





Abstract
Mutations identified in zebrafish genetic screens allow the dissection of a wide array of problems in vertebrate biology. Most screens have examined mutations induced by treatment of spermatogonial (premeiotic) cells with the chemical mutagen N-ethyl-N-nitrosourea (ENU). Treatment of postmeiotic gametes with ENU induces specific-locus mutations at a higher rate than premeiotic regimens, suggesting that postmeiotic mutagenesis protocols could be useful in some screening strategies. Whereas there is extensive evidence that ENU induces point mutations in premeiotic cells, the range of mutations induced in postmeiotic zebrafish germ cells has been less thoroughly characterized. Here we report the identification and analysis of five mutations induced by postmeiotic ENU treatment. One mutation, snhst1, is a translocation involving linkage group (LG) 11 and LG 14. The other four mutations, oepst2, knyst3, Df(LG 13)st4, and cycst5, are deletions, ranging in size from less than 3 cM to greater than 20 cM. These results show that germ cell stage is an important determinant of the type of mutations induced. The induction of chromosomal rearrangements may account for the elevated frequency of specific-locus mutations observed after treatment of postmeiotic gametes with ENU.
GENETIC screens in the zebrafish (Danio rerio) provide the means to identify vertebrate genes with essential functions (Eisen 1996; Schier and Talbot 1998; Detrich et al. 1999). Large-scale screens employing two-generation breeding schemes have obtained mutations in hundreds of genes required for embryonic viability (Driever et al. 1996; Haffter et al. 1996). In these screens, embryos were examined at several embryonic timepoints, and a wide array of mutant phenotypes was detected. A number of smaller screens have also been conducted, some taking advantage of single-generation haploid and parthenogenetic diploid screening protocols, to identify mutations disrupting specific processes such as hindbrain patterning, neural crest development, and visual system function (Brockerhoff et al. 1995; Henion et al. 1996; Moens et al. 1996; Beattie et al. 1999). Thus mutations discovered in zebrafish genetic screens allow the genetic dissection of vertebrate development, physiology, and behavior.
A variety of agents have been employed to induce mutations in the zebrafish germ line, including γ-rays, insertional vectors, and chemicals such as 4,5′,8-trimethylpsoralen (TMP) and N-ethyl-N-nitrosourea (ENU; Mullins et al. 1994; Solnica-Krezel et al. 1994; Riley and Grunwald 1995; Gaiano et al. 1996; Ando and Mishina 1998; Walker 1999). Of these, the DNA alkylating agent ENU is by far the most widely employed. Most zebrafish mutations have been induced by ENU mutagenesis of spermatogonial stem cells (Mullins et al. 1994; Solnica-Krezel et al. 1994; van Eeden et al. 1999), accomplished by several treatments of adult males at weekly intervals followed by a waiting period to eliminate affected postmeiotic germ cells. In specificlocus tests with pigmentation genes, spermatogonial ENU treatment induces 1–3 mutations per locus per 1000 mutagenized haploid genomes, a rate high enough to permit efficient screens. Extensive genetic and molecular analyses indicate that the great majority of lesions resulting from spermatogonial ENU mutagenesis are point mutations, which are suitable for defining the functions of individual genes (Mullins et al. 1994; Solnica-Krezel et al. 1994; see also Table 2). Because this protocol induces mutations in germ-line stem cells, mutations are transmitted at this rate for at least several months after the treatment. Another important advantage is that mutations are induced prior to meiosis, so that the initial mutation, usually an ethylation of a base on one DNA strand, is fixed by DNA replication prior to production of differentiated sperm, leading to nonmosaic offspring in the next generation.
Some recent screens have employed postmeiotic ENU mutagenesis, accomplished by mating males within 2 wk of a single ENU treatment (Riley and Grunwald 1995; Alexander et al. 1998; Appel et al. 1999). Postmeiotic 10-fold increase in the frequency of induced mutations in specific-locus tests (Riley and Grunwald 1995). The mosaicism in F1 individuals and resulting non-Mendelian transmission from the F1 to F2 generations hinder some screening protocols. However, mosaicism in the F1 generation is not a great impediment for single-generation screens of haploid F2 embryos, which can detect mutations present in only a few percent of progeny (Alexander et al. 1998). Genetic and, in one case, molecular tests show that postmeiotic ENU treatment can induce point mutations (Riley and Grunwald 1995; Appel et al. 1999), but the range of mutations induced has not been extensively analyzed.
Here we report the identification and analysis of five zebrafish mutations induced by postmeiotic ENU treatment. Genetic mapping and analysis of the mutant chromosomes show that each of the five mutations is a chromosomal rearrangement. Four alleles are deletions, ranging in size from <3 cM to ≥20 cM, and the fifth mutation is a translocation. These results indicate that ENU induces different spectrums of mutations in premeiotic and postmeiotic germ cells and that the elevated specific-locus mutation frequency of postmeiotic mutagenesis may result from the production of chromosomal rearrangements.
MATERIALS AND METHODS
ENU mutagenesis: Twenty males were exposed to 0.8 mm ENU in 3 mm 4-morpholineethanesulfonic acid, 0.08% NaCl for 1 hr at 28°, essentially as described by Riley and Grunwald (1995). Two days after ENU exposure (d2), mutagenized males were crossed in natural matings to albino (alb)−/+ females. An additional cross with alb−/+ females was performed on d11, and crosses with wild-type females were performed on d7 and d15. Mutations induced in postmeiotic cells are transmitted for 2–3 wk following ENU treatment; after 2–3 wk mutations induced in premeiotic cells are transmitted (Mullins et al. 1994; Solnica-Krezel et al. 1994; Riley and Grunwald 1995). Unfertilized eggs and dead embryos were removed on the first and second days of development, and surviving progeny were examined on the third day. About 30% (1860 of 6234) of F1 progeny developed as severely abnormal embryos, displaying a range of malformations. To determine the frequency of ENU-induced mutations at alb, progeny from crosses of ENU males to alb−/+ females were scored for the presence of nonpigmented cells within the eye (progeny with severe abnormalities were excluded). Of these embryos, 1.5% (56 of 3699) had alb mutant cells in the eye, corresponding to mutation rate of 3.0% (56/1849.5 haploid genomes screened). Viability of F1 progeny was also monitored. Nearly all severely abnormal embryos died prior to the free-feeding stage. Of progeny without severe abnormalities, one-third (619 of 1841 monitored) survived to the free-feeding stage, and survival rates beyond this stage were very high.
Nomenclature: According to nomenclature of Drosophila rearrangements (Ashburner 1989) and previous use in zebrafish (Talbot et al. 1998), the st1 reciprocal translocation is described as T(LG 11;LG 14)st1, and the two elements of the translocation are termed T(LG 11;LG 14)st1,11U14L and T(LG 11;LG 14)st1,14U11L (where U and L refer to the upper and lower segments of the affected linkage groups shown in standard orientation). Segregation of the rearranged LG 11-LG 14 chromosomes and their standard order counterparts results in euploid and aneuploid meiotic products, including snhst1 and Df(LG 14)st1 (see Figure 7). snhst1 mutants bear a deletion of ~40 cM on LG 11 and a duplication of ~60 cM on LG 14. Df(LG 14)st1 mutants bear a deletion of ~30 cM on LG 14 and a duplication of ~35 cM on LG 11.
