a proxy for DSB repair by HR, (2) sensitivity to UV light, which requires functional DSB repair (Khan and Kuzminov 2012), (3) a flow-cytometric assay for the ability to induce the SOS response, which uses an SOS-regulated promoter fused to gfp (Pennington and Rosenberg 2007), and (4) an assay for genes that promote MBR by contributing to induction of the RpoS general stress response (Al Mamun et al. 2012), which measures the ability to bypass a specific requirement for an MBR gene by artificial upregulation of RpoS (Al Mamun et al. 2012), achieved by deletion of the RpoS negative regulator rssB (Battesti et al. 2013). Finally, we tested for genes that promote MBR via activation of the SOS response, as in Al Mamun et al. (2012). SOS functions in the indel and base-substitution pathway of MBR by transcriptional upregulation of DinB error-prone DNA polymerase (Galhardo et al. 2009). Because the requirement for SOS is fully suppressed by constitutive expression of dinB at SOS-like levels, using a dinB operator-constitutive (oc) allele (Galhardo et al. 2009), genes that promote MBR solely via SOS induction are not needed for MBR in dinB(oc) cells. In addition, we determined relative changes in F′ plasmid copy number by qPCR, comparing the ratio of each three sites in the F′ plasmid with a chromosomal position.
Stationary-phase NAP Dps inhibits mutagenesis
We found that Dps inhibited mutagenesis (Figure 1A). Deletion of dps led to a 3.5-fold increase in Lac+ mutation rates (Figure 1A). Dps-mediated inhibition of MBR was seen for both point (indel) mutation and amplification rates (Figure 1B). The increased mutagenesis in ∆dps cells required known MBR proteins RpoS, RecA, RuvC, and DinB [reviewed in Rosenberg et al. (2012)] (Figure 2A). We conclude that increased mutagenesis in Δdps cells reflects increased MBR, not an increase in an alternative mutagenesis mechanism/pathway.
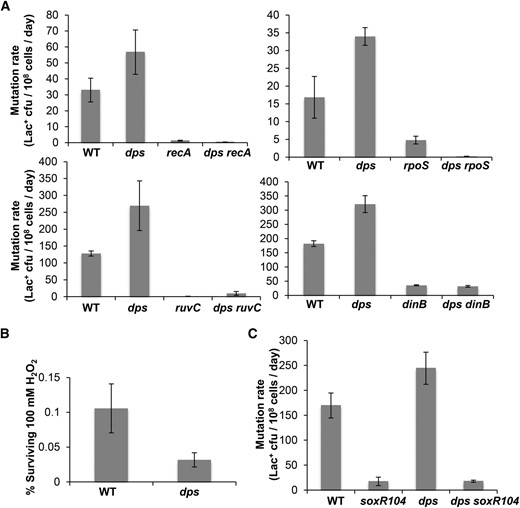
Dps inhibits the canonical MBR pathway. (A) Hypermutation in Δdps cells requires known MBR proteins RpoS, RecA, RuvC, and DinB. Thus, the MBR pathway (Rosenberg et al. 2012; Rogers et al. 2015), not some other mutagenic mechanism, was increased in ∆dps cells. Rates of three experiments (mean ± SEM) compared with those of Dps-proficient and Δdps single-mutant cells. (B) Δdps cells are sensitive to hydrogen peroxide. Relative survival of Dps-proficient and Δdps cells after short-term exposure to 100 mM H2O2 showed 70% lower viability of Δdps than Dps-proficient cells (72 ± 0.5%; n = 3 experiments). (C) Constitutive activation of the SoxR (anti-ROS) regulon reduces MBR. The soxR104 allele constitutively activates genes encoding ROS-detoxifying enzymes, reduces basal intracellular ROS content (Nunoshiba and Demple 1994), and here reduced MBR rate in Δdps and Dps-proficient strains (240 ± 30 vs. 18 ± 2 Lac+ cfu/108 cells/day for Δdps vs. soxR104 Δdps; 169 ± 25 vs. 17 ± 8 Lac+ cfu/108 cells/day for wild-type cells vs. soxR104; n = 3 experiments in both cases). These data imply that ROS promote MBR (explored in a separate study: J. M. Moore, S. M. Rosenberg, and P. J. Hastings, unpublished manuscript).
