





Abstract
The grass genus Zea contains the domesticate maize and several wild taxa indigenous to Central and South America. Here we study the genetic consequences of speciation and domestication in this group by sampling DNA sequences from four taxa—maize (Zea mays ssp. mays), its wild progenitor (Z. mays ssp. parviglumis), a more distant species within the genus (Z. luxurians), and a representative of the sister genus (Tripsacum dactyloides). We sampled a total of 26 sequences from the glb1 locus, which encodes a nonessential seed storage protein. Within the Zea taxa sampled, the progenitor to maize contains the most sequence diversity. Maize contains 60% of the level of genetic diversity of its progenitor, and Z. luxurians contains even less diversity (32% of the level of diversity of Z. mays ssp. parviglumis). Sequence variation within the glb1 locus is consistent with neutral evolution in all four taxa. The glb1 data were combined with adh1 data from a previous study to make inferences about the population genetic histories of these taxa. Comparisons of sequence data between the two morphologically similar wild Zea taxa indicate that the species diverged ∼700,000 years ago from a common ancestor of intermediate size to their present populations. Conversely, the domestication of maize was a recent event that could have been based on a very small number of founding individuals. Maize retained a substantial proportion of the genetic variation of its progenitor through this founder event, but diverged rapidly in morphology.
MUCH of the process of speciation remains a mystery, yet some aspects ofthe genetics of speciation are coming to light with new approaches. For example, comparisons of DNA sequence variation between closely related species have provided insight into the amount of divergence between sibling species, the date of divergence between sibling species, and the ancestral population size of sibling species. This genealogical approach has been applied to speciation events among Drosophila species (Hey and Kliman 1993; Kliman and Hey 1993; Hilton et al. 1994; Hilton and Hey 1996, 1997; Wang and Hey 1996; Wang et al. 1997), but has not been applied broadly to studies of plant speciation. In plants, the genealogical approach has the potential to elucidate the processes of both “natural” speciation events between wild taxa and “artificial” speciation events such as crop domestication. Here we present a genealogical study of the genus Zea, with the explicit goal of contrasting an artificial speciation event to a natural speciation event.
The grass genus Zea contains the domesticate maize (Zea mays ssp. mays) and six wild taxa. Here we focus on maize; its closest wild relative, Z. mays ssp. parviglumis (hereafter also referred to as “parviglumis”), and the more distantly related species Z. luxurians. The close relationship between maize and parviglumis has been established by a number of genetic and systematic studies (Doebley et al. 1984, 1987a,b; Doebley 1990a; Buckler and Holtsford 1996), and it is thought that maize was domesticated from parviglumis ∼7500 years ago in southern or central Mexico (Iltis 1983; Doebley et al. 1984). The present range of parviglumis extends to several areas of southern and central Mexico (Doebley 1990b). The species Z. luxurians is placed taxonomically in a different section of the genus than parviglumis and maize (Doebley 1990a). It is found in a restricted range, primarily in Guatemala, and is largely geographically isolated from both maize and parviglumis (Doebley 1990b). We have also included individuals from Tripsacum dactyloides in this study; the genus Tripsacum is the sister genus to Zea (Kellogg and Watson 1993).
We collected DNA variation data from a 1.2-kb portion of the glb1 locus from multiple individuals of the four taxa. GLB1 is one of the most abundant proteins in maize embryos (Kriz and Schwartz 1986) and is encoded by a single gene located on the long arm of chromosome 1 (Schwartz 1979). Expression of the gene is limited to seed tissues (Belanger and Kriz 1989). However, the GLB1 protein is apparently not essential for seedling growth or survival because homozygosity for the null allele has no effect on embryo development or germination (Schwartz 1979). It has
Individuals sampled for glb1
| Taxon | Land race or accession no. | Location | Seed source | Sequence abbrev. | GenBank no. |
|---|---|---|---|---|---|
| Maize | IL high protein | Illinois | — | glb-hb | U28017 |
| VA236 | United States | — | glb-s | X59084 | |
| Araguito | Lowland Venezuala | M. M. Goodmana | Arag | AF064212 | |
| Chococeno | Colombia | M. M. Goodman | Choc | AF064213 | |
| Conico | Northern Mexico | M. M. Goodman | Coni | AF064214 | |
| Coroico | Amazon basin | M. M. Goodman | Coro | AF064215 | |
| Karapampa | Andean Mountains | M. M. Goodman | Kara | AF064216 | |
| Nal-tel | Latin America | M. M. Goodman | Nal | AF064217 | |
| Tuxpeno | Latin America | M. M. Goodman | Tuxp | AF064218 | |
| Z. mays ssp. parviglumis | 331783 | Guerrero, Mexico | USDA-ARSb | Parv8c | AF064219 |
| 331785 | Michoacan, Mexico | USDA-ARS | Parv1 | AF064220 | |
| 331786 | Mexico, Mexico | USDA-ARS | Parv2 | AF064221 | |
| 351787 | Mexico, Mexico | USDA-ARS | Parv9 | AF064222 | |
| 384062 | Guerrero, Mexico | USDA-ARS | Parv10 | AF064223 | |
| 384064 | Guerrero, Mexico | USDA-ARS | Parv4 | AF064224 | |
| M046 | Jalisco, Mexico | J. F. Doebleyd | Parv5 | AF064225 | |
| M106 | Guerrero, Mexico | J. F. Doebley | Parv7 | AF064226 | |
| Z. luxurians | 21863 | Guatemala | USDA-ARS | Lux1 | AF064227 |
| 21866 | Guatemala | USDA-ARS | Lux2 | AF064228 | |
| 21879 | Chiquimula, Guatemala | USDA-ARS | Lux3 | AF064229 | |
| 306615 | Jutiapa, Guatemala | USDA-ARS | Lux5 | AF064230 | |
| 311282 | Chiquimula, Guatemala | USDA-ARS | Lux6 | AF064231 | |
| M027 | Jutiapa, Guatemala | J. F. Doebley | Lux8 | AF064232 | |
| T. dactyloides | Trip2061 | Arkansas | C. deWalde | Trip1 | AF064233 |
| Trip2065f | Missouri | C. deWald | Trip2a | AF064234 | |
| Trip 2b | AF064235 |
| Taxon | Land race or accession no. | Location | Seed source | Sequence abbrev. | GenBank no. |
|---|---|---|---|---|---|
| Maize | IL high protein | Illinois | — | glb-hb | U28017 |
| VA236 | United States | — | glb-s | X59084 | |
| Araguito | Lowland Venezuala | M. M. Goodmana | Arag | AF064212 | |
| Chococeno | Colombia | M. M. Goodman | Choc | AF064213 | |
| Conico | Northern Mexico | M. M. Goodman | Coni | AF064214 | |
| Coroico | Amazon basin | M. M. Goodman | Coro | AF064215 | |
| Karapampa | Andean Mountains | M. M. Goodman | Kara | AF064216 | |
| Nal-tel | Latin America | M. M. Goodman | Nal | AF064217 | |
| Tuxpeno | Latin America | M. M. Goodman | Tuxp | AF064218 | |
| Z. mays ssp. parviglumis | 331783 | Guerrero, Mexico | USDA-ARSb | Parv8c | AF064219 |
| 331785 | Michoacan, Mexico | USDA-ARS | Parv1 | AF064220 | |
| 331786 | Mexico, Mexico | USDA-ARS | Parv2 | AF064221 | |
| 351787 | Mexico, Mexico | USDA-ARS | Parv9 | AF064222 | |
| 384062 | Guerrero, Mexico | USDA-ARS | Parv10 | AF064223 | |
| 384064 | Guerrero, Mexico | USDA-ARS | Parv4 | AF064224 | |
| M046 | Jalisco, Mexico | J. F. Doebleyd | Parv5 | AF064225 | |
| M106 | Guerrero, Mexico | J. F. Doebley | Parv7 | AF064226 | |
| Z. luxurians | 21863 | Guatemala | USDA-ARS | Lux1 | AF064227 |
| 21866 | Guatemala | USDA-ARS | Lux2 | AF064228 | |
| 21879 | Chiquimula, Guatemala | USDA-ARS | Lux3 | AF064229 | |
| 306615 | Jutiapa, Guatemala | USDA-ARS | Lux5 | AF064230 | |
| 311282 | Chiquimula, Guatemala | USDA-ARS | Lux6 | AF064231 | |
| M027 | Jutiapa, Guatemala | J. F. Doebley | Lux8 | AF064232 | |
| T. dactyloides | Trip2061 | Arkansas | C. deWalde | Trip1 | AF064233 |
| Trip2065f | Missouri | C. deWald | Trip2a | AF064234 | |
| Trip 2b | AF064235 |
North Carolina State University.