Screening: Haploid embryos were generated as described (Ransom and Zon 1999; Walker 1999). Embryos were staged according to standard morphological criteria (Kimmel et al. 1995). Whole mount in situ hybridization was done as described (Schier et al. 1997) using a goosecoid (gsc) probe (Stachel et al. 1993).
Genetic mapping and markers: Mapping methods and PCR conditions have been described (Postlethwait and Talbot 1997; Gates et al. 1999; Talbot and Schier 1999). Primers for simple sequence length polymorphism (SSLP) markers (Knapik et al. 1998; Shimoda et al. 1999) were obtained from Research Genetics (Huntsville, AL) or synthesized based on sequences from the MGH/CVRC Zebrafish Server (http://zebrafish.mgh.harvard.edu/). The following primer pairs were used to amplify DNA fragments from oep, BAC165D4, bmp7, and cyclops (Rebagliati et al. 1998; Sampath et al. 1998; Zhang et al. 1998; Dick et al. 2000; Schmid et al. 2000). oep: 5′-GAGATGGAGATGTTCTAATGGTG-3′ and 5′-ATTCAAGA ATTCAGCGTATTGC-3′; BAC165D4: 5′-GATAAATCAAACA CCTTTGTTTA-3′ and 5′-TCCCAAAACTGTATTATAGTTCC-3′, bmp7: 5′-TTCCACTTTGATAGTGGCTTG-3′ and 5′-ATCC AGCATGTACATGGGAGC-3′, and cyclops: 5′-GGTGGACATGCATGTGGATTT-3′ and 5′-CCGTTCTCGTAGTACAGCATA-3′. The WIK line (Rauch et al. 1997a) was used to establish polymorphic mapping crosses.
RESULTS
Genetic screens for embryonic lethal mutations induced by postmeiotic mutagenesis with ENU: To identify mutations that disrupt embryonic development, we used morphological criteria and marker gene expression to screen haploid progeny of F1 mutagenized females, as described below. Mutations were induced in adult males with a single 1-hr treatment of 0.8 mm ENU (Riley and Grunwald 1995). Treated males were crossed to untreated females to produce the F1 generation. Haploid F2 progeny of F1 females were screened for mutants with abnormal morphology at approximately 12 and 24 hr postfertilization, corresponding to the early segmentation and pharyngula periods, respectively (Kimmel et al. 1995). In addition, mutations in genes required for the normal pattern of goosecoid (gsc) expression were sought using whole-mount in situ hybridization to analyze gsc transcripts in haploid progeny at the early gastrula stage (6 hr). Because F1 individuals produced by postmeiotic treatment of males with ENU are mosaic (Riley and Grunwald 1995), we examined at least 25 haploid F2 embryos from each F1 female screened. F1 females whose offspring included putative mutants were rescreened and, if the phenotype was observed in a second batch of embryos, outcrossed to normal males to produce a diploid F2 generation. Mutations were recovered by analyzing the haploid progeny of
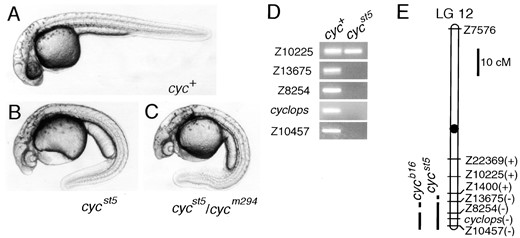
The cycst5 mutation causes cyclopia and deletes a segment of LG 12. Morphology of (A) wild-type, (B) cycst5 homozygous, and (C) cycst5/cycm294 diploid embryos at 36 hr. (D) Pools of DNA from cyc+ and cycst5 diploid embryos were tested with SSLP markers on LG 12. All SSLP markers were also tested in individual embryos to confirm the presence or absence of the markers (data not shown). (D) Linkage map of LG 12 is shown with the presence (+) or absence (−) of the SSLP markers in cycst5. Primers for the cyc gene (Rebagliati et al. 1998; Sampath et al. 1998) were also tested. The cycb16 mutation, already known to have a deletion on LG 12 (Rebagliati et al. 1998; Sampath et al. 1998; Talbot et al. 1998) was also tested with the SSLP markers for comparison. cycb16 deletes Z10457 and Z8254 but does not remove Z13675, which is deleted in cycst5. Marker distances are based on Shimoda et al. (1999).
F2 females, a fraction of which are heterozygous for mutations present in the germ line of the F1 female. Fourteen putative mutations were identified among the haploid progeny of the 284 F1 females that were screened. Six of these mutations were recovered, and analysis revealed that all of these were chromosomal rearrangements (see below; Y. Imai, A. F. Schier and W. S. Talbot, unpublished results). Here we report a phenotypic and genetic analysis of four mutations recovered in this screen, oepst2, knyst3, Df(LG 13)st4, and snhst1.
We also describe a fifth mutation, cycst5, which was identified in a noncomplementation screen for new mutant alleles of cyclops and squint. In this screen, the same F1 fish produced after postmeiotic ENU mutagenesis were crossed to doubly heterozygous sqtcz35/+; cycm294/+ tester fish. The resulting embryos were scored for cyclopic phenotypes by morphological inspection. In most cases, F1 fish whose progeny included cyclopic embryos were crossed again to sqtcz35/+; cycm294/+ testers. To establish stocks for the recovery of putative mutations, F1 individuals were outcrossed to wild type. We recovered two mutations from a screen of 228 F1 fish. One of these was cycst5, described below, and preliminary analysis of the second suggests that it is a translocation involving LG 21 (Y. Imai, unpublished results), the location of the squint gene (Feldman et al. 1998). One other F1 produced cyclopic progeny in the initial test and the rescreen, but the recovery of this putative mutation was not pursued.
cycst5 deletes the cyc region of LG 12: The cycst5 mutation was identified by crossing a mosaic F1 individual to a sqtcz35/+; cycm294/+ double heterozygote (8% mutant, 3/38 total). cycst5 is a Mendelian recessive mutation (23.0% of cycst5/+ intercross progeny are mutants, 105/456 total) that produces a cyclopic phenotype (Figure 1B). Crosses of cycst5/+ and cycm294/+ individuals yielded cyclopic embryos (24.9% mutant, 140/562 total) that were morphologically indistinguishable from cycm294 homozygotes (Figure 1C). As previous work showed that cyclops maps to LG 12 (Rebagliati et al. 1998; Sampath et al. 1998; Talbot et al. 1998), we analyzed markers from this linkage group in cycst5 mutants. Primers corresponding to the cyc gene and nearby markers, including Z10457, Z8254, and Z13675, did not amplify products from cycst5 genomic DNA. Thus the cycst5 mutation deletes about 10 cM of the distal region of LG 12, including cyclops (Figure 1, D and E).