We investigated whether Dps inhibited mutagenesis by protection from ROS (reviewed earlier). If it did, we would expect that reduction in ROS would decrease MBR at least in Δdps cells and perhaps generally. In support of this possibility, we found that constitutive activation of the sox regulon via the soxR104 allele (Nunoshiba and Demple 1994), which reduces ROS levels, reduced MBR in otherwise wild-type cells and in Δdps cells (Figure 2C). The sox regulon includes sodA and sodB, which encode superoxide dismutases (Nunoshiba and Demple 1994). This result implies that MBR generally and increased MBR in Δdps cells specifically require ROS. We conclude that Dps inhibits MBR and suggest that MBR is promoted by ROS and that Dps inhibits MBR by protection against ROS. Further work is required to test this hypothesis.
Canonical NAPs H-NS, Fis, CspE, and CbpA are required for stress-induced MBR
Deletion of the genes encoding four NAPs, H-NS, Fis, CspE, and CbpA, caused significant reductions in mutagenesis, showing that they are wholly or partially required for MBR. Deletions of hns, fis, cspE, and cbpA resulted in 90, 50, 50, and 60% reduction in MBR rates, respectively (Figure 1A, P = 0.005, 0.03, 0.03, and 0.049, Student’s two-tailed t-test). Point mutation and amplification mechanisms were reduced roughly equally (Figure 1B). Further, we showed that the reduction in mutant colonies was not caused by a slow colony-growth phenotype of these mutants under these experimental conditions [reviewed in Rosenberg (2001)] (Figure 1C). We conclude that these NAPs are needed for mutagenesis, not for colony formation.
CspE is a known upstream regulator of RpoS stability (Shenhar et al. 2012), which is required for MBR (Layton and Foster 2003; Lombardo et al. 2004; Ponder et al. 2005; Shee et al. 2011; Al Mamun et al. 2012) and so was not studied further here.
CbpA promotes F′ plasmid maintenance
The following data imply that CbpA is not required for the DSB repair or SOS or RpoS response components of MBR but rather contributes to F′ plasmid maintenance. We assayed for defects in HR at DSB ends by quantitative P1 transduction (Gibson et al. 2010) (Figure 3A), a HR-dependent process (Rouviere-Yaniv et al. 1979). We infected ∆cbpA cells with P1 grown on donor bacteria carrying a tetracycline (Tet) resistance cassette and then determined the frequency of TetR recombinant colonies. ΔcbpA did not affect TetR transductant frequency, whereas the HR-blocking recA mutation did (Figure 3A), indicating that ΔcbpA cells are HR proficient. DSB repair proficiency also was supported by the finding that ΔcbpA strains were UV resistant (Figure 3B), unlike the DSB repair–defective ∆recA strain (Figure 3B) or recB (Kushner 1974). These data rule out the possibility that CbpA promotion of MBR is via promotion of HR, which is required for MBR.
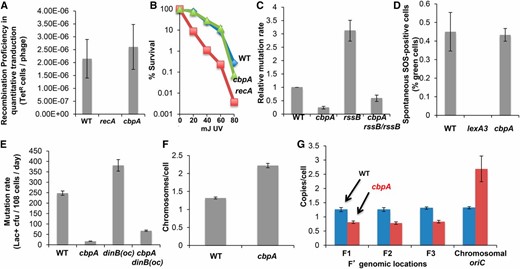
CbpA is required for F′ plasmid maintenance. (A) HR proficiency of ΔcbpA strains in P1-transduction HR assays (2.6 × 10−6 ± 7 × 10−7 TetR phages for wild-type cells vs. 2.6 × 10−6 ± 8 × 10−7 for ΔcbpA; n = 3 experiments). (B) UV resistance of ΔcbpA cells (P = 0.98, Student’s two-tailed t-test for significant difference between survival curves). Representative experiment of three experiments using three cultures per strain. (C) Deletion of RpoS-negative regulator rssB does not restore MBR ΔcbpA cells. MBR rates from three experiments ± SEM. (D) ΔcbpA cells are SOS response-induction proficient. Percentage of ΔcbpA cells showing green fluorescence in a population of 105 cells in strains with gfp fused to an SOS response-inducible promoter. lexA3 (lex-noninducible) was included as a positive control. Data from three experiments ± SEM. (E) dinB(oc) did not suppress the ΔcbpA MBR phenotype, showing that CbpA is not required in MBR for upregulation of the SOS response. Data from three experiments ± SEM. (F) ΔcbpA cells contain roughly 2.3 chromosomes per cell compared with 1.3 in wild-type cells. Comparison of fluorescence intensity due to DAPI straining. Data from three experiments ± SEM. (G) ΔcbpA strains are defective in maintaining the F′ plasmid. qPCR at oriC on the chromosome and at three sites on the F′ conjugative plasmid shows one F′ per chromosome in wild-type cells and 0.38 F′ per chromosome in ΔcbpA cells. This equates to an average of 0.87 F′ per cell. Data from three experiments ± SEM.