USDA Agricultural Research Station, Iowa State University, Ames, IA.
Numbers were chosen to coincide with the study of sequence diversity of the adh1 locus and, therefore, are not contiguous.
University of Minnesota.
USDA-ARS, Woodward, OK.
Both alleles were sequenced from this heterozygote.
Individuals sampled for glb1
| Taxon | Land race or accession no. | Location | Seed source | Sequence abbrev. | GenBank no. |
|---|---|---|---|---|---|
| Maize | IL high protein | Illinois | — | glb-hb | U28017 |
| VA236 | United States | — | glb-s | X59084 | |
| Araguito | Lowland Venezuala | M. M. Goodmana | Arag | AF064212 | |
| Chococeno | Colombia | M. M. Goodman | Choc | AF064213 | |
| Conico | Northern Mexico | M. M. Goodman | Coni | AF064214 | |
| Coroico | Amazon basin | M. M. Goodman | Coro | AF064215 | |
| Karapampa | Andean Mountains | M. M. Goodman | Kara | AF064216 | |
| Nal-tel | Latin America | M. M. Goodman | Nal | AF064217 | |
| Tuxpeno | Latin America | M. M. Goodman | Tuxp | AF064218 | |
| Z. mays ssp. parviglumis | 331783 | Guerrero, Mexico | USDA-ARSb | Parv8c | AF064219 |
| 331785 | Michoacan, Mexico | USDA-ARS | Parv1 | AF064220 | |
| 331786 | Mexico, Mexico | USDA-ARS | Parv2 | AF064221 | |
| 351787 | Mexico, Mexico | USDA-ARS | Parv9 | AF064222 | |
| 384062 | Guerrero, Mexico | USDA-ARS | Parv10 | AF064223 | |
| 384064 | Guerrero, Mexico | USDA-ARS | Parv4 | AF064224 | |
| M046 | Jalisco, Mexico | J. F. Doebleyd | Parv5 | AF064225 | |
| M106 | Guerrero, Mexico | J. F. Doebley | Parv7 | AF064226 | |
| Z. luxurians | 21863 | Guatemala | USDA-ARS | Lux1 | AF064227 |
| 21866 | Guatemala | USDA-ARS | Lux2 | AF064228 | |
| 21879 | Chiquimula, Guatemala | USDA-ARS | Lux3 | AF064229 | |
| 306615 | Jutiapa, Guatemala | USDA-ARS | Lux5 | AF064230 | |
| 311282 | Chiquimula, Guatemala | USDA-ARS | Lux6 | AF064231 | |
| M027 | Jutiapa, Guatemala | J. F. Doebley | Lux8 | AF064232 | |
| T. dactyloides | Trip2061 | Arkansas | C. deWalde | Trip1 | AF064233 |
| Trip2065f | Missouri | C. deWald | Trip2a | AF064234 | |
| Trip 2b | AF064235 |
| Taxon | Land race or accession no. | Location | Seed source | Sequence abbrev. | GenBank no. |
|---|---|---|---|---|---|
| Maize | IL high protein | Illinois | — | glb-hb | U28017 |
| VA236 | United States | — | glb-s | X59084 | |
| Araguito | Lowland Venezuala | M. M. Goodmana | Arag | AF064212 | |
| Chococeno | Colombia | M. M. Goodman | Choc | AF064213 | |
| Conico | Northern Mexico | M. M. Goodman | Coni | AF064214 | |
| Coroico | Amazon basin | M. M. Goodman | Coro | AF064215 | |
| Karapampa | Andean Mountains | M. M. Goodman | Kara | AF064216 | |
| Nal-tel | Latin America | M. M. Goodman | Nal | AF064217 | |
| Tuxpeno | Latin America | M. M. Goodman | Tuxp | AF064218 | |
| Z. mays ssp. parviglumis | 331783 | Guerrero, Mexico | USDA-ARSb | Parv8c | AF064219 |
| 331785 | Michoacan, Mexico | USDA-ARS | Parv1 | AF064220 | |
| 331786 | Mexico, Mexico | USDA-ARS | Parv2 | AF064221 | |
| 351787 | Mexico, Mexico | USDA-ARS | Parv9 | AF064222 | |
| 384062 | Guerrero, Mexico | USDA-ARS | Parv10 | AF064223 | |
| 384064 | Guerrero, Mexico | USDA-ARS | Parv4 | AF064224 | |
| M046 | Jalisco, Mexico | J. F. Doebleyd | Parv5 | AF064225 | |
| M106 | Guerrero, Mexico | J. F. Doebley | Parv7 | AF064226 | |
| Z. luxurians | 21863 | Guatemala | USDA-ARS | Lux1 | AF064227 |
| 21866 | Guatemala | USDA-ARS | Lux2 | AF064228 | |
| 21879 | Chiquimula, Guatemala | USDA-ARS | Lux3 | AF064229 | |
| 306615 | Jutiapa, Guatemala | USDA-ARS | Lux5 | AF064230 | |
| 311282 | Chiquimula, Guatemala | USDA-ARS | Lux6 | AF064231 | |
| M027 | Jutiapa, Guatemala | J. F. Doebley | Lux8 | AF064232 | |
| T. dactyloides | Trip2061 | Arkansas | C. deWalde | Trip1 | AF064233 |
| Trip2065f | Missouri | C. deWald | Trip2a | AF064234 | |
| Trip 2b | AF064235 |
North Carolina State University.