oepst2 deletes the oep region of LG 10: Homozygous oepst2 mutants display cyclopia at 24 hr (Figure 2B). In addition, gsc expression is strongly reduced in haploid embryos at the early gastrula stage (6 hr, data not shown). Because cyclopia and reduced gsc expression are characteristic of the previously described oep mutant phenotype (Schier et al. 1997), we tested whether oepst2 complemented the oeptz57 mutation. The oepst2 mutation is a Mendelian recessive allele of oep: approximately one-fourth (22 mutants among 93 progeny) of offspring from an oeptz57/+ × oepst2/+ cross displayed a cyclopic phenotype morphologically indistinguishable from oeptz57 homozygotes (Figure 2, C and D). At 24 hr, oepst2 homozygotes show signs of neural degeneration (Figure 2B), a phenotype not characteristic of oeptz57. By molecular criteria, oeptz57 is a null allele (Zhang et al. 1998), suggesting that the neural degeneration in oepst2 homozygotes results from the loss of one or more genes linked to oep. To determine whether the oepst2 mutant chromosome bears a deletion in the oep region, we analyzed the oep gene and neighboring markers in oepst2 mutants. A pair of primers for the oep gene amplified the expected fragment from genomic DNA prepared from wild-type haploid embryos, but not their oepst2 mutant siblings (Figure 2E). SSLP markers in the oep region, including Z7316, Z8146, and Z13632, were also deleted, whereas
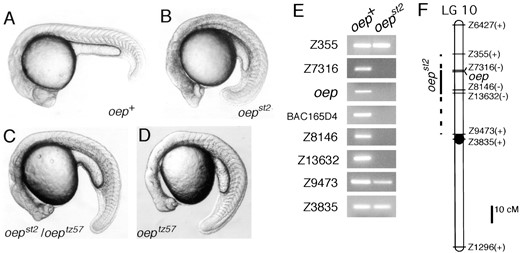
The oepst2 mutation causes cyclopia and deletes the oep region of LG 10. Morphology of (A) wild-type, (B) oepst2 homozygous, (C) oepst2/oeptz57, and (D) oeptz57 homozygous diploid embryos at 24 hr. (B) The oepst2 mutant shows a neural degeneration phenotype (note the dark region of brain) in addition to cyclopia. (E) Pools of DNA from wild-type (oep+) and oepst2 haploid siblings were tested with SSLP markers on LG 10. Primers for the oep gene and an STS from BAC165D4 (Zhang et al. 1998), a clone isolated in the oep chromosomal walk, were also tested. All SSLP markers were also tested in individual haploid embryos to confirm the presence or absence of the markers (data not shown). (F) Linkage map of LG 10 with the presence (+) or absence (−) of markers in oepst2. Marker distances are based on Shimoda et al. (1999). The oep gene was placed in reference to the other markers using data from the T51 RH panel (Geisler et al. 1999) and the heat-shock diploid genetic mapping panel (http://zebrafish.stanford.edu).
more distal (Z6427 and Z355) and proximal (Z9473 and Z3835) LG 10 markers were present on the oepst2 mutant chromosome (Figure 2, E and F). Thus oepst2 deletes a 12- to 33-cM segment of LG 10 that includes the oep gene.
knyst3 deletes a segment of LG 14: Homozygous knyst3 embryos have a short axis (Figure 3B), a phenotype characteristic of the convergent extension mutants trilobite and knypek (Solnica-Krezel et al. 1996). In addition, some knyst3 haploid embryos were cyclopic (Figure 3D), but this phenotype was not evident in homozygous knyst3 diploid embryos. knyst3 failed to complement knym119 (28% mutant, 37/132 total), whereas all progeny from knyst3/+ × tritk50/+ crosses were wild type. knyst3 was inherited as a simple Mendelian recessive mutation (Table 1). We mapped the knyst3 mutation by analyzing SSLP markers covering the genome in pools of genomic DNA from wild-type and knyst3 mutant siblings (Figure 3F; data not shown). SSLPs on LG 14 were linked to knyst3, and analysis of haploid individuals (Figure 3E) in knyst3 mapping crosses placed the mutation near markers Z9057 and Z22107 (0 and 5 recombinants, respectively, among 141 haploid progeny). These experiments also revealed that the knyst3 mutant chromosome harbors a deletion on LG 14, as markers Z8801, Z20663, and Z3091 did not amplify from knyst3 mutant genomic DNA (Figure 3F). Neighboring LG 14 markers, including Z9057, Z6667, Z20545, and Z13401, are present on the mutant chromosome, indicating that knyst3 covers <3 cM of the LG 14 genetic map (Figure 3, F and G).
Df(LG 13)st4 deletes a segment of LG 13: Df(LG 13)st4 is a Mendelian recessive mutation that causes curvature of the body axis and widespread degeneration (Figure 4B; Table 1). Analysis of SSLP markers localized Df(LG 13)st4 to LG 13 and revealed that a 20- to 30-cM region of this linkage group is deleted from the mutant chromosome (Figure 4, C and D).
snhst1 is a translocation involving LG 11 and LG 14: As we have described previously (Dick et al. 2000), the snhst1 allele was identified as a mutation causing a dorsalized phenotype visible by morphological inspection during the early segmentation period (Figure 5B). Analysis of SSLP markers localized snhst1 to LG 11, and snhst1 fails to complement snhty68. Markers spanning about 40 cM of LG 11 were deleted in snhst1 mutants (Figure 6). Previous work showed that the bmp7 gene resides within the deleted segment and that its absence causes the dorsalized phenotype of snhst1 mutants (Dick et al. 2000).
Non-Mendelian inheritance (Table 1) and the presence of a class of progeny with a distinct neural degeneration phenotype (Figure 5D) among the haploid progeny of snhst1/+ females suggested that the snhst1 mutation was a translocation. Further support came from analysis of markers Z22206 and Z21385 (Figure 6B; data not shown), which showed that a region of LG 11 was duplicated in the neural degeneration mutants (note the presence of two alleles in haploid individuals in Figure 6B). The duplication and deletion of regions of LG 11 provided strong evidence that snhst1 is a translocation involving LG 11.