The following data suggest that CbpA promotes MBR other than or in addition to by promoting the RpoS stress response. RssB is a negative regulator of RpoS that targets RpoS for proteolytic degradation (Battesti et al. 2013). Deleting rssB increases RpoS levels and can suppress the MBR deficiencies of genes required in MBR for RpoS upregulation (Al Mamun et al. 2012). We found that removal of RssB from ΔcbpA strains did not suppress their MBR deficiency (Figure 3C), suggesting that CbpA does not promote MBR by upregulation of RpoS. MBR rates were 3.6 times higher in wild-type cells than ∆cbpA cells and 1.7 times higher in rssB cells than in ∆rssB ΔcbpA cells—ratios that did not quite differ significantly (P = 0.052, Student’s t-test).
Loss of CbpA did not affect the frequencies of spontaneous DNA damage or SOS induction, as shown with the flow cytometric assay of Pennington and Rosenberg (2007) for fluorescent SOS-induced cells (Figure 3D) carrying a chromosomal SOS-regulated PsulA-gfp transgene. Isogenic PsulA-gfp cells with the SOS-blocking lexA3(Ind−) mutation demonstrate the SOS dependence of fluorescence in this assay (Figure 3D) (Pennington and Rosenberg 2007). These data imply that CbpA is not required generally for SOS-response activation. Further, upregulation of dinB can substitute for the SOS response in MBR (Galhardo et al. 2009) such that dinB(oc) mutant alleles, which express SOS-induced levels of dinB constitutively, suppress the MBR deficiency of SOS-noninducible lexA(Ind−) mutants (Galhardo et al. 2009). We found that the dinB(oc) mutation did not alleviate the requirement for CbpA in MBR (Figure 3E). We conclude that CbpA promotes MBR other than or in addition to by upregulation of the SOS response.
We found that CbpA promotes maintenance of the F′ plasmid carrying the lac mutation-reporter gene as follows: in wild-type cells, chromosome copy number is two in ∼40% and one in ∼60% of stationary-phase cells (Akerlund et al. 1995). We used DAPI staining and flow cytometry to determine chromosome copy number and qPCR to determine the ratio of F′ plasmids to chromosomes. In wild-type cells, chromosome copy number averaged 1.3 chromosomes per cell (Figure 3F), similar to that seen previously (Akerlund et al. 1995), and the F′-to-chromosome ratio was ∼1:1 (Figure 3G; compare three F′ loci with the chromosomal locus). Whereas deletion of cbpA increased chromosome copy number to ∼2.3 chromosomes per cell (Figure 3F), ∆cbpA significantly reduced the F′-to-chromosome ratio to 0.38 (Figure 3, F and G, P = 0.00003, Student’s t-test). These data indicate that ΔcbpA cells had, on average, only 0.9 copies of the F′ per cell (2.3 × 0.38 = 0.87), suggesting that reduced MBR in ΔcbpA cells may result from an inability to maintain the F′ plasmid carrying the reporter allele either via fewer mutation targets or via fewer cells with a sister F′ plasmid to engage in DSB repair. We infer that reduced MBR in ΔcbpA cells probably results from reduced F′ copy number, which reduces MBR opportunities. The slight and not quite significant restoration of MBR in RpoS-elevated ∆rssB cells (Figure 3C) might affect maintenance of plasmid copy number—a hypothesis that remains to be tested.