USDA Agricultural Research Station, Iowa State University, Ames, IA.
Numbers were chosen to coincide with the study of sequence diversity of the adh1 locus and, therefore, are not contiguous.
University of Minnesota.
USDA-ARS, Woodward, OK.
Both alleles were sequenced from this heterozygote.
been suggested that glb1 would be an excellent marker for the analysis of genetic variation because the gene is single copy and because it is highly variable (Belanger and Kriz 1991).
The purpose of this study is to explore the population genetic history of domestication and speciation in maize and its wild relatives. To do this, we have sampled glb1 sequences from 26 individuals representing maize, parviglumis, Z. luxurians, and T. dactyloides. These same taxa have also been studied at the adh1 locus (Eyre-Walker et al. 1998). Here we analyze glb1 and adh1 data to (1) examine the evolutionary dynamics of the glb1 gene, (2) investigate the genetic consequences of speciation between two wild taxa, (3) provide further insights into genetic diversity within maize, and (4) contrast a natural speciation event with a crop domestication event.
MATERIALS AND METHODS
DNA sequences: We PCR amplified ∼200 bases of the glb1 promoter and the first 1000 bp of the glb1 gene from seven maize individuals, eight parviglumis individuals, and six Z. luxurians individuals (Table 1). Three alleles from T. dactyloides were also sequenced. The individuals were chosen both to represent a fairly large geographic area and to overlap with the individuals studied in Eyre-Walker et al. (1998). The glb1 gene was amplified with Taq polymerase using primers specific to the AGGA element of the promoter and to the third exon (Figure 1). The DNA sequence for the forward primer was 5′ CCGGATAAGCACGGTAAGGA 3′, and the sequence for the reverse primer was 5′ CTTGCTGAAGCTCGACAGGA 3′. PCR consisted of 32 cycles of 1 min at 94°, 1 min at 65° and 2 min at 72°. Amplified products were cloned into pGEM-T and sequenced with T7 polymerase, using several primers internal to the glb1 gene. The sequence reactions were analyzed on
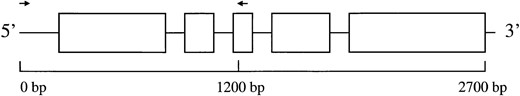
—A schematic diagram of the glb1 locus and the region sequenced for this study. Open boxes represent exons, and the primers used for PCR amplification are represented by arrows.
an ALFexpress automated sequencer (Pharmacia, Piscataway, NJ). Sequence alignment was done manually.
Many of the glb1 sequences contained “singletons,” a single basepair change relative to the remainder of the sequences. Because we used a cloning technique that isolates a single copy of a single allele, singletons can either represent true sequence variation or Taq polymerase artifact. (In contrast, polymorphisms shared among more than one sequence have a negligible probability of being produced by Taq polymerase error.) The singletons were double checked by reamplifying and resequencing the appropriate glb1 allele from all Zea individuals, except parviglumis line 384,064, whose DNA was degraded on reamplification. Most individuals were heterozygous, but determining the appropriate allele was straighforward because of high amounts of variation within taxa. We found that 29% (18 of 62) of the original singletons were the result of Taq polymerase error, and these singletons were corrected. This method of error verification also helped ensure that our sequences were not interallelic PCR recombinants (e.g., Bradley and Hillis 1997). One PCR recombinant was found and removed from the data set.
Sequence analysis: To summarize genetic diversity within Zea, we calculated both , the average pairwise difference between sequences (Tajima 1983), and , Watterson's estimator of θ (Watterson 1975). Both have expected values of 4Nμ, where N is the population size and μ is the mutation rate per locus per generation. To estimate the divergence time and the ancestral θ between species, we used the isolation model of Wakeley and Hey (1997), as modified by Wang et al. (1997), for combined adh1 and glb1 data. Estimation of θ and π was based on all sites, but divergence dates were based on only silent sites (defined here as intron and third position sites). The dates of divergence between species were estimated under the assumption that the mutation rate is 6.5 × 10–9 mutations per silent site per annual generation, which is equal to the average rate of substitutions per synonymous site per year at adh1 and adh2 in grasses (Gaut et al. 1996). Sequence analysis was performed, in part, with the program SITES (Hey and Wakeley 1997). We tested for deviation from the neutral equilibrium model of evolution using the tests of Tajima (1989), McDonald and Kreitman (1991), and Hudson et al. (1987; the HKA test). The HKA testwas modified slightly from the original, by using the average divergence between taxa when more than one line was sequenced in both taxa. Rates of recombination were estimated with the methods of Hudson and Kaplan (1985) and Hey and Wakeley (1997). Phylogenetic reconstruction was performed with the neighbor-joining method (Saitou and Nei 1987) with Kimura two-parameter distances (Kimura 1980), as implemented in PHYLIP 3.5 (Felsenstein 1990). One-thousand bootstrap replicates were used to assess confidence in the phylogeny. All nucleotide sites were used for investigation of phylogeny, recombination, and selection.
Coalescent simulations: We used DNA sequence data from maize and parviglumis to estimate Nb, the bottleneck population size during maize domestication. We followed the methods detailed in Eyre-Walker et al. (1998) and used coalescent model II as described in that study. For this coalescent model, estimation of Nb was conditioned on the number of shared polymorphisms between maize and parviglumis. Estimation of Nb was also conditioned upon estimates of θ from the progenitor (in this case parviglumis), the current θ of the domesticate, a time of domestication of 7500 YBP, and a mutation rate of 6.5 × 10–9 per silent site per annual generation. Under the assumption that maize went through a bottleneck (and therefore has not evolved in accordance with a neutral equilibrium model), the current θ of maize cannot be estimated easily from DNA sequence data. The study of Eyre-Walker et al. (1998), however, found that estimation of Nb was relatively insensitive to assumed values of the current θ of maize. Here we use from parviglumis to represent the current θ of maize, but we must accentuate that the estimation of Nb varies little over a wide range of values for the current θ of maize. [Readers are directed to Eyre-Walker et al. (1998) for greater detail.]
RESULTS
DNA sequence variation and test for deviation from neutrality: Table 2 provides a summary of the glb1 sequence data in terms of the number of sequences, the number of polymorphic sites, and the number of insertion-deletion events within each taxon. (A full table of the polymorphic sites is available from the authors.) Table 3 provides two estimates of variation: per base pair and per base pair. These estimates of variation indicate that parviglumis contains a very high level of variation relative to maize and Z. luxurians. As calculated by relative values of per base pair, maize contains ∼60% of the amount of variation found in parviglumis, while Z. luxurians contains ∼32% of the amount of variation found in parviglumis. Most sequence haplotypeswere unique; only two haplotypes were found more than once (one haplotype was shared between Coni and Parv5, and one haplotype was shared between Lux1 and Lux8; see Table 1 for abbreviations).
Measures of variation can be used to examine the history of natural selection. and have the same expected value, but is more greatly influenced by low-frequency polymorphisms. Tajima's D measures the discrepancy between thesetwo measures (Tajima 1989). Estimates of D are negative in maize and parviglumis and almost zero in Z. luxurians (Table 3). None of these values differ significantly from zero, and neutrality cannot be rejected. However, it should be noted that statistical power to detect deviation from neutrality with Tajima's D may be low with the sample sizes used here (Simonsen et al. 1995).