To identify the other linkage group involved in the snhst1 translocation, we assayed SSLP markers covering the genome to determine if any were deleted in neural degeneration mutants. This analysis revealed a deficiency of approximately 30 cM of LG 14 (Figure 6). In addition, the region of LG 14 that is not deleted in neural degeneration mutants is duplicated in haploid embryos with the dorsalized snh phenotype, as indicated
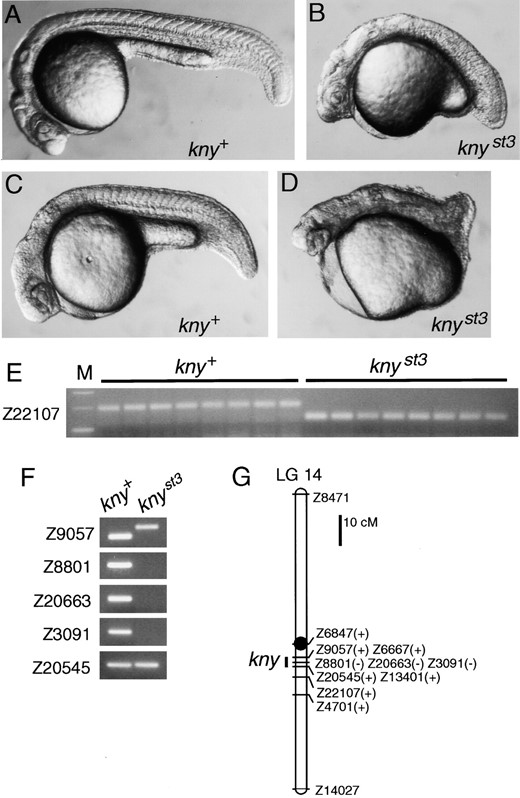
The knyst3 mutant has a short axis and deletes a segment of LG 14. Morphology of (A and C) wild-type and (B and D) knyst3 embryos at 24 hr. (A and B) Diploid and (C and D) haploid embryos are shown. Some knyst3 haploids are partially cyclopic (D). (E) Eight wild-type (kny+) and eight knyst3 haploid siblings were tested with the LG 14 SSLP marker Z22107. Approximately 200-bp and ~130-bp alleles segregate with kny+ and knyst3 alleles, respectively. (F) Pools of DNA from wild type (kny+) and knyst3 haploid siblings were tested with SSLP markers on LG 14. For Z9057, alleles of different sizes are apparent in wild-type and mutant pools. These SSLP markers were also tested in individual haploid embryos to confirm the presence or absence of the amplified fragments (data not shown). (G) Linkage map of LG 14 with the presence (+) or absence (−) of SSLP markers in knyst3. Marker distances are based on Shimoda et al. (1999).
by the presence of two alleles of LG 14 markers Z11725, Z7030, Z5435, and Z14027 (Figure 6B; data not shown). In light of the evidence that a region of LG 14 is deleted in the neural degeneration mutants, we referred to this as the Df(LG 14)st1 phenotype.
Analysis of LG 11 and LG 14 markers in individual haploid embryos with wild-type, snhst1 (dorsalized), and Df(LG 14)st1 (neural degeneration) phenotypes confirmed the presence of rearranged LG 11-LG 14 chromosomes. Positions of breakpoints were determined from the extents of the deletions of LG 11 and LG 14 in snhst1 and Df(LG 14)st1 mutants, respectively (Figure 6, A and C). The LG 11 breakpoint is at or near the position of the centromere (Knapik et al. 1998), since the snhst1 deletion covers one arm of LG 11 (Figure 6C). Markers flanking the inferred breakpoint positions were analyzed for cosegregation of LG 11 and LG 14 loci, the hallmark of a translocation. As shown in Figure 6B, one allele of the LG 11 marker Z22206 was coupled to one allele of the LG 14 marker Z11725: there were 12 recombinants among the 100 wild-type embryos scored. [snhst1 and Df(LG 14)st1 individuals were excluded from these calculations because both alleles of one of the markers were present in these mutant embryos, so that some recombinant chromosomes could not be distinguished from parentals.] This rearranged LG 11-LG 14 chromosome is designated 11U14L in Figure 7 (U and L refer to the upper and lower segments of the affected linkage groups shown in standard orientation). Mapping experiments also indicated the presence of a reciprocally rearranged 14U11L chromosome, which combined the regions of LG 11 and LG 14 not present on 11U14L (Figures 6 and 7). There were 17 recombinants among 100 wild-type haploid progeny for the alleles of the LG 11 marker Z10464 and the LG 14 marker Z9720 (Figure 6B) that are coupled on the rearranged 14U11L chromosome. (As above, mutant embryos were excluded from these calculations because either the LG 11 or LG 14 marker was deleted in these individuals.)
Identification of alleles associated with the rearranged and standard order chromosomes showed which chromosomes were associated with wild-type and mutant individuals (Figure 6) and suggested a model explaining how inheritance of the rearranged chromosomes causes the observed duplications and deletions (Figure 7). Among the haploid progeny of a T(LG 11; LG 14)st1/+ translocation heterozygote (the genotype diagrammed in Figure 7B), some wild-type embryos inherited a standard order LG 11 and a standard order LG 14 (Figure 7D). The other phenotypically wild-type embryos, designated WT* in Figure 7D, inherited both rearranged chromosomes and so had a euploid genotype. (The absence of detectable morphological abnormalities in WT* haploid embryos at 24 hr shows that the breakpoints do not disrupt genes with essential functions in the early embryo, but does not preclude inactivation of genes with later essential functions.) These euploid genotypes would result from the segregation of alternate LG 11 and LG 14 centromeres to the same pole (see postulated configuration of paired chromosomal segments in Figure 7C). Embryos with the Df(LG 14)st1 phenotype inherit 11U14L and a standard order LG 11 (Figure 7D), so that they lack the ~30-cM segment of LG 14 that is not present on 11U14L. The Df(LG 14)st1 genotype is one result of segregation of adjacent LG 11 and LG 14 centromeres to the same pole (adjacent-1 segregation pattern). Haploid embryos inheriting a
Mutant frequencies among haploid and diploid embryos
| % mutant of haploid progeny of F1 founders (no. mutant/total)a | % mutant of diploid progeny of heterozygote intercrosses (no. mutant/total)b | |
|---|---|---|
| snhst1 | 6.0 (13/217) | 0.6 (7/1211) |
| Df(LG 14)st1 | – | 3.3 (40/1211) |
| oepst2 | 22 (22/102) | 27.5 (107/389) |
| knyst3 | 16 (21/131) | 27 (38/140) |
| Df(LG 13)st4 | 20 (43/211) | 27.0 (104/386) |
| % mutant of haploid progeny of F1 founders (no. mutant/total)a | % mutant of diploid progeny of heterozygote intercrosses (no. mutant/total)b | |
|---|---|---|
| snhst1 | 6.0 (13/217) | 0.6 (7/1211) |
| Df(LG 14)st1 | – | 3.3 (40/1211) |
| oepst2 | 22 (22/102) | 27.5 (107/389) |
| knyst3 | 16 (21/131) | 27 (38/140) |
| Df(LG 13)st4 | 20 (43/211) | 27.0 (104/386) |
F2 haploid embryos were produced from the original F1 mosaic females screened for mutations. In all cases, the frequency is less than the Mendelian expectation of 50% mutant embryos from nonmosaic females. No value is given for the Df (LG 14)st1 frequency because these mutants were not observed in the initial screen.
Diploid embryos were obtained from intercrosses of nonmosaic F2 or F3 diploid heterozygotes. oepst2, knyst3, and Df 13)st4 are transmitted as Mendelian recessive mutations.