Fis is required for SOS induction in MBR
We found that Fis promotes SOS induction, which upregulates DinB error-prone DNA polymerase during MBR. First, loss of Fis did not confer a significant reduction in HR assayed via quantitative P1 transduction (Figure 4A), which implies that HR at DSB ends is unaffected. Similarly, Δfis strains were not UV sensitive (Figure 4B), also indicating HR proficiency. Second, deletion of rssB did not restore MBR to ∆fis cells in that Δfis conferred similar MBR reductions in rssB and wild-type cells (Figure 4C, Fis/WT = 0.53, Fis RssB/RssB = 0.62, P = 0.119, Student’s two-tailed t-test). Third, ∆fis caused an insignificant increase in chromosome copy number (Figure 4D) and an increased F′-to-chromosome ratio (Figure 4E), which indicated more, not fewer, F′ plasmids per cell. Thus, Fis promotes MBR other than by decreasing homologous recombination, RpoS upregulation, or F′ plasmid copy number. However, Δfis conferred a threefold decrease in both spontaneous and UV-induced SOS induction, measured as GFP+ cells by flow cytometry of the fluorescent SOS-reporter strain (Figure 4F). Spontaneous SOS/GFP+ cells were ≤0.1% of ∆fis cells compared with 0.45% of wild-type cells (Figure 4F, P = 0.012, Student’s two-tailed t-test). After UV irradiation, only ∼20% of Δfis cells showed SOS induction, whereas ∼77% of wild-type cells did (Figure 4F, P = 0.031, Student’s two-tailed t-test). Moreover, the dinB(oc) allele completely restored MBR to Δfis cells (Figure 4G), indicating that SOS-induced levels of DinB created by this mutant allele (Galhardo et al. 2009) substituted fully for the role of Fis in MBR. These data indicate that Fis promotes MBR by allowing activation of the SOS response, which upregulates DinB.
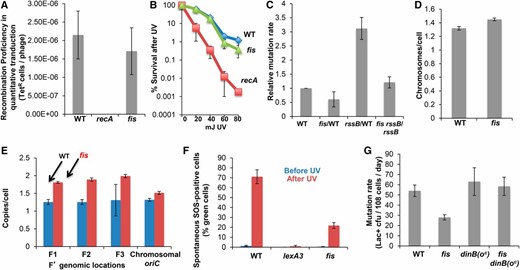
Fis promotes MBR by allowing the SOS response and DinB induction. (A) Δfis cells were as proficient as wild-type cells at recombinational DSB repair, as assayed by quantitative P1 transduction. Data from three experiments ± SEM. (B) Similar survival of Δfis cells and wild-type cells after UV irradiation. Representative experiment of three experiments using three cultures per strain. (C) The MBR defect of Δfis cells is not alleviated by ΔrssB upregulation of RpoS. Deletion of the rssB, a negative regulator of RpoS, did not suppress the fis MBR phenotype. Data from three experiments ± SEM. (D) Δfis cells did not alter chromosomal copy number, showing the same fluorescence intensity due to DAPI strain as wild-type cells. Data from three experiments ± SEM. (E) Δfis cells are not defective in plasmid maintenance, determined by qPCR at oriC and at three plasmid locations. Three experiments ± SEM. (F) Fis is required for SOS induction. UV irradiation induced GFP under an SOS-inducible promoter in Δfis cells only about one-quarter as well as in wild-type cells. (G) SOS-like levels of DinB provided by the dinB(oc) mutation completely suppresses the fis MBR defect. We conclude that Fis is required in MBR for induction of the SOS response that upregulates DinB. Data from 3 experiments ± SEM.
H-NS can be substituted in MBR by increasing ROS levels
Following the findings with Dps reported earlier, we discovered that H-NS also affects MBR by controlling ROS levels. Δhns cells showed no significant reduction in HR measured by quantitative transduction (Figure 5A) and were UV resistant (Figure 5B), indicating that the Δhns MBR defect is not due to loss of DSB repair ability. Similarly, the proportion of spontaneously SOS-induced cells was unchanged in Δhns cells, which indicates an intact SOS response (Figure 5C). Δhns caused higher chromosome copy numbers (Figure 5D) and higher F′-to-chromosome ratios (Figure 5E), which show that neither F′ maintenance nor availability of sister DNA molecules for repair account for H-NS function in MBR.