Selection can also be examined by intertaxon comparisons of nucleotide substitutions. We applied the McDonald and Kreitman (1991) test of selection to Zea glb1 data and found no evidence of selection across any pair of taxa (maize-Z. luxurians, χ2 = 0.18, P = 0.68; parviglumis-Z. luxurians, χ2 = 0.00, P = 0.98; maize-T. dactyloides, χ2 = 1.41, P = 0.24; parviglumis-T. dactyloides χ2 = 1.45, P = 0.23; Z. luxurians-T. dactyloides, χ2 = 0.05, P = 0.83). Maize cannot be contrasted to parviglumis with the McDonald-Kreitman test because there were zero fixed differences between the two taxa.
The amount of sequence variation in glb1 is higher than in adh1 (Table 3; Eyre-Walker et al. 1998), probably in part because of the relatively high amount of amino-acid-replacing polymorphisms in glb1 (see below). Levels of variation in these two loci were examined for deviation from neutral equilibrium evolution using the HKA test (Hudson et al. 1987). The HKA test did not reject the neutral model in any comparison (maize-Z. luxurians,
The number of polymorphic sites within species
| Promoter and 5′ leader | Exons | Introns | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| n | Subs | Indels | Avg. length | Syn. | Rep. | Indels | Avg. length | Subs | Indels | Avg. length | |
| Maize | 9 | 6 | 6 | 181 | 18 | 19 | 4 | 756 | 13 | 9 | 208 |
| Parviglumis | 8 | 13 | 11 | 181 | 28 | 26 | 5 | 755 | 22 | 10 | 200 |
| Z. luxurians | 6 | 8 | 5 | 180 | 4 | 7 | 0 | 756 | 6 | 2 | 195 |
| Zeaa | 23 | 20 | 17 | 181 | 42 | 54 | 6 | 756 | 34 | 17 | 195 |
| T. dactyloides | 3 | 10 | 5 | 199 | 4 | 11 | 1 | 762 | 1 | 2 | 192 |
| Promoter and 5′ leader | Exons | Introns | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| n | Subs | Indels | Avg. length | Syn. | Rep. | Indels | Avg. length | Subs | Indels | Avg. length | |
| Maize | 9 | 6 | 6 | 181 | 18 | 19 | 4 | 756 | 13 | 9 | 208 |
| Parviglumis | 8 | 13 | 11 | 181 | 28 | 26 | 5 | 755 | 22 | 10 | 200 |
| Z. luxurians | 6 | 8 | 5 | 180 | 4 | 7 | 0 | 756 | 6 | 2 | 195 |
| Zeaa | 23 | 20 | 17 | 181 | 42 | 54 | 6 | 756 | 34 | 17 | 195 |
| T. dactyloides | 3 | 10 | 5 | 199 | 4 | 11 | 1 | 762 | 1 | 2 | 192 |
n, the number of lines examined in each taxa; Subs, the number of base substitutions at the sequence level; Indels, the number of insertion-deletion events; Avg. length, the average sequence length of the lines (these lengths vary slightly among lines because of indel variation); Syn., the number of synonymous polymorphisms in exons; Rep., the number of replacement or nonsynonymous polymorphisms in exons.
The group Zea refers to all sequences from maize, parviglumis, and Z. luxurians.
The number of polymorphic sites within species
| Promoter and 5′ leader | Exons | Introns | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| n | Subs | Indels | Avg. length | Syn. | Rep. | Indels | Avg. length | Subs | Indels | Avg. length | |
| Maize | 9 | 6 | 6 | 181 | 18 | 19 | 4 | 756 | 13 | 9 | 208 |
| Parviglumis | 8 | 13 | 11 | 181 | 28 | 26 | 5 | 755 | 22 | 10 | 200 |
| Z. luxurians | 6 | 8 | 5 | 180 | 4 | 7 | 0 | 756 | 6 | 2 | 195 |
| Zeaa | 23 | 20 | 17 | 181 | 42 | 54 | 6 | 756 | 34 | 17 | 195 |
| T. dactyloides | 3 | 10 | 5 | 199 | 4 | 11 | 1 | 762 | 1 | 2 | 192 |
| Promoter and 5′ leader | Exons | Introns | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| n | Subs | Indels | Avg. length | Syn. | Rep. | Indels | Avg. length | Subs | Indels | Avg. length | |
| Maize | 9 | 6 | 6 | 181 | 18 | 19 | 4 | 756 | 13 | 9 | 208 |
| Parviglumis | 8 | 13 | 11 | 181 | 28 | 26 | 5 | 755 | 22 | 10 | 200 |
| Z. luxurians | 6 | 8 | 5 | 180 | 4 | 7 | 0 | 756 | 6 | 2 | 195 |
| Zeaa | 23 | 20 | 17 | 181 | 42 | 54 | 6 | 756 | 34 | 17 | 195 |
| T. dactyloides | 3 | 10 | 5 | 199 | 4 | 11 | 1 | 762 | 1 | 2 | 192 |
n, the number of lines examined in each taxa; Subs, the number of base substitutions at the sequence level; Indels, the number of insertion-deletion events; Avg. length, the average sequence length of the lines (these lengths vary slightly among lines because of indel variation); Syn., the number of synonymous polymorphisms in exons; Rep., the number of replacement or nonsynonymous polymorphisms in exons.
The group Zea refers to all sequences from maize, parviglumis, and Z. luxurians.
χ2 = 0.031, P = 0.86; parviglumis-Z. luxurians, χ2 = 0.139, P = 0.71; maize-T. dactyloides, χ2 = 0.032, P = 0.85; Z. luxurians-T. dactyloides, χ2 = 0.023, P = 0.88; parviglumis-T dactyloides, χ2 = 0.002, P = 0.96). Overall, tests for selection do not detect any deviation from neutral equilibrium evolution at the glb1 locus.
The footprint of selection can be more difficult to detect in regions of high recombination. We estimated the minimum number of recombination events within the sample of maize glb1 sequences by the method of Hudson and Kaplan (1985). By this method, the maize sample contains a minimum of seven recombination events, the parviglumis sample contains nine recombination events, and the Z. luxurians sample contains one event. We also estimated the quantity c/μ, where c is the recombination rate per generation per base pair, and μ is the mutation rate per base pair (Hey and Wakeley 1997). For maize and parviglumis, c/μ was 1.8 and 2.1, respectively, suggesting recombination at a rate twice that of mutation. Z. luxurians had a lower estimate of 0.3. In general, recombination appears to be common in the glb1 locus, although either recombination or our ability to detect recombination is somewhat reduced in Z. luxurians relative to the other Zea taxa.