Mutant frequencies among haploid and diploid embryos
| % mutant of haploid progeny of F1 founders (no. mutant/total)a | % mutant of diploid progeny of heterozygote intercrosses (no. mutant/total)b | |
|---|---|---|
| snhst1 | 6.0 (13/217) | 0.6 (7/1211) |
| Df(LG 14)st1 | – | 3.3 (40/1211) |
| oepst2 | 22 (22/102) | 27.5 (107/389) |
| knyst3 | 16 (21/131) | 27 (38/140) |
| Df(LG 13)st4 | 20 (43/211) | 27.0 (104/386) |
| % mutant of haploid progeny of F1 founders (no. mutant/total)a | % mutant of diploid progeny of heterozygote intercrosses (no. mutant/total)b | |
|---|---|---|
| snhst1 | 6.0 (13/217) | 0.6 (7/1211) |
| Df(LG 14)st1 | – | 3.3 (40/1211) |
| oepst2 | 22 (22/102) | 27.5 (107/389) |
| knyst3 | 16 (21/131) | 27 (38/140) |
| Df(LG 13)st4 | 20 (43/211) | 27.0 (104/386) |
F2 haploid embryos were produced from the original F1 mosaic females screened for mutations. In all cases, the frequency is less than the Mendelian expectation of 50% mutant embryos from nonmosaic females. No value is given for the Df (LG 14)st1 frequency because these mutants were not observed in the initial screen.
Diploid embryos were obtained from intercrosses of nonmosaic F2 or F3 diploid heterozygotes. oepst2, knyst3, and Df 13)st4 are transmitted as Mendelian recessive mutations.
standard order LG 14 and the 11U14L chromosome lack the portion of LG 11 not present on 11U14L and therefore display the dorsalized snhst1 phenotype associated with loss of the bmp7 gene. The snhst1 genotype results from adjacent-2 segregation, in which homologous centromeres (e.g., those of LG 14 and 11U14L) migrate to the same pole.
The haploid genotypes described so far represent
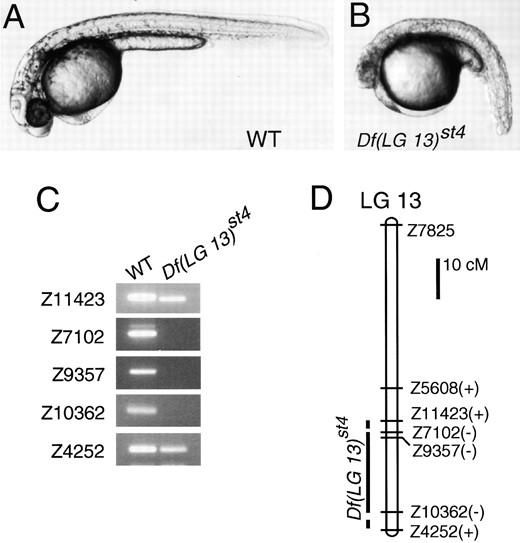
The Df(LG 13)st4 mutation causes a widespread degeneration phenotype and deletes a segment of LG 13. (A) Wild-type and (B) Df(LG 13)st4 homozygote at 36 hr. (C) Pools of DNA from wild-type (WT) and Df(LG 13)st4 haploid siblings were tested with SSLP markers on LG 13. All SSLP markers were also tested in individual haploid embryos to confirm the presence or absence of the markers (data not shown). (D) Linkage map of LG 13 with the presence (+) or absence (−) of the SSLP markers in Df(LG 13)st4. Marker distances are based on Shimoda et al. (1999).
four of the six possible segregation products of the heterozygous T(LG 11; LG 14)st1 translocation (Figure 7D). Both products of alternate segregation (WT and WT*) are included, but only one result of adjacent-1 segregation [Df(LG 14)st1] and one result of adjacent-2 segregation (snhst1) are represented. Because the missing adjacent-1 and adjacent-2 products are aneuploid, it was possible that these embryos had early lethal phenotypes, degenerating prior to stages at which we typically scored embryonic phenotypes and prepared genomic DNA for mapping studies. To examine this possibility, we produced a clutch of 88 haploid progeny from a translocation heterozygote and prepared genomic DNA from embryos at different stages, according to their phenotypes. DNA was collected from dorsalized embryos and those that appeared to be degenerating at 12 hr. For the remaining embryos, phenotypes were scored and genomic DNA collected on the following day. We scored markers distinguishing the rearranged and standard order chromosomes to determine the genotypes of the 88 embryos (Figure 7D). Among the embryos degenerating at 12 hr were 12 that inherited LG 14 and 14U11L, the missing adjacent-1 product, and 5 embryos that inherited LG 11 and 14U11L, the missing adjacent-2 product. These results confirmed that some of the aneuploid products of adjacent-1 and adjacent-2 segregation have early lethal phenotypes.
One noteworthy aspect of the T(LG 11; LG 14)st1 translocation is the incidence of adjacent-2 segregation products, which are typically recovered only rarely because homologous centromeres do not usually migrate to the same pole (Ashburner 1989). Among the most important factors affecting the incidence of adjacent-2 segregation in other species is the length of the interstitial region, the interval between the centromere and the translocation breakpoint (see Ashburner 1989 for review of factors affecting disjunction in translocation heterozygotes). Adjacent-2 segregation is infrequent in translocations with long interstitial regions, but adjacent-2 can be more prevalent than adjacent-1 segregation
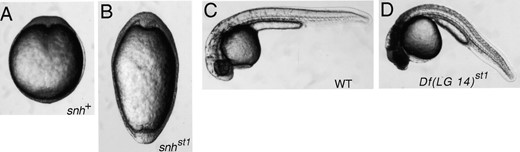
Phenotypes of snhst1 and Df(LG 14)st1 mutants. Dorsal view at the early segmentation stage (12 hr) of wild-type (A) and snhst1 (B) embryos. Lateral view of wild-type (C) and Df(LG 14)st1 (D) embryos at 36 hr. Diploid embryos are shown.
in translocations with short interstitial segments. This relationship presumably exists because interstitial crossovers prevent the migration of homologous centromeres to the same pole and thus favor adjacent-1 and alternate segregation (Ashburner 1989). Because the LG 11 breakpoint is at or near the centromere, the 14U11L chromosome is predicted to have a very short interstitial segment, consistent with previous models and the substantial incidence of adjacent-2 segregation products.