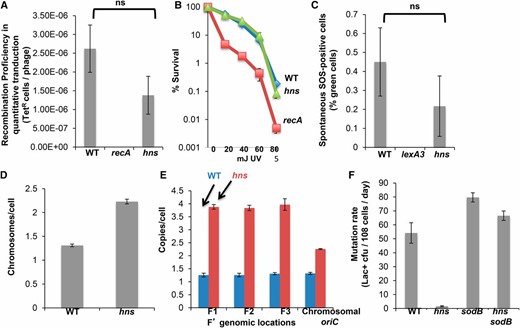
H-NS promotes MBR via maintenance of normal ROS levels. (A) Δhns cells are not defective in homologous recombination, as determined by quantitative P1 transduction. The mean of three experiments (± SEM) shows no significant difference from wild-type cells (P = 0.198, Student’s two-tailed t-test). (B) UV resistance of Δhns cells confirms no difference from wild-type cells in homologous recombination. Representative example of three experiments with three cultures per strain. (C) Δhns cells are proficient at SOS induction. Percentage of green fluorescent cells in populations of 105 cells of Δhns and wild-type cells with gfp fused to an SOS response-inducible promoter. Δhns cells showed no significant deficiency in spontaneous activation of the SOS response (P = 0.167, Student’s two-tailed t-test) measured by flow cytometry. (D and E) Δhns cells are not defective in plasmid maintenance. Deletion of hns led to an increase in both the average chromosomal content per cell (2.3 chromosomes per cell shown by DAPI staining) and F′-to-chromosome ratio (1.7 F′ per chromosome). qPCR was used to measure copy number of the chromosomal oriC region compared with multiple F′ sites. (F) H-NS promotes MBR by repressing SodB and maintaining normal ROS levels required for mutagenesis. Deletion of superoxide dismutase gene sodB completely suppressed the MBR defect of Δhns cells, implying that deletion of sodB and the resulting increase in ROS promotes MBR. Data are from three experiments ± SEM.
The following data indicate that the known H-NS role in transcriptional repression of sodB (Niederhoffer et al. 1990; Dubrac and Touati 2002; Zhang et al. 2005), a superoxide dismutase that detoxifies ROS, can explain H-NS function in MBR. We found that deletion of sodB, which increases superoxide levels (Liochev and Fridovich 1997), completely substituted for H-NS in MBR (Figure 5F). The Δhns ΔsodB strains showed higher MBR than the Δhns single mutant (Figure 5F, P = 0.00004, Student’s two-tailed t-test). Moreover, as observed previously (Farr et al. 1986), ΔsodB conferred only slight mutator activity in otherwise wild-type cells, which was not significant at the 5% level (less than twofold, P = 0.06, Student’s two-tailed t-test). That is, ∆sodB is not a general mutator but rather specifically relieves the MBR defect of ∆hns cells (Figure 5F). These data suggest that H-NS promotes MBR by repressing the ROS-detoxifying enzyme sodB, allowing ROS to persist within the cell, which promotes mutagenesis (also shown in Figure 2C). How ROS promote MBR is explored in a separate work (J. M. Moore, S. M. Rosenberg, and P. J. Hastings, unpublished manuscript).
Discussion
We found that the canonical NAPs Fis, H-NS, CspE, and CbpA are required for MBR and that Dps, the major stationary-phase NAP, inhibits MBR. Of 15 NAPs, 7 studied here and 5 studied previously (reviewed later), only 2, StpA, an analog of H-NS (Lucchini et al. 2009), and Hha did not affect MBR (Figure 1). Surprisingly, the various NAPs appear to modulate MBR by different mechanisms (summarized in Table 2) nearly all positively, and many, but not all, centered on stress-response regulation. Regulation of stress responses comprised the major allocation of genes in a large 93-gene network that underlies MBR in E. coli, highlighting the importance of stress-response activation to MBR (Al Mamun et al. 2012) (extended later).