Summary of DNA sequence variation glb1 and adh1
| glb1 | adh1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| n | S | D | n | S | D | |||||
| Maize | 9 | 56 | 0.0173 | 0.0189 | –0.5284 | 9 | 50 | 0.0136 | 0.01491 | 0.2835 |
| Parviglumis | 8 | 89 | 0.0259 | 0.0319 | –1.1102 | 8 | 64 | 0.0195 | 0.01791 | –0.4464 |
| Z. luxurians | 6 | 25 | 0.0102 | 0.0101 | 0.0678 | 7 | 26 | 0.0077 | 0.0081 | 0.0386 |
| T. dactyloides | 3 | 26 | 0.0146 | 0.0158 | NA | NA | NA | NA | NA | NA |
| glb1 | adh1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| n | S | D | n | S | D | |||||
| Maize | 9 | 56 | 0.0173 | 0.0189 | –0.5284 | 9 | 50 | 0.0136 | 0.01491 | 0.2835 |
| Parviglumis | 8 | 89 | 0.0259 | 0.0319 | –1.1102 | 8 | 64 | 0.0195 | 0.01791 | –0.4464 |
| Z. luxurians | 6 | 25 | 0.0102 | 0.0101 | 0.0678 | 7 | 26 | 0.0077 | 0.0081 | 0.0386 |
| T. dactyloides | 3 | 26 | 0.0146 | 0.0158 | NA | NA | NA | NA | NA | NA |
Estimates are based on all nucleotide sites. The last three columns are from the adh1 data of Eyre-Walker et al. (1998). n, the number of sequences; S, the number of segregating sites; D, Tajima's D; NA, not available.
Summary of DNA sequence variation glb1 and adh1
| glb1 | adh1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| n | S | D | n | S | D | |||||
| Maize | 9 | 56 | 0.0173 | 0.0189 | –0.5284 | 9 | 50 | 0.0136 | 0.01491 | 0.2835 |
| Parviglumis | 8 | 89 | 0.0259 | 0.0319 | –1.1102 | 8 | 64 | 0.0195 | 0.01791 | –0.4464 |
| Z. luxurians | 6 | 25 | 0.0102 | 0.0101 | 0.0678 | 7 | 26 | 0.0077 | 0.0081 | 0.0386 |
| T. dactyloides | 3 | 26 | 0.0146 | 0.0158 | NA | NA | NA | NA | NA | NA |
| glb1 | adh1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| n | S | D | n | S | D | |||||
| Maize | 9 | 56 | 0.0173 | 0.0189 | –0.5284 | 9 | 50 | 0.0136 | 0.01491 | 0.2835 |
| Parviglumis | 8 | 89 | 0.0259 | 0.0319 | –1.1102 | 8 | 64 | 0.0195 | 0.01791 | –0.4464 |
| Z. luxurians | 6 | 25 | 0.0102 | 0.0101 | 0.0678 | 7 | 26 | 0.0077 | 0.0081 | 0.0386 |
| T. dactyloides | 3 | 26 | 0.0146 | 0.0158 | NA | NA | NA | NA | NA | NA |
Estimates are based on all nucleotide sites. The last three columns are from the adh1 data of Eyre-Walker et al. (1998). n, the number of sequences; S, the number of segregating sites; D, Tajima's D; NA, not available.
Molecular evolution of the glb1 gene: Within exons, the glb1 data contain more amino acid-replacing polymorphisms than synonymous polymorphisms. For example, we found 54 replacement changes compared to 42 synonymous changes within exons of the Zea sequences (Table 2). This is a relatively high proportion for genes from these taxa. In contrast, the ratio of replacement to synonymous polymorphism in Zea sequences was 2:34 in adh1 (Eyre-Walker et al. 1998) and 2:3 in c1 (Hanson et al. 1996). To confirm that this high rate of replacements was neither an artifact nor a population level phenomenon, one line of maize, parviglumis, Z. luxurians, and Tripsacum were compared individually to the glb1 sequence of barley (Heck et al. 1993). In 390 bases of alignable coding region, there were, on average, 37 replacement changes compared with 21 synonymous changes. This result indicates that the relatively high ratio of replacement changes to synonymous changes is not limited to our Zea data.
The high proportion of replacement polymorphisms in glb1 sequences raises the question as to whether gbl1 is evolving without selective constraint on amino acid replacements. If there is no constraint, the ratio of the number of substitutions per nonsynonymous nucleotide site to the number of substitutions per synonymous nucleotide
Shared polymorphisms and fixed differences at glb1 and adh1
| Fixed differences | Shared polymorphisms | |||
|---|---|---|---|---|
| glb1 | adh1a | glb1 | adh1 | |
| Maize-parviglumis | 0 | 0 | 34 | 35 |
| Maize-Z. luxurians | 4 | 5 | 4 | 9 |
| Parviglumis-Z. luxurians | 2 | 6 | 3 | 11 |
| Zea-Tripsacumb | 77 | 53 | 0 | 0 |
| Fixed differences | Shared polymorphisms | |||
|---|---|---|---|---|
| glb1 | adh1a | glb1 | adh1 | |
| Maize-parviglumis | 0 | 0 | 34 | 35 |
| Maize-Z. luxurians | 4 | 5 | 4 | 9 |
| Parviglumis-Z. luxurians | 2 | 6 | 3 | 11 |
| Zea-Tripsacumb | 77 | 53 | 0 | 0 |
The adh1 data are from Eyre-Walker et al. (1998).
Numbers in this row refer to the average between each of the Zea taxa and T. dactyloides.
Shared polymorphisms and fixed differences at glb1 and adh1
| Fixed differences | Shared polymorphisms | |||
|---|---|---|---|---|
| glb1 | adh1a | glb1 | adh1 | |
| Maize-parviglumis | 0 | 0 | 34 | 35 |
| Maize-Z. luxurians | 4 | 5 | 4 | 9 |
| Parviglumis-Z. luxurians | 2 | 6 | 3 | 11 |
| Zea-Tripsacumb | 77 | 53 | 0 | 0 |
| Fixed differences | Shared polymorphisms | |||
|---|---|---|---|---|
| glb1 | adh1a | glb1 | adh1 | |
| Maize-parviglumis | 0 | 0 | 34 | 35 |
| Maize-Z. luxurians | 4 | 5 | 4 | 9 |
| Parviglumis-Z. luxurians | 2 | 6 | 3 | 11 |
| Zea-Tripsacumb | 77 | 53 | 0 | 0 |
The adh1 data are from Eyre-Walker et al. (1998).
Numbers in this row refer to the average between each of the Zea taxa and T. dactyloides.
site should not differ significantly from 1.0 (Hughes and Nei 1988; Zhang et al. 1998). We used the distance measure of Nei and Gojobori (1986) to estimate this ratio between the barley glb1 sequence and a glb1 sequence from each of the Zea taxa. The ratio varied from 0.48 to 0.53, and this was significantly less than 1.0 in all cases [data not shown; the G-test was used to test the hypothesis that the ratio is equal to 1.0, per Zhang et al. (1998)]. Thus, amino acid replacements in the glb1 gene are subject to some selective constraint, but this constraint appears to be somewhat low.