DISCUSSION
Although ENU is generally considered to induce point mutations and other intragenic lesions, our results demonstrate that postmeiotic mutagenesis with ENU
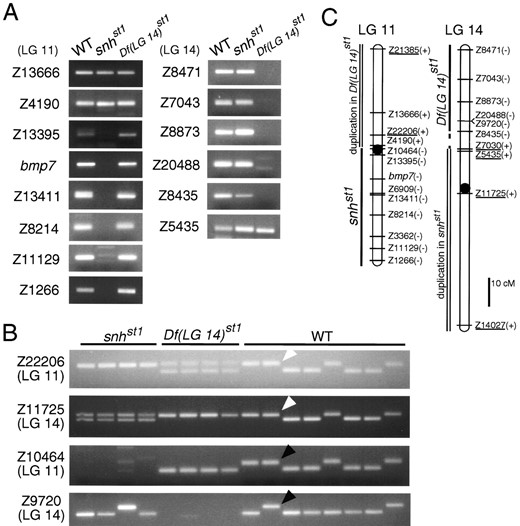
Analysis of LG 11 and LG 14 markers in the snhst1 and Df(LG 14)st1 mutants. (A) Pools of DNA from wild-type (WT), snhst1, and Df(LG 14)st1 haploid progeny from a heterozygous T(LG 11; LG 14)st1/+ female were tested with SSLP markers and the bmp7 gene (Dick et al. 2000; Schmid et al. 2000). All SSLP markers were also tested in individual haploid embryos to confirm the presence or absence of the markers (data not shown). (B) Individual haploid progeny from a T(LG 11; LG 14)st1/+ heterozygous female were tested with SSLP markers. snhst1 haploids inherit no Z10464 (LG 11) allele but two different alleles of Z11725 (LG 14). Df(LG 14)st1 haploids inherit no Z9720 allele (LG 14) but two different alleles of Z22206 (LG 11). One allele of Z22206 is coupled to one allele of Z11725 (white arrowheads), and one allele of Z10464 is coupled to one allele of Z9720 (black arrowheads). (C) Linkage maps of LG 11 and LG 14 with the presence (+) or absence (−) of the SSLP markers in the snhst1 or Df(LG 14)st1. LG 11 and LG 14 markers known to be duplicated in Df(LG 14)st1 and in snhst1 haploids, respectively, are underlined. Marker distances are based on Shimoda et al. (1999). Centromere positions (circles) were determined by Knapik et al. (1998).
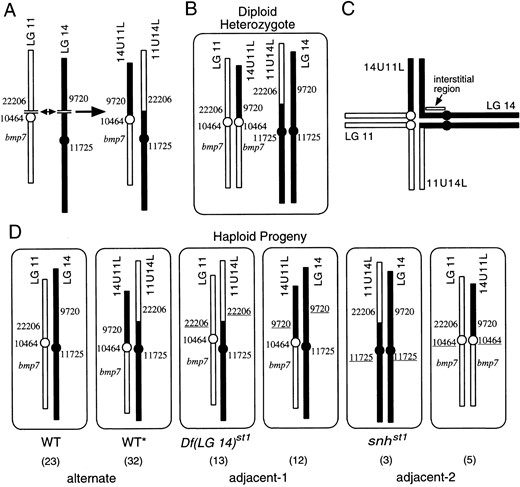
Model of chromosomal rearrangement and segregation in T(LG 11:LG 14)st1 translocation. (A) Reciprocal rearrangement at breakpoints in LG 11 and LG 14 produced 11U14L and 14U11L rearranged chromosomes. The LG 11 breakpoint is at or near the centromere (circle). Approximate positions of bmp7 gene and some markers used for analysis in Figure 6 are shown. (B) Heterozygous T(LG 11;LG 14)st1/+ diploid has a standard order LG 11, a standard order LG 14, and the rearranged chromosomes 11U14L and 14U11L. (C) Postulated configuration of meiotic pairing of chromosomal segments in T(LG 11;LG 14)st1/+ heterozygote. In meiosis I, each centromere is attached to two chromatids, which are represented by a single line for clarity. (D) Two euploid and four aneuploid products are generated from meiosis in a T(LG 11;LG 14)st1/+ heterozygote. Segregation of alternate LG 11 and LG 14 centromeres to the same pole produces euploid WT and WT* haploid genotypes. Adjacent-1 (segregation of adjacent LG 11 and LG 14 centromeres to the same pole) and adjacent-2 (segregation of homologous centromeres to the same pole) segregation produce aneuploid genotypes. Under each meiotic product is the number of haploid embryos observed with that genotype among 88 haploid progeny from a T(LG 11;LG 14)st1 heterozygote. Genotypes were tested with SSLP markers Z22206, Z10464, Z9720, and Z11725, which allow the aneuploid genotypes to be identified by absence of one of the four SSLP markers and duplication of another. Duplicated SSLP markers are underlined. We did not test for duplication of bmp7, but infer that it is duplicated in one adjacent-2 product (right) from the analysis of the linked marker Z10464. The length of the interstitial region correlates with the frequency of adjacent-2 segregation (see text).
can induce chromosomal rearrangements in zebrafish. Indeed, none of the five new mutations that we analyzed in this study was a point mutation. Four are deletions, and the fifth is a translocation involving LG 11 and LG 14. In addition, preliminary analysis shows that the three additional mutations recovered in the screens we describe are chromosomal rearrangements (Y. Imai, A. F. Schier and W. S. Talbot, unpublished observations).
Despite the absence of point mutations among the mutations we have so far analyzed, previous work shows that postmeiotic ENU treatment can induce point mutations. In their report on postmeiotic ENU mutagenesis, Riley and Grunwald (1995) showed that all of five new alleles of pigmentation loci were homozygous viable and that some were hypomorphic rather than null alleles. Although genetic markers near the affected genes were not available for analysis at the time of that study, the evidence suggests that these mutations were not large deficiencies. In addition, three of five mutations induced by a higher concentration of ENU (1.0 mm instead of 0.8 mm) were homozygous viable, whereas the other two were dominant lethal, likely the result of chromosomal rearrangements. Direct evidence for the induction of point mutations by postmeiotic ENU treatment derives from Appel et al. (1999), who identified a single base-pair mutation in a mutant allele of the deltaA gene.
Our results together with previous studies show that postmeiotic ENU treatment of male germ cells can induce a wide spectrum of lesions, ranging from point mutations to small deletions to extensive chromosomal rearrangements. The relatively small number of mutations characterized in our study and previous ones precludes a precise evaluation of the frequencies of different types of mutations induced. Furthermore, different screens may be biased for different types of mutations in the spectrum produced by postmeiotic ENU mutagenesis. Thus our screen for phenotypes at early stages may have identified a higher proportion of deletions and translocations than a screen for phenotypes at later stages. It is also possible that subtle differences in the parameters of mutagenesis (e.g., ENU concentration or activity, time of mating after treatment) can lead to important differences in the mutational spectrum. Nevertheless, our finding that the spectrum of induced mutations includes multilocus chromosomal rearrangements suggests that such rearrangements, rather than an increased occurrence of point mutations, account for the elevated mutation frequency observed in specific-locus tests after postmeiotic ENU treatment.
Effect of germ cell stage on mutations induced by ENU: In contrast to our finding of chromosomal rearrangements induced by postmeiotic treatment, many previous studies indicate that ENU predominantly induces point mutations in zebrafish spermatogonial stem cell mutagenesis regimens. Analysis of new alleles of the pigmentation loci albino, golden, sparse, and brass shows that the great majority are homozygous viable, with only 1 lethal allele among more than 20 mutations analyzed (Mullins et al. 1994; Solnica-Krezel et al. 1994). Moreover, sequence analysis of 39 mutations induced by premeiotic ENU treatment shows that all but 1 are single-base mutations, the sole exception being an 8-bp insertion (Table 2). Thus it seems that the spectrum of mutations induced by ENU depends on whether premeiotic or postmeiotic germ cells are exposed.