Summary of NAP effects in MBR
| Mutant gene(s) | Effect on MBR | RpoS activationd | SOS activatione | RpoE activationf | HR proficiencyg | UV | F′ copy numberh | ROS levels |
|---|---|---|---|---|---|---|---|---|
| hns | Downa | ND | Downa | NCa | Downa | NCa | Upa | Downa |
| fis | Downa | NCa | Downa | NCa | Downa | NCa | NCa | |
| cbpA | Downa | NCa | NCa | NCa | NCa | NCa | Downa | |
| dps | Upa | NCa | NCa | NCa | NCa | NCa | Upa | |
| ihfA | Downb | NCb | Sb | NCb | ||||
| hupA hupB | Downc | NCc | NCc | NCc | ||||
| lrp | Downb | NC | Sb | NCb | ||||
| cspC | Downb | Downb | NCb | NCb | ||||
| cspE | Downa | Downi | ||||||
| hfq | Downb | Downb | NCb | NCb | Sb | |||
| hha | No effecta | |||||||
| stpA | No effecta |
| Mutant gene(s) | Effect on MBR | RpoS activationd | SOS activatione | RpoE activationf | HR proficiencyg | UV | F′ copy numberh | ROS levels |
|---|---|---|---|---|---|---|---|---|
| hns | Downa | ND | Downa | NCa | Downa | NCa | Upa | Downa |
| fis | Downa | NCa | Downa | NCa | Downa | NCa | NCa | |
| cbpA | Downa | NCa | NCa | NCa | NCa | NCa | Downa | |
| dps | Upa | NCa | NCa | NCa | NCa | NCa | Upa | |
| ihfA | Downb | NCb | Sb | NCb | ||||
| hupA hupB | Downc | NCc | NCc | NCc | ||||
| lrp | Downb | NC | Sb | NCb | ||||
| cspC | Downb | Downb | NCb | NCb | ||||
| cspE | Downa | Downi | ||||||
| hfq | Downb | Downb | NCb | NCb | Sb | |||
| hha | No effecta | |||||||
| stpA | No effecta |
Bold text, probable cause of MBR phenotype; NC, not changed; S, sensitive; UV, ultraviolet light sensitivity.
This work.
Epistasis with rssB.
Determined by flow cytometry.
Detergent sensitivity.
Quantitative transduction.
qPCR.
| Mutant gene(s) | Effect on MBR | RpoS activationd | SOS activatione | RpoE activationf | HR proficiencyg | UV | F′ copy numberh | ROS levels |
|---|---|---|---|---|---|---|---|---|
| hns | Downa | ND | Downa | NCa | Downa | NCa | Upa | Downa |
| fis | Downa | NCa | Downa | NCa | Downa | NCa | NCa | |
| cbpA | Downa | NCa | NCa | NCa | NCa | NCa | Downa | |
| dps | Upa | NCa | NCa | NCa | NCa | NCa | Upa | |
| ihfA | Downb | NCb | Sb | NCb | ||||
| hupA hupB | Downc | NCc | NCc | NCc | ||||
| lrp | Downb | NC | Sb | NCb | ||||
| cspC | Downb | Downb | NCb | NCb | ||||
| cspE | Downa | Downi | ||||||
| hfq | Downb | Downb | NCb | NCb | Sb | |||
| hha | No effecta | |||||||
| stpA | No effecta |
| Mutant gene(s) | Effect on MBR | RpoS activationd | SOS activatione | RpoE activationf | HR proficiencyg | UV | F′ copy numberh | ROS levels |
|---|---|---|---|---|---|---|---|---|
| hns | Downa | ND | Downa | NCa | Downa | NCa | Upa | Downa |
| fis | Downa | NCa | Downa | NCa | Downa | NCa | NCa | |
| cbpA | Downa | NCa | NCa | NCa | NCa | NCa | Downa | |
| dps | Upa | NCa | NCa | NCa | NCa | NCa | Upa | |
| ihfA | Downb | NCb | Sb | NCb | ||||
| hupA hupB | Downc | NCc | NCc | NCc | ||||
| lrp | Downb | NC | Sb | NCb | ||||
| cspC | Downb | Downb | NCb | NCb | ||||
| cspE | Downa | Downi | ||||||
| hfq | Downb | Downb | NCb | NCb | Sb | |||
| hha | No effecta | |||||||
| stpA | No effecta |
Bold text, probable cause of MBR phenotype; NC, not changed; S, sensitive; UV, ultraviolet light sensitivity.
This work.
Epistasis with rssB.
Determined by flow cytometry.
Detergent sensitivity.
Quantitative transduction.
qPCR.