In elite lines of maize, glb1 is rich in glutamate, glutamine, and arginine (Kriz 1989; Belanger and Kriz 1991). Over the three Zea taxa examined here, we found that ∼25% of codons encode Gln, Glu, or Arg, and that this percentage does not differ among either Zea taxa (range 24.2-24.8%) or between Zea taxa and the glb1-hb and glb1-s sequences from elite maize lines. The glb1 gene is also GC rich—the GC content is 64% over all sites and >90% at third position sites.
Genetic relationships among taxa based on sequence polymorphisms: We used glb1 sequences to assess genetic relationships among the four taxa—maize, parviglumis, Z. luxurians, and T. dactyloides. We assessed relationships in two ways: (1) we compared the numbers of shared polymorphisms and fixed differences between taxa and (2) we built a gene tree of glb1 sequences.
A fixed difference is a site where all the individuals in one taxon have one base and all the individuals in a second taxon have a different base (Hey 1991). The accumulation of fixed differences between taxa reveals that they do not share genetic drift and are, therefore, evolving independently. A shared polymorphism occurs when two taxa have the same two bases segregating at the same site. Shared polymorphisms reveal a history of polymorphism that has not yet been erased by genetic drift; they reflect either a short divergence time between taxa or historically large population sizes.
The numbers of fixed differences and shared polymorphisms for both glb1 and adh1 are listed in Table 4. The average of the three Zea taxa to T. dactyloides is listed in the last row. Maize and parviglumis have no fixed differences and a large amount of shared polymorphism. This is consistent with very recent (or no) divergence between the taxa. In contrast, there are many fixed differences and no shared polymorphisms between the Zea taxa and T. dactyloides, indicating that the taxa are genetically distinct. On the basis of net divergence (Nei 1987) at silent sites between Zea and T. dactyloides gbl1 sequences, we estimate that the two genera diverged roughly 4.8 mya. Similarly, sequence data from the adh1 locus suggest the two genera diverged 4.5 mya.
There are several fixed differences between Z. luxurians and both maize and parviglumis, but Z. luxurians still shares polymorphisms with these two taxa (Table 4). The presence of both fixed differences and shared polymorphisms can be found only in loci with recombination or in comparisons of multiple unlinked loci (Wakeley and Hey 1997). The fixed differences reveal that there has been some level of divergence between Z. luxurians and both maize and parviglumis, but the presence of shared polymorphisms reveals that genetic drift has not been so strong that it erased all the variation that existed in the common ancestor of the three taxa. Incidentally, the ratio of fixed to shared polymorphisms among taxa does not differ significantly between adh1 and glb1 (data not shown), which suggests that these loci provide similar information about the genetic history of these taxa.
Wakeley and Hey (1997) devised a method to estimate both the ancestral population size and the speciation time for species that share polymorphisms and have fixed differences between them. Using the modifications of Wang et al. (1997), we combined the data from glb1 and adh1 to estimate the population parameters listed in Table 5. Note that we could not contrast maize to parviglumis because of a lack of fixed differences. We compare maize and Z. luxurians because the ancestor of these two species should be the same as the ancestor of parviglumis and Z. luxurians. However, the domestication of maize almost certainly violates the constant population size assumptions of the Wakeley and Hey (1997) model, so results based on maize and Z. luxurians should be treated with caution.
The ancestral θ is estimated to be ∼40 for both comparisons (Table 5). This estimate is lower than the estimated θ of parviglumis based on this model (Table 5), suggesting that parviglumis may have undergone a population expansion after its divergence from Z. luxurians. Estimates of Tajima's D for glb1 and adh1 (Eyre-Walker et al. 1998) are both negative for parviglumis and, therefore, consistent with an expanding population size for parviglumis (Table 3). In contrast, the ancestral θ is estimated to be larger than the current size of Z. luxurians, which is consistent with the positive value of Tajima's D for both glb1 and adh1.
The model of Wakeley and Hey (1997) also facilitates
Estimates of ancestral population parameters using combined gbl1 and adh1 data
| Species pair | Divergence time (yr) | |||
|---|---|---|---|---|
| Parviglumis-Z. luxurians | 111.4 | 15.7 | 43.1 | 700,000 |
| Maize-Z. luxurians | 36.2 | 14.5 | 40.3 | 630,000 |
| Species pair | Divergence time (yr) | |||
|---|---|---|---|---|
| Parviglumis-Z. luxurians | 111.4 | 15.7 | 43.1 | 700,000 |
| Maize-Z. luxurians | 36.2 | 14.5 | 40.3 | 630,000 |
, the estimate of the population parameter for the first species listed, i.e., parviglumis or maize; , the estimate for Z. luxurians; , the estimate for the ancestral population of the two taxa.
Estimates of ancestral population parameters using combined gbl1 and adh1 data
| Species pair | Divergence time (yr) | |||
|---|---|---|---|---|
| Parviglumis-Z. luxurians | 111.4 | 15.7 | 43.1 | 700,000 |
| Maize-Z. luxurians | 36.2 | 14.5 | 40.3 | 630,000 |
| Species pair | Divergence time (yr) | |||
|---|---|---|---|---|
| Parviglumis-Z. luxurians | 111.4 | 15.7 | 43.1 | 700,000 |
| Maize-Z. luxurians | 36.2 | 14.5 | 40.3 | 630,000 |
, the estimate of the population parameter for the first species listed, i.e., parviglumis or maize; , the estimate for Z. luxurians; , the estimate for the ancestral population of the two taxa.
estimation of divergence times between species. Using only silent sites to estimate divergence times, we estimate that Z. luxurians diverged from parviglumis and maize roughly 630,000-700,000 years ago (Table 5).