A similar dependence on the stage of mutagenized germ cells has been observed in mouse and the medaka
Molecularly characterized mutations induced by spermatogonial ENU treatment
| Mutant allele | Lesion | Reference |
|---|---|---|
| flhtk241 | TTG → TAG | Talbot et al. (1995) |
| flytm229 | AAA → TAA | |
| noith44 | GAA → TAA | Brand et al. (1996) |
| noitu29 | CGA → TGA | Lun and Brand (1998) |
| noity31 | GGC → GTC | |
| noitm243 | AG → TG (splice acceptor) | |
| dintt250 | GT → AT (splice donor) | Schulte-Merker et al. (1997); Dick et al. (2000) |
| swr3ta7 | TGA → TGG | Kishimoto et al. (1997); Nguyen et al. (1998) |
| swrtc300 | TGT → TGG | |
| pptta98 | TGC → CGC | Rauch et al. (1997b) |
| pptth278 | TGC → AGC | |
| pptti265 | AAG → TAG | |
| oeptz57 | ATG → GTG | Zhang et al. (1998) |
| oepm134 | TCG → TAG | |
| Valb337 | CAG → TAG | Moens et al. (1998) |
| aceti282 | GT → AT (splice donor) | Reifers et al. (1998) |
| syutq252 | G → A (promoter) | Schauerte et al. (1998) |
| Syutbx392 | GT → AT (splice donor) | |
| cyctf219 | ATG → ATA | Rebagliati et al. (1998) |
| cycm294 | TGC → CGC | Sampath et al. (1998) |
| sptb433 | 8-bp insertion | Grifian et al. (1998) |
| sptm423 | TAT → TAA | |
| sautb223 | GTC → GAC | Brownlie et al. (1998) |
| sauty121 | CTG→CAG | |
| yqetp61 | ATG → AGG | Wang et al. (1998) |
| yofty119 | CGA → TGA | Karlstrom et al. (1999) |
| yotty17 | CAG → TAG | |
| bozm168 | TGG → TGA | Fekany et al. (1999) |
| sbntc24 | ACA → ATA | Hild et al. (1999) |
| mfntm124 | GGA→ TGA | Connors et al. (1999) |
| mfnty130 | TGT → TGG | |
| mfntb241 | GGA → TGA | |
| mfntn217 | CTG→ CCG | |
| mfntf215 | CGA → TGA | |
| spajle1 | TTG → TAG | Parichy et al. (1999) |
| spajle56 | TAT → TAA | |
| spajle41 | TAT → TAA | |
| nacw2 | CAA → TAA | Lister et al. (1999) |
| snhty68 | GTG → GGG | Dick et al. (2000); Schmid et al. (2000) |
| Mutant allele | Lesion | Reference |
|---|---|---|
| flhtk241 | TTG → TAG | Talbot et al. (1995) |
| flytm229 | AAA → TAA | |
| noith44 | GAA → TAA | Brand et al. (1996) |
| noitu29 | CGA → TGA | Lun and Brand (1998) |
| noity31 | GGC → GTC | |
| noitm243 | AG → TG (splice acceptor) | |
| dintt250 | GT → AT (splice donor) | Schulte-Merker et al. (1997); Dick et al. (2000) |
| swr3ta7 | TGA → TGG | Kishimoto et al. (1997); Nguyen et al. (1998) |
| swrtc300 | TGT → TGG | |
| pptta98 | TGC → CGC | Rauch et al. (1997b) |
| pptth278 | TGC → AGC | |
| pptti265 | AAG → TAG | |
| oeptz57 | ATG → GTG | Zhang et al. (1998) |
| oepm134 | TCG → TAG | |
| Valb337 | CAG → TAG | Moens et al. (1998) |
| aceti282 | GT → AT (splice donor) | Reifers et al. (1998) |
| syutq252 | G → A (promoter) | Schauerte et al. (1998) |
| Syutbx392 | GT → AT (splice donor) | |
| cyctf219 | ATG → ATA | Rebagliati et al. (1998) |
| cycm294 | TGC → CGC | Sampath et al. (1998) |
| sptb433 | 8-bp insertion | Grifian et al. (1998) |
| sptm423 | TAT → TAA | |
| sautb223 | GTC → GAC | Brownlie et al. (1998) |
| sauty121 | CTG→CAG | |
| yqetp61 | ATG → AGG | Wang et al. (1998) |
| yofty119 | CGA → TGA | Karlstrom et al. (1999) |
| yotty17 | CAG → TAG | |
| bozm168 | TGG → TGA | Fekany et al. (1999) |
| sbntc24 | ACA → ATA | Hild et al. (1999) |
| mfntm124 | GGA→ TGA | Connors et al. (1999) |
| mfnty130 | TGT → TGG | |
| mfntb241 | GGA → TGA | |
| mfntn217 | CTG→ CCG | |
| mfntf215 | CGA → TGA | |
| spajle1 | TTG → TAG | Parichy et al. (1999) |
| spajle56 | TAT → TAA | |
| spajle41 | TAT → TAA | |
| nacw2 | CAA → TAA | Lister et al. (1999) |
| snhty68 | GTG → GGG | Dick et al. (2000); Schmid et al. (2000) |
Molecularly characterized mutations induced by spermatogonial ENU treatment
| Mutant allele | Lesion | Reference |
|---|---|---|
| flhtk241 | TTG → TAG | Talbot et al. (1995) |
| flytm229 | AAA → TAA | |
| noith44 | GAA → TAA | Brand et al. (1996) |
| noitu29 | CGA → TGA | Lun and Brand (1998) |
| noity31 | GGC → GTC | |
| noitm243 | AG → TG (splice acceptor) | |
| dintt250 | GT → AT (splice donor) | Schulte-Merker et al. (1997); Dick et al. (2000) |
| swr3ta7 | TGA → TGG | Kishimoto et al. (1997); Nguyen et al. (1998) |
| swrtc300 | TGT → TGG | |
| pptta98 | TGC → CGC | Rauch et al. (1997b) |
| pptth278 | TGC → AGC | |
| pptti265 | AAG → TAG | |
| oeptz57 | ATG → GTG | Zhang et al. (1998) |
| oepm134 | TCG → TAG | |
| Valb337 | CAG → TAG | Moens et al. (1998) |
| aceti282 | GT → AT (splice donor) | Reifers et al. (1998) |
| syutq252 | G → A (promoter) | Schauerte et al. (1998) |
| Syutbx392 | GT → AT (splice donor) | |
| cyctf219 | ATG → ATA | Rebagliati et al. (1998) |
| cycm294 | TGC → CGC | Sampath et al. (1998) |
| sptb433 | 8-bp insertion | Grifian et al. (1998) |
| sptm423 | TAT → TAA | |
| sautb223 | GTC → GAC | Brownlie et al. (1998) |
| sauty121 | CTG→CAG | |
| yqetp61 | ATG → AGG | Wang et al. (1998) |
| yofty119 | CGA → TGA | Karlstrom et al. (1999) |
| yotty17 | CAG → TAG | |
| bozm168 | TGG → TGA | Fekany et al. (1999) |
| sbntc24 | ACA → ATA | Hild et al. (1999) |
| mfntm124 | GGA→ TGA | Connors et al. (1999) |
| mfnty130 | TGT → TGG | |
| mfntb241 | GGA → TGA | |
| mfntn217 | CTG→ CCG | |
| mfntf215 | CGA → TGA | |
| spajle1 | TTG → TAG | Parichy et al. (1999) |
| spajle56 | TAT → TAA | |
| spajle41 | TAT → TAA | |
| nacw2 | CAA → TAA | Lister et al. (1999) |
| snhty68 | GTG → GGG | Dick et al. (2000); Schmid et al. (2000) |
| Mutant allele | Lesion | Reference |
|---|---|---|
| flhtk241 | TTG → TAG | Talbot et al. (1995) |
| flytm229 | AAA → TAA | |
| noith44 | GAA → TAA | Brand et al. (1996) |
| noitu29 | CGA → TGA | Lun and Brand (1998) |
| noity31 | GGC → GTC | |
| noitm243 | AG → TG (splice acceptor) | |
| dintt250 | GT → AT (splice donor) | Schulte-Merker et al. (1997); Dick et al. (2000) |
| swr3ta7 | TGA → TGG | Kishimoto et al. (1997); Nguyen et al. (1998) |
| swrtc300 | TGT → TGG | |
| pptta98 | TGC → CGC | Rauch et al. (1997b) |
| pptth278 | TGC → AGC | |
| pptti265 | AAG → TAG | |