Perhaps the most surprising aspect of our results and those of another recent study (Warnecke et al. 2012) is the absence of detectable direct cis effects of NAPs on mutagenesis in the DNA sites that those NAPs bind. Instead, our study shows important trans effects on regulation of regulatory and other proteins and components of MBR (Table 2). Warnecke et al. (2012) used Chip-Seq to map chromosomal distributions of four NAPs—H-NS, Fis, IhfA, and IhfB—at different growth phases and compared those distributions with patterns of mutations across the genomes of 54 sequenced E. coli strains. They found a small but significant correlation of NAP occupancy with genome variation between strains (Warnecke et al. 2012). By contrast, we found robust evidence that most NAPs modulate genome evolution/mutagenesis positively in trans by virtue of their regulatory functions (summarized next).
Dps, the sole NAP inhibitor of MBR (Table 2 and Figure 1B), compacts and cocrystalizes with DNA in the stationary-phase nucleoid and protects DNA/cells from ROS (Ali Azam et al. 1999). We discovered that ROS are required for MBR in that constitutive expression of the SoxR regulon, which encodes proteins that detoxify ROS, reduced MBR (Figure 2C). We suggest that Dps might inhibit MBR by reduction of ROS or their effects on DNA. The role of ROS in MBR is explored in a separate study (J. M. Moore, S. M. Rosenberg, and P. J. Hastings, unpublished manuscript).
We found that Fis promotes MBR by allowing activation of the SOS DNA-damage response, which promotes MBR by upregulation of DinB error-prone DNA polymerase (Figure 4, F and G). Providing SOS-like DinB levels with a dinB operator-constitutive allele (Galhardo et al. 2009) wholly substituted for Fis in MBR (Figure 4G).
CspE is required for MBR (Figure 1, A and B). CspE is a known positive regulator of RpoS (Shenhar et al. 2012), suggesting that CspE, like Fis, might exert its influence on MBR through stress-response regulation—SOS for Fis and RpoS for CspE.
H-NS is also required for MBR (Figure 1, A and B) apparently via its transcriptional repression of sodB (Niederhoffer et al. 1990; Dubrac and Touati 2002; Zhang et al. 2005), which encodes a superoxide dismutase that detoxifies ROS. Deletion of sodB completely and specifically alleviated the need for H-NS in MBR (Figure 5F), presumably by allowing the accumulation of ROS. ∆sodB caused no such increase in MBR in otherwise wild-type (H-NS+) cells, implying that sufficient ROS for MBR occur in H-NS-proficient cells.
The reduction in mutagenesis in ΔcbpA cells seems likely to result from decreased numbers of mutation reporter-bearing F′ plasmids in those cells. The calculated average of 0.9 plasmids per ΔcbpA cell implies that many cells do not carry more than one copy of the plasmid. This would make HR repair of F′-located DSBs unlikely in most cells and thus reduce MBR in the F′. Increased F′ copy number was previously proposed to cause increased MBR (Foster and Rosche 1999). Because the error-prone polymerase DinB is encoded in both the F′ and the chromosome in these strains, it is possible that both chromosomal and F′ mutations might be influenced by F′ copy number, as seen previously by comparing mutations in F− and F′ cells—more copies of dinB gave more mutations additively (Shee et al. 2011).
Of the five NAPs previously shown to promote MBR [Lrp, Hfq, CspC, IHF (Al Mamun et al. 2012), and HU (Williams and Foster 2007); reviewed Table 2], Hfq, CspC, and Lrp were shown to act upstream of activation of stress-response regulators—RpoS for Hfq and CspC and RpoE for Lrp (Al Mamun et al. 2012). Additionally, ihfA mutants mimicked the sensitivity to detergent with EDTA of RpoE response-defective cells but showed normal expression of the RpoE-regulated rpoH P3 promoter (Al Mamun et al. 2012), perhaps because the rpoH-P3 assay is not conducted under similar starvation-stress conditions. The fifth NAP, HU, affected MBR possibly by promoting HR (Williams and Foster 2007). Thus, four of these five NAPs appear to promote MBR via stress-response regulation. Because of their essential nature, IciA and DnaA (Chodavarapu et al. 2008a) were not included in this study. CbpB, which, like IciA and DnaA, binds the replication origin (Skarstad et al. 1993), also was not studied.