In addition to examining the distribution of fixed and shared differences, we also assessed relationships among species by estimating the genealogy of sequences. Figure 2 diagrams the estimated gene genealogy for glb1 and provides an adh1 genealogy for comparison (Eyre-Walker et al. 1998). Four features of these two trees are remarkably consistent. First, sequences from T. dactyloides form a distinct outgroup, which is consistent with the estimated divergence time of 4.5-4.8 myr between genera. Second, Z. luxurians sequences form a distinct monophyletic group that is supported by high bootstrap values in both trees (93% in glb1 and 99% in adh1). The monophyly of Z. luxurians sequences is consistent with the accumulation of fixed differences between Z. luxurians and other taxa. Third, some allelic lineages of parviglumis are basal to the Z. luxurians group. This last observation suggests that parviglumis allelic lineages date to the common ancestor of the
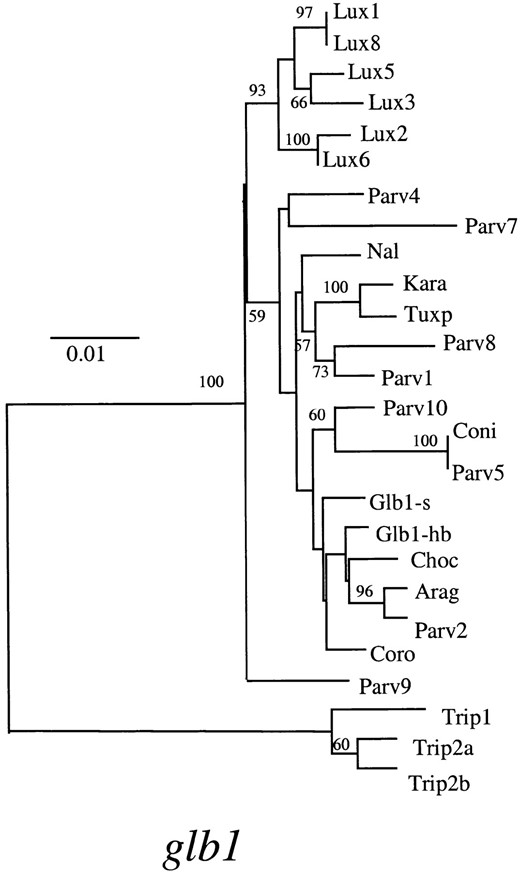
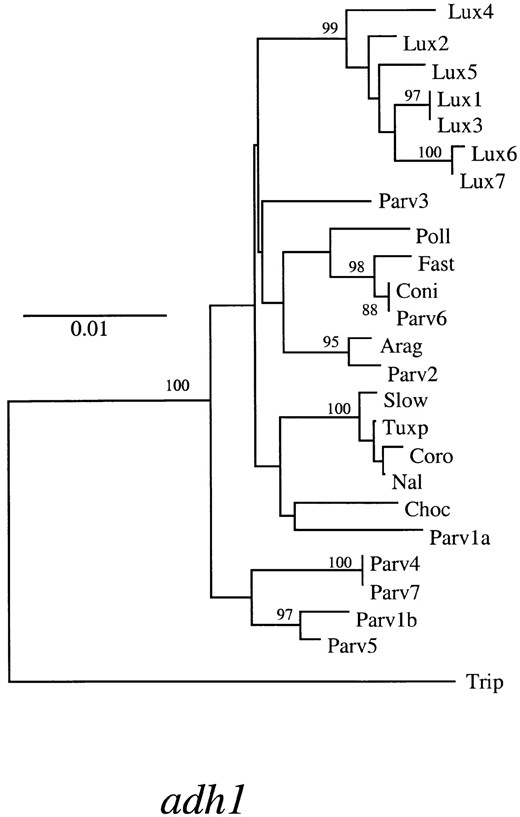
—Neighbor-joining distance tree for glb1 sequences (this study) and adh1 sequences (Eyre-Walker et al. 1998). Bootstrap values >50% are given, and scale bars denote levels of sequence divergence. Labels on the two trees coincide by individual, e.g., the sequence Parv4 on the glb1 tree was isolated from the same parviglumis individual as the Parv4 adh1 sequence.
three Zea taxa; this suggestion is corroborated by high nucleotide diversity in parviglumis relative to maize and Z. luxurians (Table 3). Finally, maize sequences do not form a distinct group. Rather, the maize and parviglumis sequences are intermixed. This intermixing is not unexpected, however, because maize was derived from parviglumis relatively recently.
Coalescent simulations and the domestication of maize: The model of Wakeley and Hey (1997) assumes constant population sizes after the divergence of species. Although we have applied this model to comparisons between maize and Z. luxurians, it is important to remember that maize probably experienced a population bottleneck at the time of its domestication from parviglumis. In a previous study of adh1, Eyre-Walker et al. (1998) estimated Nb, the size of the population during a bottleneck associated with domestication, as a function of the duration of the bottleneck. Here we estimate Nb using both glb1 data separately and glb1 and adh1 data combined. The purpose of estimating Nb is to try to gain some insight into the number of individuals that initially contributed to the maize gene pool.
Estimates of Nb are given in Table 6. Four points should be made about these estimates. First, the glb1 data are similar to the adh1 data in indicating that maize could have been founded by a very small population. For example, if the population bottleneck associated with domestication were only 10 generations in length, the glb1 data suggest that about seven parviglumis individuals comprise enough sequence diversity at the glb1 locus to explain the diversity currently found in maize (Table 6). Second, Nb increases with increasing duration of the bottleneck. The maximum duration of a bottleneck associated with domestication, based on fossil evidence, is 2800 generations (Eyre-Walker et al. 1998). For this duration, the estimate of Nb based on gbl1 data is roughly 3000 individuals. Third, the estimates based on combined data fall between estimates based on adh1 and glb1. These are probably the best estimates because they are based on the most data. Finally, 95% confidence intervals on these estimates are quite large (see Eyre-Walker et al. 1998). Thus, on the basis of these estimates, we cannot (1) conclude that the two genes are providing incongruent information about Nb or (2) reject scenarios of maize domestication that include either very large Nb or substantial introgression between maize and other wild relatives. Nonetheless, these data indicate that a founding population of very few parviglumis individuals could capture the breadth of genetic diversity found in adh1 and glb1 of maize.
DISCUSSION
We have sampled a 1.2-kb region of the glb1 locus from 26 individuals representing maize, parviglumis, Z. luxurians, and T. dactyloides. It does not appear that the pattern of variation in this region of the glb1 locus has been strongly affected by natural selection, based on several lines of evidence. First, neither Tajima's D nor the McDonald-Kreitman test detect any deviation from neutrality. Also, recombination has occurred at glb1, reducing the length of tightly linked nucleotide sites, which in turn decreases the probability that any particular portion of the sequence is tightly linked to a site under selection (Maynard Smith and Haigh 1974). Finally, we combined our glb1 data with similar data
Estimates of Nb, the size of a bottlenecked population during maize domestication
Duration of the bottleneck, in generations.
Estimates based on data from both glb1 and adh1.
Estimates of Nb, the size of a bottlenecked population during maize domestication
Duration of the bottleneck, in generations.
Estimates based on data from both glb1 and adh1.
from the adh1 locus and performed HKA tests, which did not detect deviation from neutrality. Altogether, tests of neutrality suggest that glb1 and adh1 were not under strong selection during speciation or domestication, which makes them useful markers for exploring the population history of the genus Zea.
While the pattern of standing sequence variation is consistent with neutral equilibrium evolution, the glb1 gene is not evolving without selective constraint on amino acid replacements. However, the ratio of replacement polymorphisms to silent polymorphisms is high in this gene relative to other genes, such as adh1. This suggests that constraint on amino acid replacements is generally low, an observation that is consistent with both the nonenzymatic function of the gene and the fact that the product of glb1 is not essential for seedling survival (Schwartz 1979). The glb1 gene also contains a high number of codons for glutamate, glutamine, and arginine. These amino acids likely provide a source of nitrogen for the germinating seed (Kriz 1989; Belanger and Kriz 1991). We detect no obvious change in the proportion of these amino acid residues between the domesticate and its wild relatives.
Inferences about wild taxa: We have studied the glb1 locus primarily to gain insight into the evolution of the genus Zea. Several insights can be gleaned from examination of the level and type of sequence variation within and between taxa. For example, information about the relationship between Zea and T. dactyloides is consistent over both glb1 and adh1. Neither gene contains shared polymorphisms between Zea and T. dactyloides (Table 4), and sequences from the two different genera form robust, distinct clades (Figure 2). We estimate the divergence of these two genera at ∼4.5-4.8 mya.