| oeptz57 | ATG → GTG | Zhang et al. (1998) |
| oepm134 | TCG → TAG | |
| Valb337 | CAG → TAG | Moens et al. (1998) |
| aceti282 | GT → AT (splice donor) | Reifers et al. (1998) |
| syutq252 | G → A (promoter) | Schauerte et al. (1998) |
| Syutbx392 | GT → AT (splice donor) | |
| cyctf219 | ATG → ATA | Rebagliati et al. (1998) |
| cycm294 | TGC → CGC | Sampath et al. (1998) |
| sptb433 | 8-bp insertion | Grifian et al. (1998) |
| sptm423 | TAT → TAA | |
| sautb223 | GTC → GAC | Brownlie et al. (1998) |
| sauty121 | CTG→CAG | |
| yqetp61 | ATG → AGG | Wang et al. (1998) |
| yofty119 | CGA → TGA | Karlstrom et al. (1999) |
| yotty17 | CAG → TAG | |
| bozm168 | TGG → TGA | Fekany et al. (1999) |
| sbntc24 | ACA → ATA | Hild et al. (1999) |
| mfntm124 | GGA→ TGA | Connors et al. (1999) |
| mfnty130 | TGT → TGG | |
| mfntb241 | GGA → TGA | |
| mfntn217 | CTG→ CCG | |
| mfntf215 | CGA → TGA | |
| spajle1 | TTG → TAG | Parichy et al. (1999) |
| spajle56 | TAT → TAA | |
| spajle41 | TAT → TAA | |
| nacw2 | CAA → TAA | Lister et al. (1999) |
| snhty68 | GTG → GGG | Dick et al. (2000); Schmid et al. (2000) |
fish. Russell et al. (1990) found that about half of the mutations induced by postmeiotic ENU and methylnitrosourea treatment in the mouse are large lesions, compared to <5% of those induced by treatment of spermatogonial stem cells. In medaka, Shimada and Shima (1998) found that dominant lethal mutations were induced by ENU treatment of sperm but not spermatogonia. In mouse, a number of other chemical mutagens also induce a high frequency of large lesions in postmeiotic mutagenesis of germ cells, suggesting that stage of treatment, rather than the nature of the chemical, determines the type of mutation (Russell et al. 1990). This may be true for zebrafish as well, since treatment of sperm with TMP induces a high frequency of dominant lethal mutations and results in specific-locus mutations at a rate of 0.5–2% (Ando and Mishina 1998), both of which are consistent with the formation of chromosomal rearrangements. The differences in mutagenic outcome for pre- and postmeiotic treatments may result from differences in the chromatin conformation or ploidy in sperm and spermatogonia, or the differential activity of DNA repair pathways on lesions induced at these stages (Russell et al. 1990). Interestingly, F1 founders were mosaic for the deletions that we identified, suggesting that some single-stranded modifications induced by ENU in sperm DNA were fixed as double-stranded breaks only after DNA replication in the zygote.
Applications for ENU-induced deletions and translocations: Chromosomal rearrangements are an important resource in zebrafish genetics. Deletions and translocations have enabled investigators to determine the null phenotypes of a number of essential genes (Talbot et al. 1995, 1998; Fisher et al. 1997; Schier et al. 1997; Fekany et al. 1999). Deletions and translocations are also useful in mapping cloned genes (and other DNA sequences) and mutations. Although cloned genes can be mapped meiotically and in radiation hybrid panels, testing whether a gene is removed by a deletion or translocation is probably the fastest and simplest way to determine if a given gene maps to a particular region of the genome. This can be of particular use in testing candidate genes for mutations and in positional cloning projects, in which one may test many newly isolated genomic clones to ensure that they map near the mutation of interest. Rearrangements can also be applied to map mutations by complementation tests—a simple assay to determine whether a mutation might correspond to any cloned genes known to lie within a particular deleted interval. Reverse genetic strategies lag far behind forward genetic approaches in zebrafish, but chromosomal rearrangements can provide a starting point to examine the functions of cloned genes. Fritz et al. (1996) showed that multiplex PCR methods can efficiently isolate rearrangements that delete cloned genes of interest. Although the rearrangements are for the most part too large to define the functions of individual genes, the phenotypes of mutants with smaller deletions may provide some insight. In addition, one successful case illustrates that screens for point mutations that fail to complement deletions can obtain informative mutations in individual genes within the deleted interval (Appel et al. 1999).
The rearrangements we have described should be useful in the analysis of genes on LG 10, LG 11, LG 12, LG 13, and LG 14. Indeed, snhst1 was helpful in characterizing the null phenotype of the snh/bmp7 locus, which was initially defined by a single point mutation retaining some bmp7 activity (Dick et al. 2000; Schmid et al. 2000). Our results also indicate that available map resources allow the structure and extent of chromosomal rearrangements to be elucidated, which will aid in the construction of a panel of deletions and translocations covering the zebrafish genome.
Acknowlegement
We thank David Kingsley, Anne Villenueve, and the members of the Talbot and Schier groups for helpful discussions and comments on the manuscript. We also thank Rory Feeney, Lauren Jow, Frankie Kimm, and Michele Mittman for technical assistance and fish care. Y.I. received support from the Yamada Science Foundation and the Uehara Memorial Foundation, and B.F. received support from National Institutes of Health (NIH) fellowship F32 GM1919302. This work was supported by NIH grants R01 RR12349 (W.S.T.), R01 GM57825 (W.S.T.), and R21 HG1704 (A.F.S.). A.F.S. is a Scholar of the McKnight Endowment Fund for Neuroscience. W.S.T. is a Pew Scholar in the Biomedical Sciences.
Footnotes
Communicating editor: N. A. Jenkins
LITERATURE CITED
Author notes
Present address: The National Institute for Medical Research, The Ridgeway, Mill Hill, London NW7 1AA, United Kingdom.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}