Our results both support and reveal the incompleteness of our previous study of a large network of genes required for MBR (Al Mamun et al. 2012). First, the screen that identified most of the MBR network detected about half the then-known MBR genes, suggesting that additional MBR genes remain to be discovered. Our discovery here of four NAPs that promote MBR (Fis, H-NS, CspE, and CbpA) (Figure 1), in addition to the four NAPs identified in the previous screen [Lrp, Hfq, CspC, and IHF (Al Mamun et al. 2012)], implies that the full MBR network is likely to be about twice as large as the 93 genes of Al Mamun et al. (2012)—∼180–190 genes. In addition to the four NAPs added in this work, we also subsequently identified the transcription protein Mfd (Wimberly et al. 2013) as MBR promoting. Also discovered after the 93-gene network of Al Mamun et al. (2012), PhoB and PhoR promote MBR by an as yet unknown mechanism (Gibson et al. 2015). This adds 7 new genes to the MBR network for a total of 100 demonstrated MBR genes (Figure 6).
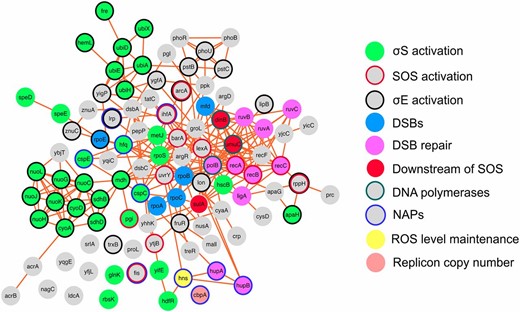
The updated mutagenic break-repair network with NAPs. Protein-protein interactions: CytoScape 3.0.2 software, unweighted force-directed layout (Saito et al. 2012), links from String 9.1 (Franceschini et al. 2013). Proteins that promote RpoS, RpoE, SOS activation, DSBs, and DSB repair are shown as solid green, black circle, red circle, solid cyan, and solid pink, respectively. Downstream of SOS, solid red; DNA polymerases, green circle; ROS level maintenance, solid yellow; replicon copy number, solid salmon; NAPs, blue circle; unknown function in MBR, solid gray (Gibson et al. 2015). This rendition includes the NAPs shown here to promote MBR, and the other proteins discovered to promote MBR after the initial description of the MBR network by Al Mamun et al. (2012): Mfd (Wimberly et al. 2013) and PhoB and PhoR (Gibson et al. 2015).
Second, more than 50% of the original network genes was implicated as acting upstream of activation stress responses, sensing and transducing stress signals to stress-response regulators that activate the effector proteins that cause mutations in DNA (Al Mamun et al. 2012). Downstream effectors constitute a small fraction of the network. As illustrated in Figure 6, our data show that most NAPs are upstream regulators. Most allow activation of stress responses (Table 2). Thus, the most important allocation of NAP genes to MBR, and of genes to MBR generally, is in the temporal regulation of mutagenesis to times of stress, when cells are poorly adapted to their environments. Modeling indicates that this strategy can accelerate evolution even in asexual bacterial populations, which do not easily lose deleterious mutations, and can be selected on this basis (Ram and Hadany 2012; Ram and Hadany 2014). The number of NAPs and other genes in this role indicates the importance of stress-response regulation to mutagenesis in E. coli.
Acknowledgments
We are very grateful to P. C. Thornton for technical assistance. This work was supported by the National Aeronautics and Space Administration through the NASA Astrobiology Institute under Cooperative Agreement No. NNA13AA91A issued through the Science Mission Directorate (P.J.H.) and National Institutes of Health grant R01-GM53158 (S.M.R.).
Author contributions: P.J.H. conceived the project; P.J.H., J.M.M., D.B., and S.M.R. directed the work; J.M.M., D.M., A.K.M., and M.A.B.N. performed the experiments; and J.M.M., S.M.R., and P.J.H. wrote the paper.
Footnotes
Communicating editor: J. A. Nickoloff
Literature Cited
Author notes
Present address: Department of Bioengineering MS-142, Rice University, Houston, TX 77005.
Present address: Undergraduate Program on Genomic Sciences, National Autonomous University of Mexico, Colonia Chamilpa, Cuernavaca, 62210, Morelos, Mexico.
Present address: Graduate School of the Stowers Institute for Medical Research, Kansas City, MO 64110.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}