This estimate is interesting for three reasons. First, this is one of few estimates of the time of divergence between sister plant genera. Second, the genus Zea has been described as “relatively young” on the basis of limited plastid genotype diversity within the genus (Larson and Doebley 1994); we can now estimate “young” at ∼5 myr. Finally, Eubanks (1997) has resurrected the theory that modern maize arose from a hybridization event between T. dactyloides and a perennial Zea species. Under this hybridization theory, maize sequences should owe their origin to both a Zea taxon and T. dactyloides. However, maize sequences neither share polymorphisms with T. dactyloides sequences (Table 4) nor cluster with T. dactyloides sequences (Figure 2). These observations are not consistent with a hybrid origin, and thus neither glb1 nor adh1 data suggest that maize arose from a hybridization event involving T. dactyloides.
The two wild Zea taxa have different levels of sequence diversity. Parviglumis is quite variable at the DNA sequence level; it has an average level of DNA sequence variation that exceeds that of other plants (Gaut and Clegg 1993; Innan et al. 1996; Miyashita et al. 1996), humans (Hey 1997), and most Drosophila species (Moriyama and Powell 1996). In contrast, Z. luxurians contains roughly one-third the level of genetic diversity of parviglumis (Table 3). Sequence data from glb1 and adh1 suggest that genetic divergence between these taxa has occurred over the last 630,000-700,000 years. Parviglumis may have undergone a population expansion since the divergence of these species, but Z. luxurians has probably experienced a reduction in population size (Table 5). Yet, these two taxa are similar morphologically (Doebley and Iltis 1980). Taken together, these observations suggest that morphological divergence between these species has been a gradual process occurring over hundreds of thousands of years.
Our estimate of the divergence time between parviglumis and Z. luxurians is much larger than the estimate of 135,000 years, based on isozyme data (Hanson et al. 1996). The latter estimate is based on potentially inaccurate assumptions about isozyme mutation rates, but it is also based on more loci. This raises an important issue: it is possible that different loci provide very different information about relationships among Zea species. For example, despite the obvious sequence divergence between parviglumis and Z. luxurians at glb1 and adh1, the limited sequence data available from both adh2 (Goloubinoff et al. 1993) and c1 (Hanson et al. 1996) suggest that there may be relatively little divergence between these taxa at these loci. At this time, it is not clear which divergence date more accurately reflects an average divergence time between species for the whole genome, but the date of 630,000-700,000 years is almost certainly more accurate for adh1 and glb1. We are currently gathering sequence polymorphism data from additional loci, with the hope of better understanding how the process of divergence varies among loci.
Drosophila is the only other system in which sequence diversity has been measured from multiple loci of closely related taxa. There are striking parallels between Zea and Drosophila, in that the relationship between parviglumis and Z. luxurians appears to be similar to the relationship between two species in the melanogaster group: Drosophila simulans and D. mauritiana. First, like parviglumis and Z. luxurians, the two Drosophila taxa differ in geographic distribution: D. simulans has a large range in eastern Africa, while D. mauritiana is largely restricted to the island of Mauritius (Lachaise et al. 1988). Second, D. simulans, the taxon with greater range, contains more sequence diversity (Kliman and Hey 1993). Third, the two species contain both shared polymorphisms and fixed differences, but the D. mauritiana sequences form a monophyletic group within D. simulans sequences. Finally, the effective population size of the common ancestor of the two Drosophila species was estimated to be intermediate to that of its descendants (Wakeley and Hey 1997). Hey and Kliman (1993) concluded that D. mauritiana arose from an ancestral species that was more like modern D. simulans. Similarly, Z. luxurians may have arisen from an ancestral species that was more like extant parviglumis.
Domestication: Our data do not permit an explicit test of the hypothesis that parviglumis is the progenitor to maize, but the data are consistent with a domesticate/progenitor relationship for three reasons. First, there are no fixed differences between maize and parviglumis, suggesting a recent divergence between taxa. Second, gene trees reveal that the maize lineages are intermixed with a subset of the parviglumis lines (Figure 2). Finally, maize contains 71% the level of variation of parviglumis over both adh1 and glb1; this reduction of variation can be interpreted as consistent with a domestication event.
The domestication of maize was evolutionarily recent; it occurred ∼7500 years ago (Iltis 1983). Despite the recent timing of domestication, maize is morphologically distinct from all members of the genus Zea, including parviglumis. In fact, the wild progenitor of maize remained a mystery throughout much of this century and was identified only after the application of molecular techniques (Doebley et al. 1984, 1987b). The morphological differences between maize and its progenitor suggest that maize probably experienced a “domestication bottleneck” resulting from selection for agronomically important traits.
We used simulation of the coalescent process to explore population sizes of the domestication bottleneck (Table 6). Our results corroborate the results of Eyre-Walker et al. (1998) in indicating that the founding population is dependent on the duration of the bottleneck, but that the population could have been quite small. For example, if domestication were completed in 10 years, joint consideration of adh1 and glb1 data indicate that the domesticate could have been based on a population of ∼10 wild individuals (Table 6). Similarly, if there were a domestication bottleneck that lasted 100 years, then the bottleneck size would correspond to 104 individuals. Unfortunately, there are no good independent estimates for the true duration of the original domestication event. Furthermore, the coalescent model is a simplistic representation of the domestication process. For example, it cannot establish whether introgression has occurred on a wide scale after domestication [see Eyre-Walker et al. (1998) for further discussion]. Nonetheless, the simulations indicate that a surprising amount of genetic variation can be retained through severe founder events [as originally demonstrated by Nei et al. (1975)].
The domestication of maize is a sharp contrast to divergence between parviglumis and Z. luxurians. Domestication led to rapid morphological divergence with the retention of high levels of genetic diversity. In contrast, divergence between parviglumis and Z. luxurians has been a slow process comprising at least 100,000 years, has been accompanied by a gradual loss of genetic diversity in Z. luxurians, but has led to relatively little morphological divergence.
It is commonly thought that crops are bereft of genetic variation compared to their wild relatives (Tanksley and McCouch 1997). In the two apparently neutral genes glb1 and adh1, maize contains 60 and 83%, respectively, of the amount of sequence diversity found in its presumed progenitor. Pearl millet is the only other crop system in which the contrast between domesticate and progenitor has been made at the sequence level, and domesticated millet contains 67% of the amount of sequence diversity found in its wild progenitor (Gaut and Clegg 1993). Taken together, these studies indicate that the domesticates contain a substantial proportion of the genetic diversity found in their wild relatives. It will be interesting to examine additional crop systems to determine whether crops have retained more genetic variation through founder events than is commonly believed.
Footnotes
Communicating editor: A. G. Clark
Acknowledgement
The authors are grateful to J. Wakeley, J. Hey, A. Eyre-Walker, J. Doebley, C. DeWald and F. Belanger for assistance and discussion and to A. Clark and an anonymous reviewer for comments. This work was supported by U.S. Department of Agriculture grant 95-37301 to B.S.G.
LITERATURE CITED

{kind=link}
{kind=link}